Bacterial Responses and Genome Instability Induced by Subinhibitory Concentrations of Antibiotics


Abstract
:1. Introduction
2. Antibiotics: Killing or Signaling Molecules?
3. Bacterial Responses to Subinhibitory Concentrations of Antibiotics
4. Subinhibitory Concentrations of Antibiotics Enhance Horizontal Gene Transfer
5. Increasing Mutation Rates in Response to Subinhibitory Concentrations of Antibiotics
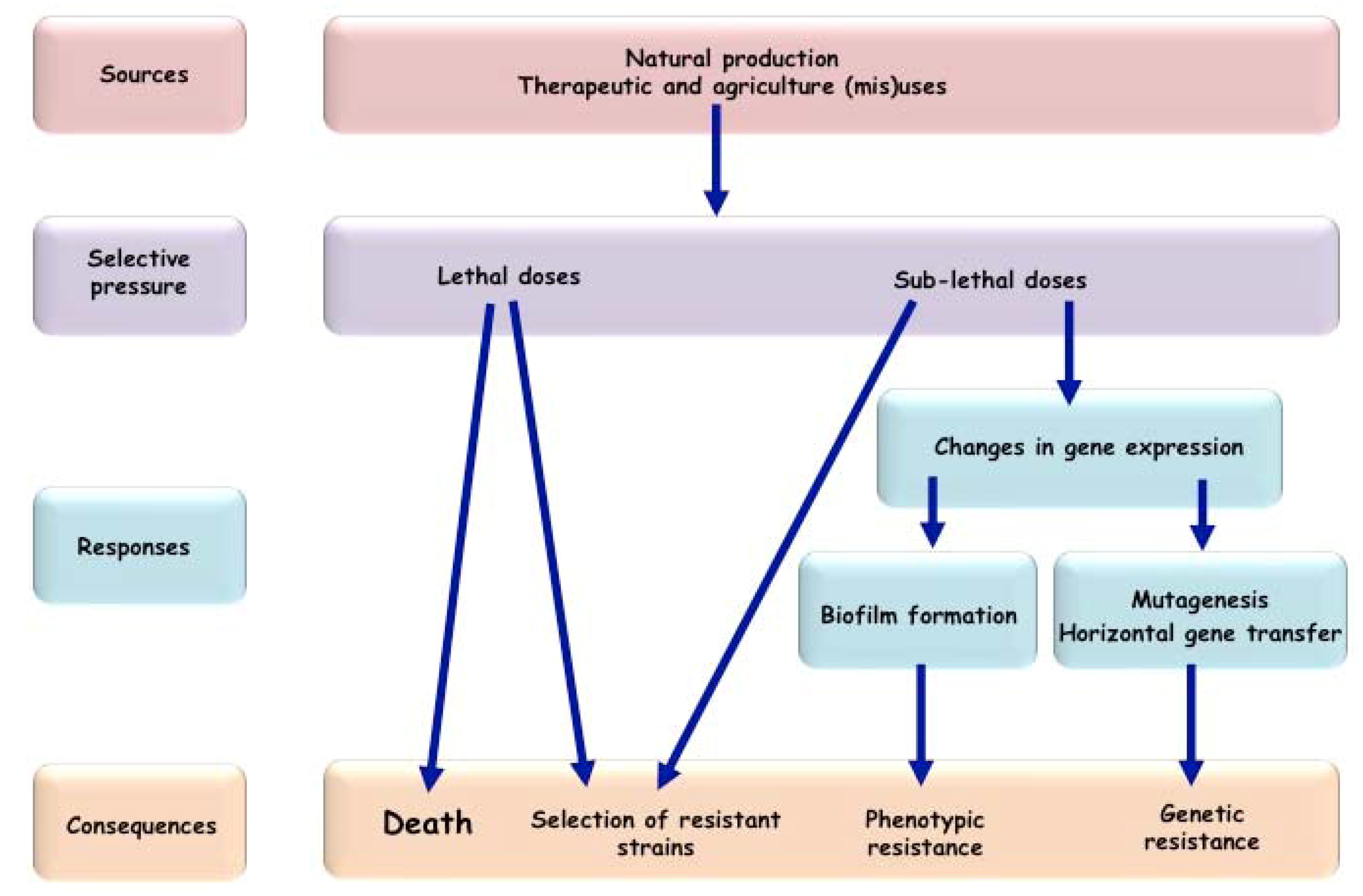

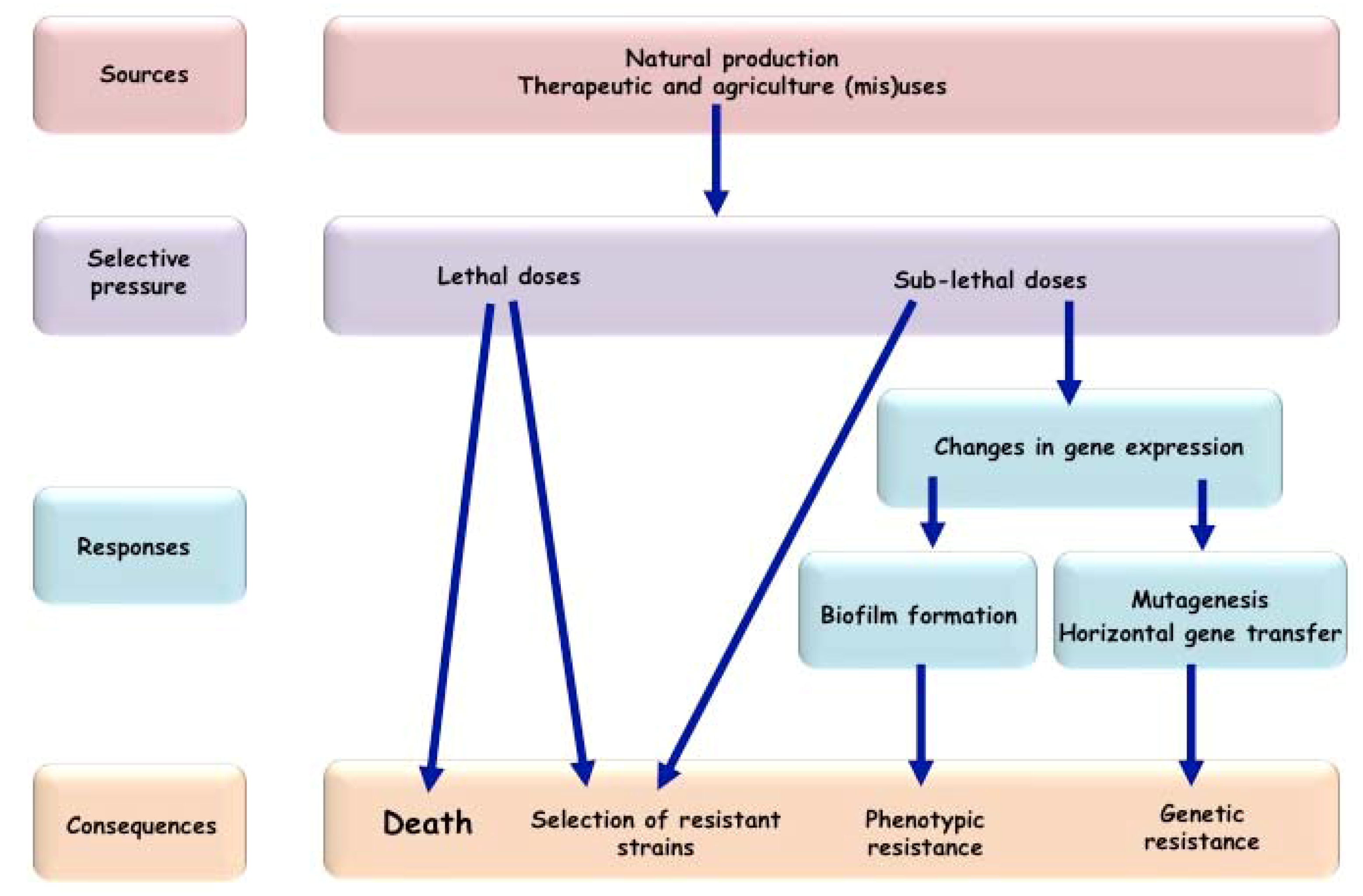{kind=link}
{kind=link}
| Organisms | β-lactam | Quinolones | Aminoglycosides | Tetracyclines |
|---|---|---|---|---|
| Escherichia coli | ROS species [55] recA, dinB [14,65] rpoS [15] | ROS species [55] recA, recBCD,lexA [14,63] polB, dinB, umuCD [66] | ROS species [55] recA, recBCD, ruvABC [67] | recA [14] |
| Streptococcus pneumoniae | dinB-independent [68] | |||
| Streptococcus uberis | umuC-independent [69] | |||
| Vibrio cholerae | lexA [13] | lexA [13] | lexA [13] | lexA [13] |
| rpoS, dinB [15] | ||||
| Staphylococcus aureus | ROS species [55] | |||
| Pseudomonas aeruginosa | rpoS, dinB [15] |
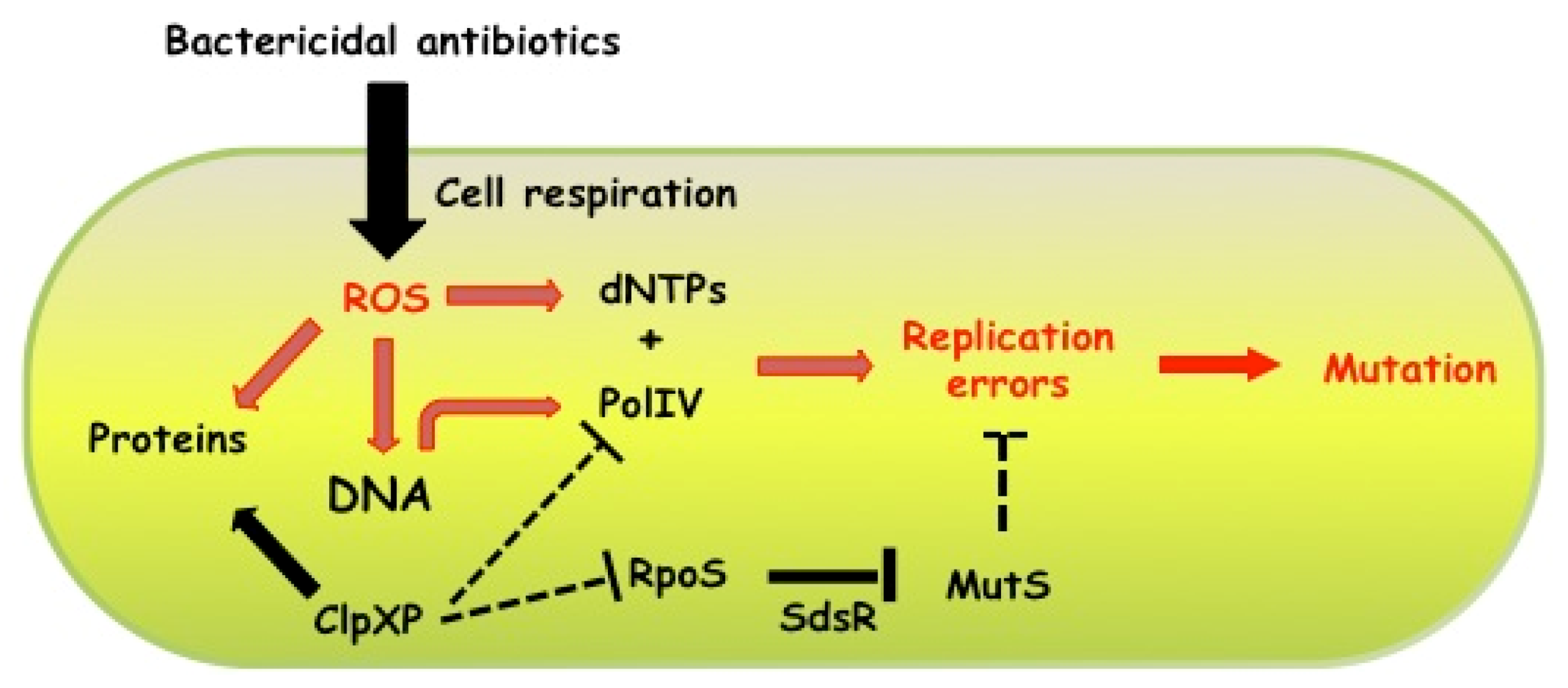
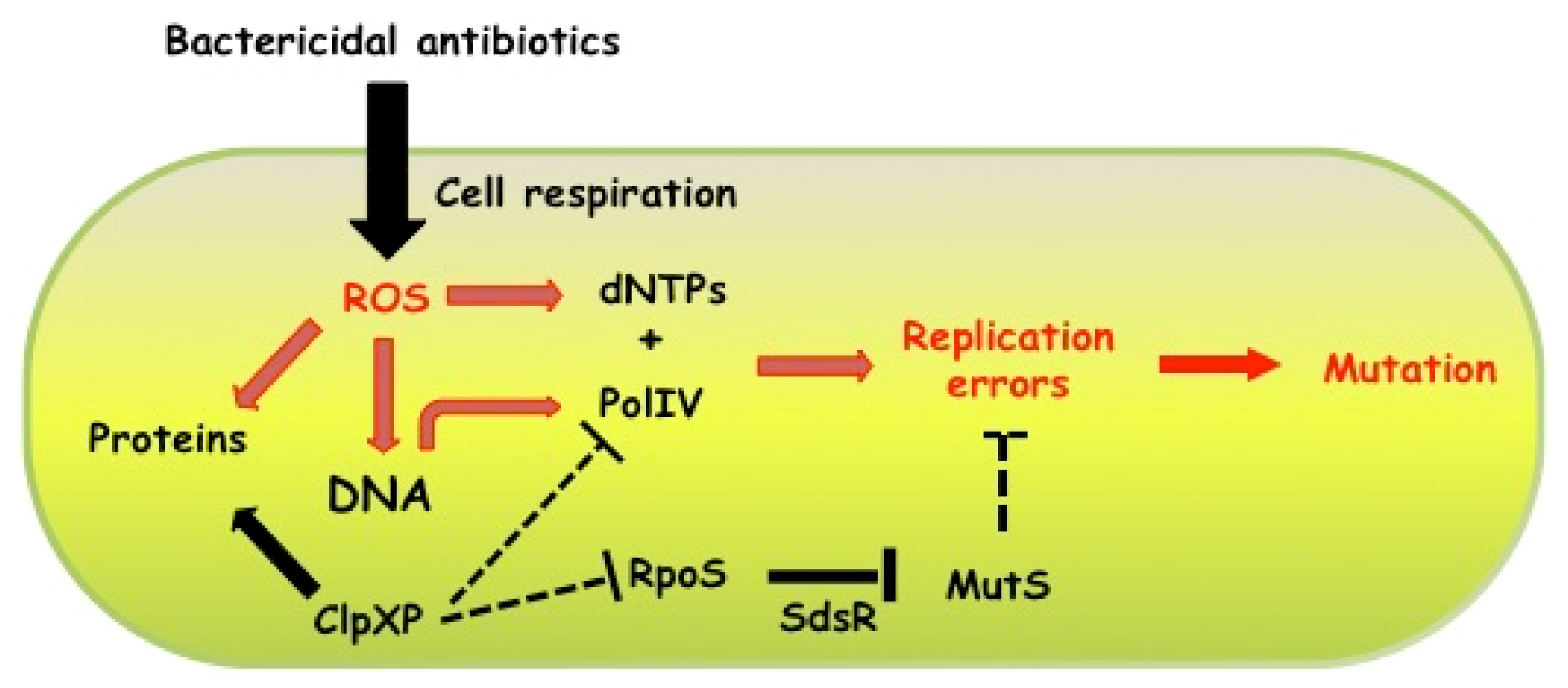
6. Concluding Remarks
Acknowledgements
References and Notes
- Davies, J. Vicious circles: Looking back on resistance plasmids. Genetics 1995, 139, 1465–1468. [Google Scholar]
- Berdy, J. Thoughts and facts about antibiotics: Where we are now and where we are heading. J. Antibiot. 2012, 65, 385–395. [Google Scholar] [CrossRef]
- Mascaretti, O.A. Bacteria Versus Antibacterial Agents. An Integrated Approach; ASM Press: Washington, D.C., USA, 2003. [Google Scholar]
- Kohanski, M.A.; Dwyer, D.J.; Hayete, B.; Lawrence, C.A.; Collins, J.J. A common mechanism of cellular death induced by bactericidal antibiotics. Cell 2007, 130, 797–810. [Google Scholar] [CrossRef]
- Dwyer, D.J.; Camacho, D.M.; Kohanski, M.A.; Callura, J.M.; Collins, J.J. Antibiotic-induced bacterial cell death exhibits physiological and biochemical hallmarks of apoptosis. Mol. Cell 2012, 46, 561–572. [Google Scholar] [CrossRef]
- Foti, J.J.; Devadoss, B.; Winkler, J.A.; Collins, J.J.; Walker, G.C. Oxidation of the guanine nucleotide pool underlies cell death by bactericidal antibiotics. Science 2012, 336, 315–319. [Google Scholar] [CrossRef]
- Marinelli, F. Chapter 2. From microbial products to novel drugs that target a multitude of disease indications. Methods Enzymol. 2009, 458, 29–58. [Google Scholar]
- Currie, C.R.; Mueller, U.G.; Malloch, D. The agricultural pathology of ant fungus gardens. Proc. Natl. Acad. Sci. 1999, 96, 7998–8002. [Google Scholar] [CrossRef]
- Neeno-Eckwall, E.C.; Kinkel, L.L.; Schottel, J.L. Competition and antibiosis in the biological control of potato scab. Can. J. Microbiol. 2001, 47, 332–340. [Google Scholar] [CrossRef]
- Davies, J.; Spiegelman, G.B.; Yim, G. The world of subinhibitory antibiotic concentrations. Curr. Opin. Microbiol. 2006, 9, 445–453. [Google Scholar] [CrossRef]
- Romero, D.; Traxler, M.F.; Lopez, D.; Kolter, R. Antibiotics as signal molecules. Chem. Rev. 2011, 111, 5492–5505. [Google Scholar] [CrossRef]
- Kummerer, K. Significance of antibiotics in the environment. J. Antimicrob. Chemother. 2003, 52, 5–7. [Google Scholar] [CrossRef]
- Baharoglu, Z.; Mazel, D. Vibriocholerae triggers SOS and mutagenesis in response to a wide range of antibiotics: A route towards multiresistance. Antimicrob. Agents Chemother. 2011, 55, 2438–2441. [Google Scholar] [CrossRef]
- Thi, T.D.; Lopez, E.; Rodriguez-Rojas, A.; Rodriguez-Beltran, J.; Couce, A.; Guelfo, J.R.; Castaneda-Garcia, A.; Blazquez, J. Effect of recA inactivation on mutagenesis of Escherichia coli exposed to sublethal concentrations of antimicrobials. J. Antimicrob. Chemother. 2011, 66, 531–538. [Google Scholar] [CrossRef]
- Gutierrez, A.; Laureti, L.; Crussard, S.; Abida, H.; Rodriguez Rojas, A.; Blazquez, J.; Baharoglu, Z.; Mazel, D.; Darfeuille, F.; Vogel, J.; et al. β-Lactam antibiotics promote mutagenesis via RpoS-mediated replication fidelity reduction. Nat. Commun. 2013, in press. [Google Scholar]
- Friedberg, E.C.; Walker, G.C.; Siede, W.; Wood, R.D.; Schultz, R.A.; Ellenberger, T. DNA Repair and Mutagenesis; ASM Press: Washington D.C., USA, 2006. [Google Scholar]
- Courcelle, J.; Khodursky, A.; Peter, B.; Brown, P.O.; Hanawalt, P.C. Comparative gene expression profiles following UV exposure in wild-type and SOS-deficient Escherichia coli. Genetics 2001, 158, 41–64. [Google Scholar]
- Drlica, K.; Zhao, X. DNA gyrase, topoisomerase IV, and the 4-quinolones. Microbiol. Mol. Biol. Rev. 1997, 61, 377–392. [Google Scholar]
- Miller, C.; Thomsen, L.E.; Gaggero, C.; Mosseri, R.; Ingmer, H.; Cohen, S.N. SOS response induction by beta-lactams and bacterial defense against antibiotic lethality. Science 2004, 305, 1629–1631. [Google Scholar]
- Kohanski, M.A.; Dwyer, D.J.; Wierzbowski, J.; Cottarel, G.; Collins, J.J. Mistranslation of membrane proteins and two-component system activation trigger antibiotic-mediated cell death. Cell 2008, 135, 679–690. [Google Scholar] [CrossRef]
- Battesti, A.; Majdalani, N.; Gottesman, S. The RpoS-mediated general stress response in Escherichia coli. Annu. Rev. Microbiol. 2011, 65, 189–213. [Google Scholar]
- Chiang, S.M.; Schellhorn, H.E. Evolution of the RpoSregulon: Origin of RpoS and the conservation of RpoS-dependent regulation in bacteria. J. Mol. Evol. 2010, 70, 557–571. [Google Scholar] [CrossRef]
- Denamur, E.; Matic, I. Evolution of mutation rates in bacteria. Mol. Microbiol. 2006, 60, 820–827. [Google Scholar] [CrossRef]
- Drummond, L.J.; Smith, D.G.; Poxton, I.R. Effects of sub-MIC concentrations of antibiotics on growth of and toxin production by Clostridium difficile. J. Med. Microbiol. 2003, 52, 1033–1038. [Google Scholar] [CrossRef]
- Grimwood, K.; To, M.; Rabin, H.R.; Woods, D.E. Inhibition of Pseudomonas aeruginosa exoenzyme expression by subinhibitory antibiotic concentrations. Antimicrob. Agents Chemother. 1989, 33, 41–47. [Google Scholar] [CrossRef]
- Joo, H.S.; Chan, J.L.; Cheung, G.Y.; Otto, M. Subinhibitory concentrations of protein synthesis-inhibiting antibiotics promote increased expression of the agr virulence regulator and production of phenol-soluble modulincytolysins in community-associated methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2010, 54, 4942–4944. [Google Scholar] [CrossRef]
- Ohlsen, K.; Ziebuhr, W.; Koller, K.P.; Hell, W.; Wichelhaus, T.A.; Hacker, J. Effects of subinhibitory concentrations of antibiotics on alpha-toxin (hla) gene expression of methicillin-sensitive and methicillin-resistant Staphylococcus aureus isolates. Antimicrob. Agents Chemother. 1998, 42, 2817–2823. [Google Scholar]
- Subrt, N.; Mesak, L.R.; Davies, J. Modulation of virulence gene expression by cell wall active antibiotics in Staphylococcus aureus. J. Antimicrob. Chemother. 2011, 66, 979–984. [Google Scholar] [CrossRef]
- Zhang, Q.; Lambert, G.; Liao, D.; Kim, H.; Robin, K.; Tung, C.K.; Pourmand, N.; Austin, R.H. Acceleration of emergence of bacterial antibiotic resistance in connected microenvironments. Science 2011, 333, 1764–1767. [Google Scholar] [CrossRef]
- Mah, T.F.; O'Toole, G.A. Mechanisms of biofilm resistance to antimicrobial agents. Trends Microbiol. 2001, 9, 34–39. [Google Scholar] [CrossRef]
- Watnick, P.; Kolter, R. Biofilm, city of microbes. J. Bacteriol. 2000, 182, 2675–2679. [Google Scholar] [CrossRef]
- Drenkard, E. Antimicrobial resistance of pseudomonas aeruginosa biofilms. Microbes Infect. 2003, 5, 1213–1219. [Google Scholar] [CrossRef]
- Hentzer, M.; Teitzel, G.M.; Balzer, G.J.; Heydorn, A.; Molin, S.; Givskov, M.; Parsek, M.R. Alginate overproduction affects Pseudomonas aeruginosa biofilm structure and function. J. Bacteriol. 2001, 183, 5395–5401. [Google Scholar] [CrossRef]
- Davies, D.G.; Parsek, M.R.; Pearson, J.P.; Iglewski, B.H.; Costerton, J.W.; Greenberg, E.P. The involvement of cell-to-cell signals in the development of a bacterial biofilm. Science 1998, 280, 295–298. [Google Scholar] [CrossRef]
- Hoffman, L.R.; D'Argenio, D.A.; MacCoss, M.J.; Zhang, Z.; Jones, R.A.; Miller, S.I. Aminoglycoside antibiotics induce bacterial biofilm formation. Nature 2005, 436, 1171–1175. [Google Scholar] [CrossRef]
- Boehm, A.; Steiner, S.; Zaehringer, F.; Casanova, A.; Hamburger, F.; Ritz, D.; Keck, W.; Ackermann, M.; Schirmer, T.; Jenal, U. Second messenger signalling governs Escherichia coli biofilm induction upon ribosomal stress. Mol. Microbiol. 2009, 72, 1500–1516. [Google Scholar] [CrossRef]
- Bagge, N.; Schuster, M.; Hentzer, M.; Ciofu, O.; Givskov, M.; Greenberg, E.P.; Hoiby, N. Pseudomonas aeruginosa biofilms exposed to imipenem exhibit changes in global gene expression and beta-lactamase and alginate production. Antimicrob. Agents Chemother. 2004, 48, 1175–1187. [Google Scholar] [CrossRef]
- Kaplan, J.B.; Izano, E.A.; Gopal, P.; Karwacki, M.T.; Kim, S.; Bose, J.L.; Bayles, K.W.; Horswill, A.R. Low levels of beta-lactam antibiotics induce extracellular DNA release and biofilm formation in Staphylococcus aureus. mBio 2012, 3, e00198–e00112. [Google Scholar]
- Rogers, P.D.; Liu, T.T.; Barker, K.S.; Hilliard, G.M.; English, B.K.; Thornton, J.; Swiatlo, E.; McDaniel, L.S. Gene expression profiling of the response of Streptococcus pneumoniae to penicillin. J. Antimicrob. Chemother. 2007, 59, 616–626. [Google Scholar] [CrossRef]
- Sailer, F.C.; Meberg, B.M.; Young, K.D. Beta-lactam induction of colanic acid gene expression in escherichia coli. FEMS Microbiol. Lett. 2003, 226, 245–249. [Google Scholar] [CrossRef]
- Aminov, R.I. Horizontal gene exchange in environmental microbiota. Front. Microbiol. 2011, 2. [Google Scholar] [CrossRef]
- Barr, V.; Barr, K.; Millar, M.R.; Lacey, R.W. Beta-lactam antibiotics increase the frequency of plasmid transfer in Staphylococcus aureus. J. Antimicrob. Chemother. 1986, 17, 409–413. [Google Scholar] [CrossRef]
- Stevens, A.M.; Shoemaker, N.B.; Li, L.Y.; Salyers, A.A. Tetracycline regulation of genes on bacteroides conjugative transposons. J. Bacteriol. 1993, 175, 6134–6141. [Google Scholar]
- Torres, O.R.; Korman, R.Z.; Zahler, S.A.; Dunny, G.M. The conjugative transposon Tn925: Enhancement of conjugal transfer by tetracycline in Enterococcus faecalis and mobilization of chromosomal genes in bacillus subtilis and E. faecalis. Mol. Gen. Genet. 1991, 225, 395–400. [Google Scholar]
- D'Costa, V.M.; McGrann, K.M.; Hughes, D.W.; Wright, G.D. Sampling the antibiotic resistome. Science 2006, 311, 374–377. [Google Scholar] [CrossRef]
- D'Costa, V.M.; Griffiths, E.; Wright, G.D. Expanding the soil antibiotic resistome: Exploring environmental diversity. Curr. Opin. Microbiol. 2007, 10, 481–489. [Google Scholar]
- Heuer, H.; Kopmann, C.; Binh, C.T.; Top, E.M.; Smalla, K. Spreading antibiotic resistance through spread manure: Characteristics of a novel plasmid type with low %g+c content. Environ. Microbiol. 2009, 11, 937–949. [Google Scholar] [CrossRef]
- Beaber, J.W.; Hochhut, B.; Waldor, M.K. SOS response promotes horizontal dissemination of antibiotic resistance genes. Nature 2004, 427, 72–74. [Google Scholar] [CrossRef]
- Ubeda, C.; Maiques, E.; Knecht, E.; Lasa, I.; Novick, R.P.; Penades, J.R. Antibiotic-induced SOS response promotes horizontal dissemination of pathogenicity island-encoded virulence factors in staphylococci. Mol. Microbiol. 2005, 56, 836–844. [Google Scholar] [CrossRef]
- Guerin, E.; Cambray, G.; Sanchez-Alberola, N.; Campoy, S.; Erill, I.; Da Re, S.; Gonzalez-Zorn, B.; Barbe, J.; Ploy, M.C.; Mazel, D. The SOS response controls integron recombination. Science 2009, 324, 1034. [Google Scholar] [CrossRef]
- Prudhomme, M.; Attaiech, L.; Sanchez, G.; Martin, B.; Claverys, J.P. Antibiotic stress induces genetic transformability in the human pathogen Streptococcus pneumoniae. Science 2006, 313, 89–92. [Google Scholar] [CrossRef]
- Salyers, A.A.; Gupta, A.; Wang, Y. Human intestinal bacteria as reservoirs for antibiotic resistance genes. Trends Microbiol. 2004, 12, 412–416. [Google Scholar] [CrossRef]
- Bahl, M.I.; Sorensen, S.J.; Hansen, L.H.; Licht, T.R. Effect of tetracycline on transfer and establishment of the tetracycline-inducible conjugative transposon Tn916 in the guts of gnotobiotic rats. Appl. Environ. Microbiol. 2004, 70, 758–764. [Google Scholar] [CrossRef]
- Doucet-Populaire, F.; Trieu-Cuot, P.; Dosbaa, I.; Andremont, A.; Courvalin, P. Inducible transfer of conjugative transposon Tn1545 from Enterococcus faecalis to Listeria monocytogenes in the digestive tracts of gnotobiotic mice. AntimicrobAgents Chemother. 1991, 35, 185–187. [Google Scholar] [CrossRef]
- Kohanski, M.A.; DePristo, M.A.; Collins, J.J. Sublethal antibiotic treatment leads to multidrug resistance via radical-induced mutagenesis. Mol. Cell 2010, 37, 311–320. [Google Scholar] [CrossRef]
- Komp Lindgren, P.; Karlsson, A.; Hughes, D. Mutation rate and evolution of fluoroquinolone resistance in Escherichia coli isolates from patients with urinary tract infections. Antimicrob. Agents Chemother. 2003, 47, 3222–3232. [Google Scholar] [CrossRef]
- Pena, C.; Albareda, J.M.; Pallares, R.; Pujol, M.; Tubau, F.; Ariza, J. Relationship between quinolone use and emergence of ciprofloxacin-resistant Escherichia coli in bloodstream infections. Antimicrob. Agents Chemother. 1995, 39, 520–524. [Google Scholar] [CrossRef]
- Gullberg, E.; Cao, S.; Berg, O.G.; Ilback, C.; Sandegren, L.; Hughes, D.; Andersson, D.I. Selection of resistant bacteria at very low antibiotic concentrations. PLoSPathog. 2011, 7, e1002158. [Google Scholar]
- Blazquez, J.; Couce, A.; Rodriguez-Beltran, J.; Rodriguez-Rojas, A. Antimicrobials as promoters of genetic variation. Curr. Opin. Microbiol. 2012, 15, 561–569. [Google Scholar]
- Chen, C.R.; Malik, M.; Snyder, M.; Drlica, K. DNA gyrase and topoisomerase IV on the bacterial chromosome: Quinolone-induced DNA cleavage. J. Mol. Biol. 1996, 258, 627–637. [Google Scholar] [CrossRef]
- Michel, B.; Grompone, G.; Flores, M.J.; Bidnenko, V. Multiple pathways process stalled replication forks. Proc. Natl. Acad. Sci. 2004, 101, 12783–12788. [Google Scholar]
- Anderson, D.G.; Kowalczykowski, S.C. Reconstitution of an sos response pathway: Derepression of transcription in response to DNA breaks. Cell 1998, 95, 975–979. [Google Scholar] [CrossRef]
- Cirz, R.T.; Chin, J.K.; Andes, D.R.; de Crecy-Lagard, V.; Craig, W.A.; Romesberg, F.E. Inhibition of mutation and combating the evolution of antibiotic resistance. PLoSBiol. 2005, 3, e176. [Google Scholar]
- Veigl, M.L.; Schneiter, S.; Mollis, S.; Sedwick, W.D. Specificities mediated by neighboring nucleotides appear to underlie mutation induced by antifolates in E. coli. Mutation Res. 1991, 246, 75–91. [Google Scholar] [CrossRef]
- Perez-Capilla, T.; Baquero, M.R.; Gomez-Gomez, J.M.; Ionel, A.; Martin, S.; Blazquez, J. SOS-independent induction of DINB transcription by beta-lactam-mediated inhibition of cell wall synthesis in Escherichia coli. J. Bacteriol. 2005, 187, 1515–1518. [Google Scholar] [CrossRef]
- Lopez, E.; Elez, M.; Matic, I.; Blazquez, J. Antibiotic-mediated recombination: Ciprofloxacin stimulates SOS-independent recombination of divergent sequences in Escherichia coli. Mol. Microbiol. 2007, 64, 83–93. [Google Scholar] [CrossRef]
- Balashov, S.; Humayun, M.Z. Mistranslation induced by streptomycin provokes a RecABC/ RuvABC-dependent mutator phenotype in Escherichia coli cells. J. Mol. Biol. 2002, 315, 513–527. [Google Scholar]
- Henderson-Begg, S.K.; Livermore, D.M.; Hall, L.M. Effect of subinhibitory concentrations of antibiotics on mutation frequency in Streptococcus pneumoniae. J. Antimicrob. Chemother. 2006, 57, 849–854. [Google Scholar] [CrossRef]
- Varhimo, E.; Savijoki, K.; Jefremoff, H.; Jalava, J.; Sukura, A.; Varmanen, P. Ciprofloxacin induces mutagenesis to antibiotic resistance independent of umuc in Streptococcus uberis. Environ. Microbiol. 2008, 10, 2179–2183. [Google Scholar] [CrossRef]
- Nagel, R.; Chan, A. Mistranslation and genetic variability: The effect of streptomycin. Mutation Res. 2006, 601, 162–170. [Google Scholar] [CrossRef]
- Murphy, H.S.; Humayun, M.Z. Escherichia coli cells expressing a mutant glyV (glycinetRNA) gene have a UVM-constitutive phenotype: Implications for mechanisms underlying the mutA or mutCmutator effect. J. Bacteriol. 1997, 179, 7507–7514. [Google Scholar]
- Ren, L.; Rahman, M.S.; Humayun, M.Z. Escherichia coli cells exposed to streptomycin display a mutator phenotype. J. Bacteriol. 1999, 181, 1043–1044. [Google Scholar]
- Slupska, M.M.; Baikalov, C.; Lloyd, R.; Miller, J.H. MutatortRNAs are encoded by the Escherichia coli mutator genes mutA and mutC: A novel pathway for mutagenesis. Proc. Natl. Acad. Sci. 1996, 93, 4380–4385. [Google Scholar]
- Kobayashi, S.; Valentine, M.R.; Pham, P.; O’Donnell, M.; Goodman, M.F. Fidelity of Escherichia coli DNA polymerase IV. Preferential generation of small deletion mutations by DNTP-stabilized misalignment. J. Biol. Chem. 2002, 277, 34198–34207. [Google Scholar]
- Petrosino, J.F.; Galhardo, R.S.; Morales, L.D.; Rosenberg, S.M. Stress-induced beta-lactam antibiotic resistance mutation and sequences of stationary-phase mutations in the Escherichia coli chromosome. J. Bacteriol. 2009, 191, 5881–5889. [Google Scholar] [CrossRef]
- Wagner, J.; Nohmi, T. Escherichia coli DNA polymerase IV mutator activity: Genetic requirements and mutational specificity. J. Bacteriol. 2000, 182, 4587–4595. [Google Scholar] [CrossRef]
- Yamada, M.; Nunoshiba, T.; Shimizu, M.; Gruz, P.; Kamiya, H.; Harashima, H.; Nohmi, T. Involvement of Y-family DNA polymerases in mutagenesis caused by oxidized nucleotides in Escherichia coli. J. Bacteriol. 2006, 188, 4992–4995. [Google Scholar] [CrossRef]
- Frohlich, K.S.; Papenfort, K.; Berger, A.A.; Vogel, J. A conserved RpoS-dependent small RNA controls the synthesis of major porin ompd. Nucleic Acids Res. 2012, 40, 3623–3640. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Laureti, L.; Matic, I.; Gutierrez, A. Bacterial Responses and Genome Instability Induced by Subinhibitory Concentrations of Antibiotics. Antibiotics 2013, 2, 100-114. https://doi.org/10.3390/antibiotics2010100
Laureti L, Matic I, Gutierrez A. Bacterial Responses and Genome Instability Induced by Subinhibitory Concentrations of Antibiotics. Antibiotics. 2013; 2(1):100-114. https://doi.org/10.3390/antibiotics2010100
Chicago/Turabian StyleLaureti, Luisa, Ivan Matic, and Arnaud Gutierrez. 2013. "Bacterial Responses and Genome Instability Induced by Subinhibitory Concentrations of Antibiotics" Antibiotics 2, no. 1: 100-114. https://doi.org/10.3390/antibiotics2010100