Synthesis, Antibacterial Evaluation and QSAR of α-Substituted-N4-Acetamides of Ciprofloxacin and Norfloxacin

 ,
,
Abstract
:
1. Introduction



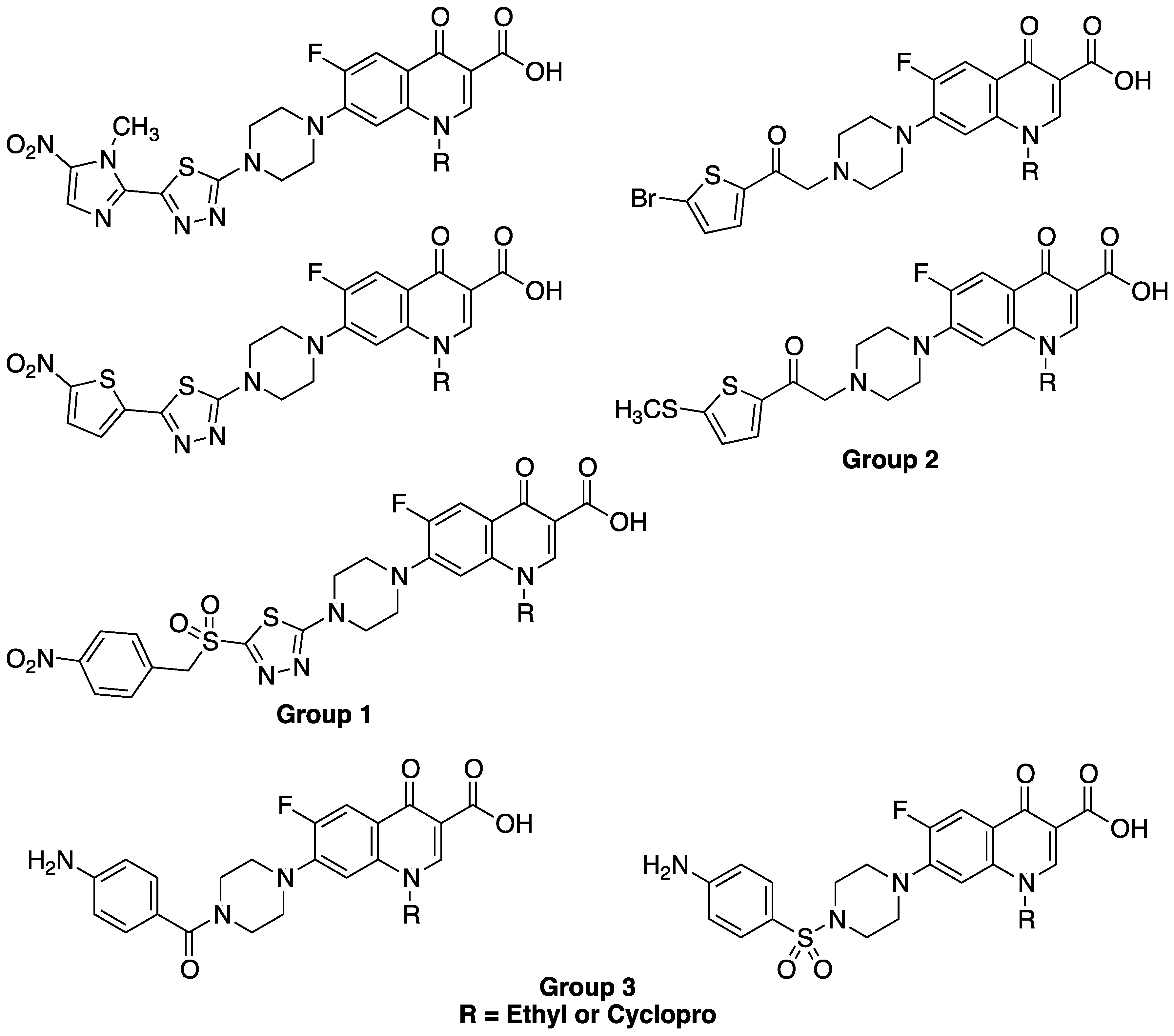
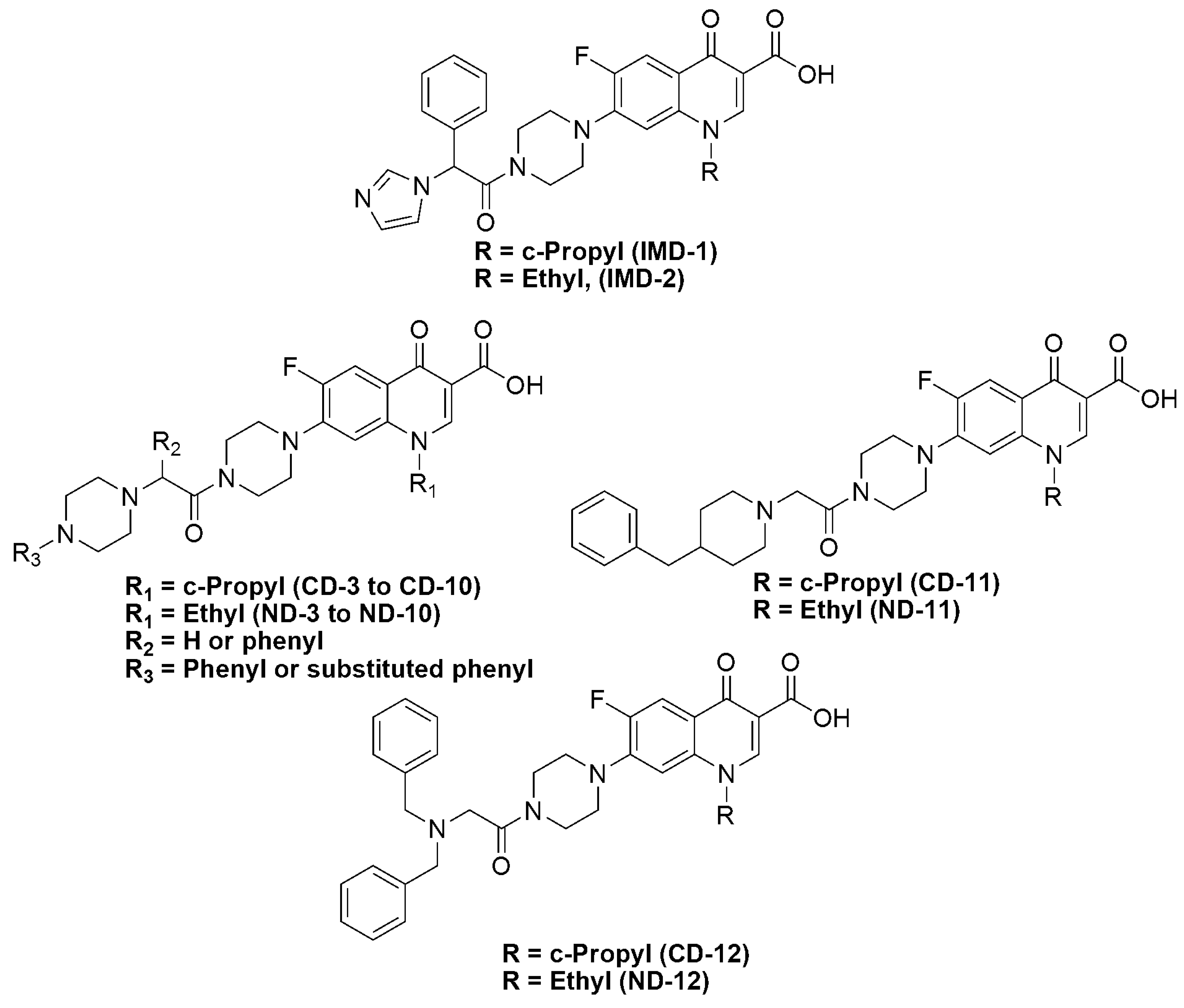
2. Results and Discussion
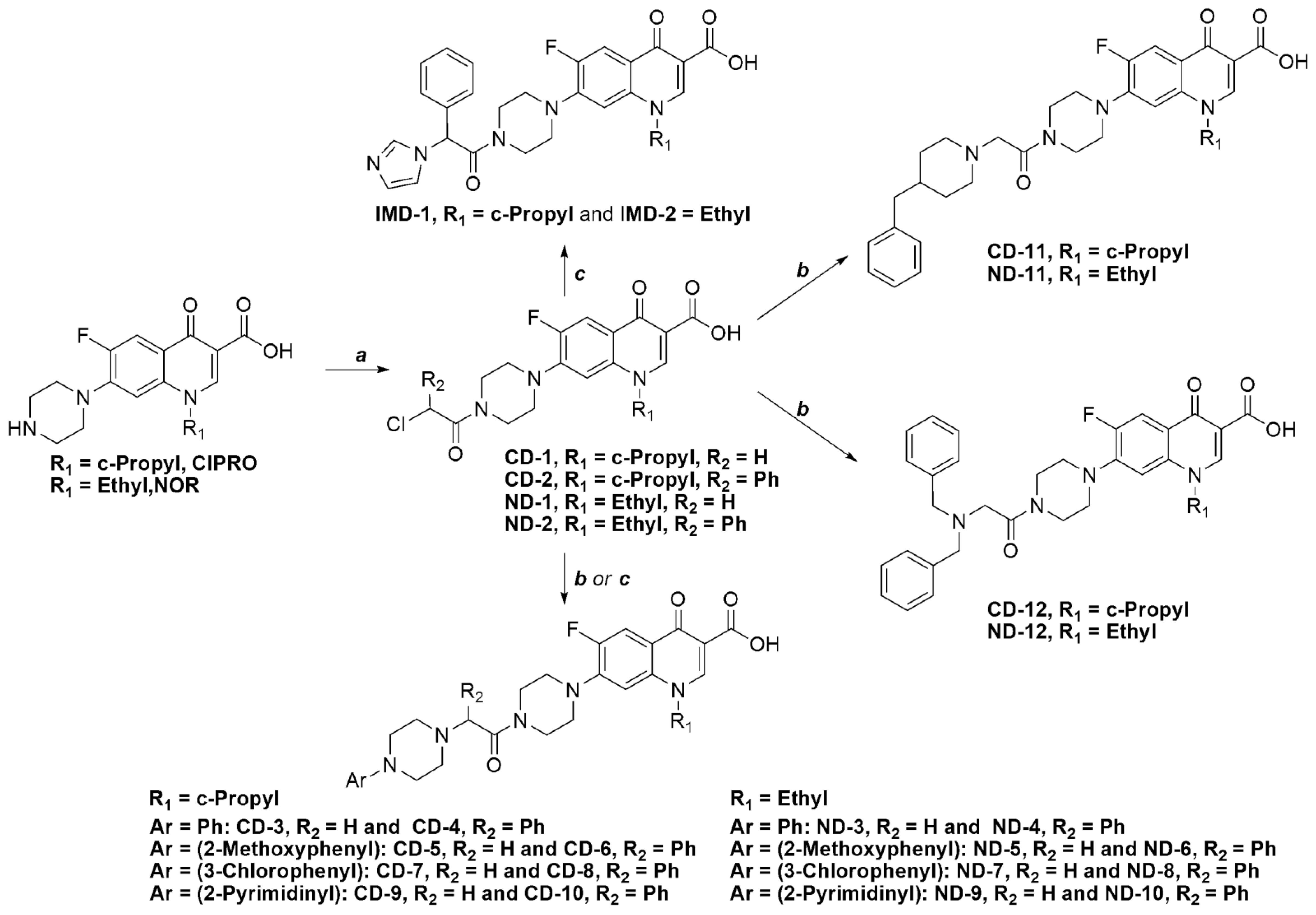
2.1. Chemistry
2.2. In Vitro Antibacterial Activity Assays

{kind=link}
{kind=link}
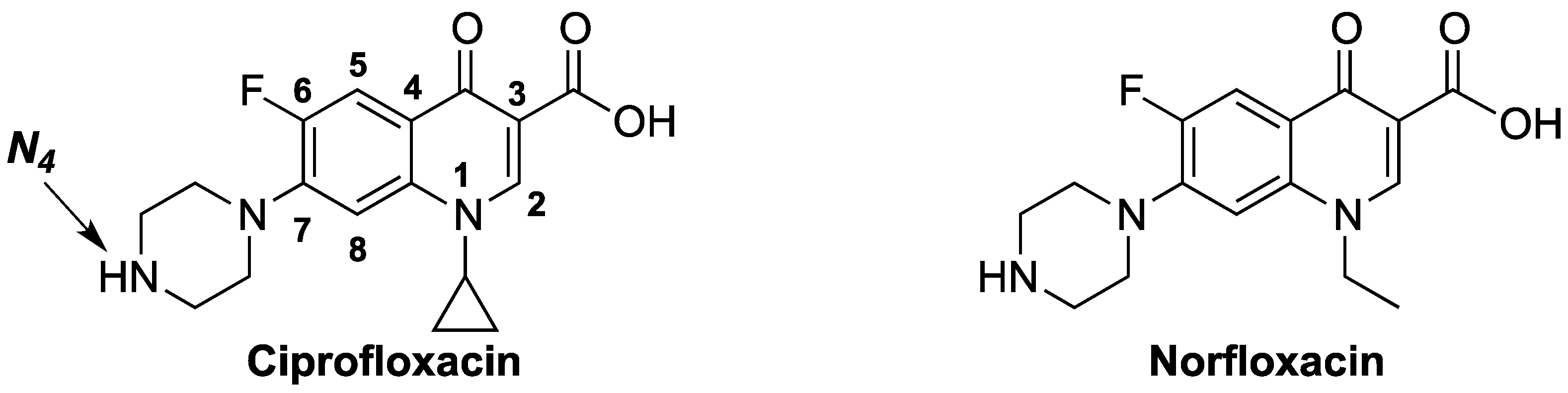{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | MIC | |||||||
|---|---|---|---|---|---|---|---|---|
| E. coli | P. aeruginosa | S. aureus | B. subtilis | |||||
| (μM) | (µg/mL) | (μM) | (µg/mL) | (μM) | (µg/mL) | (μM) | (µg/mL) | |
| IMD-1 | 8.5 | 4.4 | 114 | 59 | 0.6 | 0.3 | 0.5 | 0.2 |
| CD-2 | 7.5 | 3.7 | 101 | 49 | 0.3 | 0.2 | 0.1 | 0.1 |
| CD-3 | 146 | 78 | 733 | 391 | 55 | 29 | 5.3 | 2.9 |
| CD-4 | 192 | 117 | 2,306 | 1,406 | 56 | 34 | 5.0 | 3.1 |
| CD-5 | 24 | 14 | NA * | NA * | 6.1 | 3.4 | 0.5 | 0.3 |
| CD-6 | 31 | 20 | 366 | 234 | 3.1 | 2.0 | 0.5 | 0.3 |
| CD-7 | 9.5 | 5.4 | 103 | 59 | 0.5 | 0.3 | 0.3 | 0.2 |
| CD-8 | NA * | NA * | NA * | NA * | 9.1 | 5.9 | 0.5 | 0.3 |
| CD-9 | 11 | 6.1 | 291 | 156 | 0.9 | 0.6 | 0.9 | 0.5 |
| CD-10 | 12 | 7.4 | 80 | 49 | 0.4 | 0.2 | 0.1 | 0.1 |
| CD-11 | 9.0 | 4.9 | 62 | 34 | 16.4 | 9.0 | 2.1 | 1.2 |
| CD-12 | 206 | 117 | NA * | NA * | 120 | 68 | 5.4 | 3.1 |
| CIPRO | 1.7 | 0.6 | 0.5 | 0.2 | 2.2 | 0.7 | 0.3 | 0.1 |
| IMD-2 | 542 | 273 | 775 | NA * | 4.7 | 2.4 | 1.2 | 0.6 |
| ND-2 | 10 | 4.9 | 103 | NA * | 0.4 | 0.2 | 0.1 | 0.1 |
| ND-3 | 112 | 58 | 1,198 | NA* | 23 | 12 | 3.5 | 1.8 |
| ND-4 | NA * | NA * | NA * | NA * | 392 | 234 | 9.2 | 5.5 |
| ND-5 | 1,133 | 625 | NA * | NA * | 42 | 23 | 8.8 | 4.9 |
| ND-6 | NA * | NA * | NA * | NA * | 1.9 | 1.2 | 0.7 | 0.4 |
| ND-7 | NA * | NA * | NA * | NA * | 1.8 | 1.0 | 0.6 | 0.3 |
| ND-8 | NA * | NA * | NA * | NA * | 4.6 | 2.9 | 1.0 | 0.6 |
| ND-9 | 130 | 68 | 3,581 | 1,875 | 13 | 7.0 | 3.7 | 2.0 |
| ND-10 | 81 | 49 | 1,042 | 625 | 2.5 | 1.5 | 0.8 | 0.5 |
| ND-11 | 165 | 88 | NA * | NA * | 7.1 | 3.8 | 4.6 | 2.4 |
| ND-12 | 105 | 59 | 983 | 547 | 123 | 68 | 5.5 | 3.1 |
| NOR | 2.3 | 0.7 | 0.6 | 0.2 | 1.1 | 0.4 | 0.6 | 0.2 |
2.3. Structure-Activity Relationships
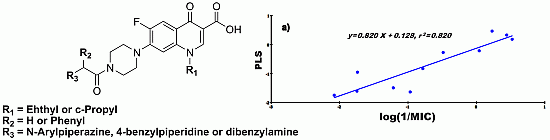
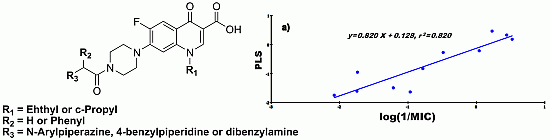
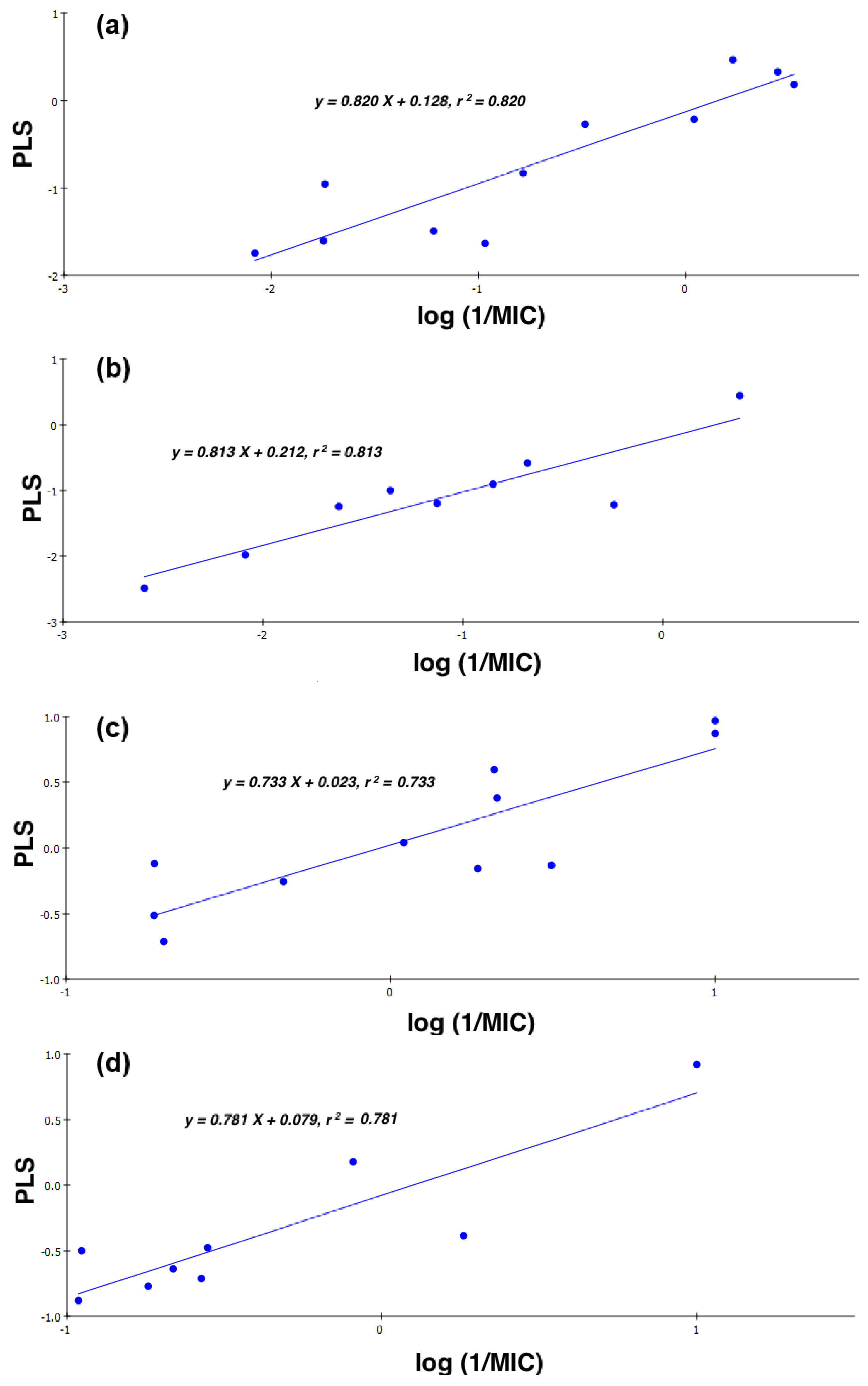
2.4. Quantitative Structure-Activity Relationships (QSAR)
| ID | Log (1/MIC) | cLog D | FPSA | ∆C-13 * | |
|---|---|---|---|---|---|
| S. aureus | B. subtilis | ||||
| IMD-1 | 0.229 | 0.328 | 2.216 | 0.216 | 0.550 |
| CD-2 | 0.523 | 1.000 | 2.009 | 0.189 | −0.480 |
| CD-3 | −1.739 | −0.728 | 1.359 | 0.185 | 1.690 |
| CD-4 | −1.746 | −0.699 | 3.111 | 0.163 | 2.770 |
| CD-5 | −0.783 | 0.268 | 1.371 | 0.191 | 1.770 |
| CD-6 | −0.486 | 0.319 | 3.094 | 0.170 | 2.250 |
| CD-7 | 0.268 | 0.495 | 2.052 | 0.177 | 1.650 |
| CD-8 | −0.968 | 0.319 | 3.775 | 0.156 | 2.740 |
| CD-9 | 0.041 | 0.041 | −1.433 | 0.236 | 1.630 |
| CD-10 | 0.444 | 1.000 | 0.286 | 0.208 | −0.510 |
| CD-11 | −1.215 | −0.330 | 0.859 | 0.173 | 2.030 |
| CD-12 | −2.079 | −0.729 | 1.469 | 0.166 | 2.510 |
| IMD-2 | −0.675 | −0.086 | 1.840 | 0.210 | 0.40 |
| ND-2 | 0.387 | 1.000 | 3.337 | 0.187 | −0.660 |
| ND-3 | −1.362 | −0.545 | 1.670 | 0.177 | 1.500 |
| ND-4 | −2.593 | −0.962 | 4.401 | 0.168 | 2.590 |
| ND-5 | −1.620 | −0.946 | 1.877 | 0.183 | 1.570 |
| ND-6 | −0.288 | 0.168 | 4.385 | 0.168 | 2.650 |
| ND-7 | −0.246 | 0.260 | 2.558 | 0.176 | 1.470 |
| ND-8 | −0.667 | 0.013 | 5.066 | 0.155 | 2.570 |
| ND-9 | −1.127 | −0.571 | −1.245 | 0.234 | 1.490 |
| ND-10 | −0.398 | 0.092 | 0.496 | 0.206 | 2.630 |
| ND-11 | −0.848 | −0.659 | 1.045 | 0.172 | 1.690 |
| ND-12 | −2.089 | −0.739 | 3.276 | 0.165 | 2.240 |
- (1)
- CD-S: Log (1/MIC) = −5.107 + 0.198 cLogD + 24.830 FPSA − 0.420 ∆C-13, n = 11, r = 0.905, r2 = 0.820, RMSEE = 0.377, q2 = 0.636 and MAE = 0.474
- (2)
- ND-S: Log (1/MIC) = 4.315 − 0.298 cLogD − 18.840 FPSA − 0.988 ∆C-13, n = 9, r = 0.902, r2 = 0.813, RMSEE = 0.374, q2 = 0.594 and MAE = 0.939
- (3)
- CD-B: Log (1/MIC) = 0.741 − 4.735 × 10−2 cLogD + 5.169 × 10−3 FPSA − 0.472 ∆C-13, n = 11, r = 0.856, r2 = 0.733, RMSEE = 0.316, q2 = 0.681 and MAE = 0.260
- (4)
- ND-B: Log (1/MIC) = 0.257 + 8.340 × 10−2 cLogD + 4.373 × 10−3 FPSA − 0.581 ∆C-13, n = 9, r = 0.884, r2 = 0.781, RMSEE = 0.285, q2 = 0.699 and MAE = 0.471

3. Experimental
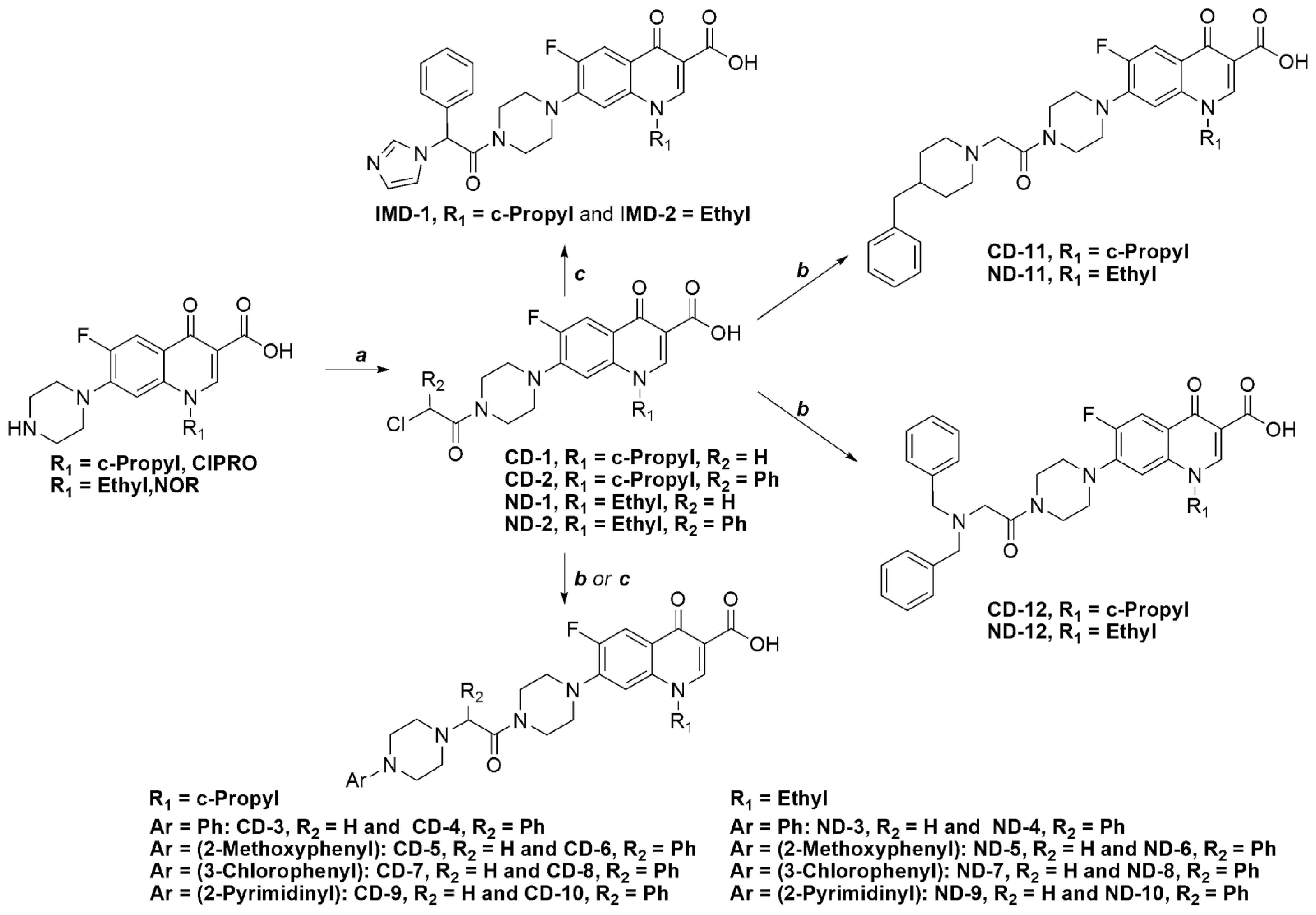
3.1. Chemistry
3.2. General Procedures for the Synthesis of Intermediate Compounds(CD-1, CD-2, ND-1 and ND-2)
3.2.1. 7-(4-(2-Chloroacetyl)piperazin-1-yl)-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid (CD-1)
3.2.2. 7-(4-(2-Chloroacetyl)piperazin-1-yl)-1-ethyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (ND-1)
3.2.3. 7-(4-(2-Chloro-2-phenylacetyl)piperazin-1-yl)-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (CD-2)
3.2.4. 7-(4-(2-Chloro-2-phenylacetyl)piperazin-1-yl)-1-ethyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (ND-2)
3.3. General Procedure for the Synthesis of Final Compounds
3.3.1. 1-Cyclopropyl-6-fluoro-7-(4-(2-(1H-imidazol-1-yl)-2-phenylacetyl)piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (IMD-1)
3.3.2. 1-Ethyl-6-fluoro-7-(4-(2-(1H-imidazol-1-yl)-2-phenylacetyl)piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (IMD-2)
3.3.3. 1-Cyclopropyl-6-fluoro-4-oxo-7-(4-(2-(4-phenylpiperazin-1-yl)acetyl)piperazin-1-yl)-1,4-dihydroquinoline-3-carboxylic acid (CD-3)
3.3.4. 1-Cyclopropyl-6-fluoro-4-oxo-7-(4-(2-phenyl-2-(4-phenylpiperazin-1-yl)acetyl)piperazin-1-yl)-1,4-dihydroquinoline-3-carboxylic acid (CD-4)
3.3.5. 1-Ethyl-6-fluoro-4-oxo-7-(4-(2-(4-phenylpiperazin-1-yl)acetyl)piperazin-1-yl)-1,4-dihydroquinoline-3-carboxylic acid (ND-3)
3.3.6. 1-Ethyl-6-fluoro-4-oxo-7-(4-(2-phenyl-2-(4-phenylpiperazin-1-yl)acetyl)piperazin-1-yl)-1,4-dihydroquinoline-3-carboxylic acid (ND-4)
3.3.7. 1-Cyclopropyl-6-fluoro-7-(4-(2-(4-(2-methoxyphenyl)piperazin-1-yl)acetyl)piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (CD-5)
3.3.8. 1-Cyclopropyl-6-fluoro-7-(4-(2-(4-(2-methoxyphenyl)piperazin-1-yl)-2-phenylacetyl)piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (CD-6)
3.3.9. 1-Ethyl-6-fluoro-7-(4-(2-(4-(2-methoxyphenyl)piperazin-1-yl)acetyl)piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (ND-5)
3.3.10. 1-Ethyl-6-fluoro-7-(4-(2-(4-(2-methoxyphenyl)piperazin-1-yl)-2-phenylacetyl)piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (ND-6)
3.3.11. 7-(4-(2-(4-(3-Chlorophenyl)piperazin-1-yl)acetyl)piperazin-1-yl)-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (CD-7)
3.3.12. 7-(4-(2-(4-(3-Chlorophenyl)piperazin-1-yl)-2-phenylacetyl)piperazin-1-yl)-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (CD-8)
3.3.13. 7-(4-(2-(4-(3-Chlorophenyl)piperazin-1-yl)acetyl)piperazin-1-yl)-1-ethyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (ND-7)
3.3.14. 7-(4-(2-(4-(3-Chlorophenyl)piperazin-1-yl)-2-phenylacetyl)piperazin-1-yl)-1-ethyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (ND-8)
3.3.15. 1-Cyclopropyl-6-fluoro-4-oxo-7-(4-(2-(4-(pyrimidin-2-yl)piperazin-1-yl)acetyl)piperazin-1-yl)-1,4-dihydroquinoline-3-carboxylic acid (CD-9)
3.3.16. 1-Cyclopropyl-6-fluoro-4-oxo-7-(4-(2-phenyl-2-(4-(pyrimidin-2-yl)piperazin-1-yl)acetyl)piperazin-1-yl)-1,4-dihydroquinoline-3-carboxylic acid (CD-10)
3.3.17. 1-Ethyl-6-fluoro-4-oxo-7-(4-(2-(4-(pyrimidin-2-yl)piperazin-1-yl)acetyl)piperazin-1-yl)-1,4-dihydroquinoline-3-carboxylic acid (ND-9)
3.3.18. 1-Ethyl-6-fluoro-4-oxo-7-(4-(2-phenyl-2-(4-(pyrimidin-2-yl)piperazin-1-yl)acetyl)piperazin-1-yl)-1,4-dihydroquinoline-3-carboxylic acid (ND-10)
3.3.19. 7-(4-(2-(4-Benzylpiperidin-1-yl)acetyl)piperazin-1-yl)-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (CD-11)
3.3.20. 7-(4-(2-(4-Benzylpiperidin-1-yl)acetyl)piperazin-1-yl)-1-ethyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (ND-11)
3.3.21. 1-Cyclopropyl-7-(4-(2-(dibenzylamino)acetyl)piperazin-1-yl)-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (CD-12)
3.3.22. 7-(4-(2-(Dibenzylamino)acetyl)piperazin-1-yl)-1-ethyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (ND-12)
3.4. In Vitro Antibacterial Activity Assays
3.5. Quantitative Structure-Activity Relationships (QSAR)
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dougherty, T.; Beaulieu, D.; Barrett, J. New quinolones and the impact on resistance. Drug Discov. Today 2001, 6, 529–536. [Google Scholar] [CrossRef]
- Appelbaum, P.; Hunter, P. The fluoroquinolone antibacterials: Past, present and future perspectives. Int. J. Antimicrob. Agents 2000, 16, 5–15. [Google Scholar] [CrossRef]
- Hawkey, P. Mechanisms of quinolone action and microbial response. J. Antimicrob. Chemother. 2003, 51, 29–35. [Google Scholar] [CrossRef]
- Andersson, M.; MacGowan, A. Development of the quinolones. J. Antimicrob. Chemother. 2003, 51, 1–11. [Google Scholar] [CrossRef]
- Nenortas, E.; Kulikowicz, T.; Burri, C.; Shapiro, T. Antitrypanosomal activities of fluoroquinolones with pyrrolidinyl substitutions. Antimicrob. Agents Chemother. 2003, 47, 3015–3017. [Google Scholar] [CrossRef]
- De Sarro, A.; de Sarro, G. Adverse reactions to fluoroquinolones. An overview on mechanistic aspects. Curr. Med. Chem. 2001, 8, 371–384. [Google Scholar] [CrossRef]
- Lipsky, B.; Baker, C. Fluoroquinolone toxicity profiles: A review focusing on newer agents. Clin. Infect. Dis. 1999, 28, 352–364. [Google Scholar]
- Akahane, K.; Kimura, Y.; Tsutomi, Y.; Hayakawa, I. Possible intermolecular interaction between quinolones and biphenylacetic acid inhibits acid receptor sites. Antimicrob. Agents Chemother. 1994, 38, 2323–2329. [Google Scholar] [CrossRef]
- Kollef, M.H. Is antibiotic cycling the answer to preventing the emergence of bacterial resistance in the intensive care unit? Clin. Infect. Dis. 2006, 43, S82–S88. [Google Scholar] [CrossRef]
- Mölstad, S.; Erntell, M.; Hanberger, H.; Melander, E.; Norman, C.; Skoog, G.; Lundborg, C.S.; Söderström, A.; Torell, E.; Cars, O. Sustained reduction of antibiotic use and low bacterial resistance: 10-year follow-up of the Swedish Strama programme. Lancet Infect. Dis. 2008, 8, 125–132. [Google Scholar] [CrossRef]
- Čižman, M. The use and resistance to antibiotics in the community. Int. J. Antimicrob. Agents 2003, 21, 297–307. [Google Scholar] [CrossRef]
- Fauci, A. Infectious diseases: Considerations for the 21st Century. Clin. Infect. Dis. 2001, 32, 675–685. [Google Scholar] [CrossRef]
- Projan, S. Why is big Pharma getting out of antibacterial drug discovery? Curr. Opin. Microbiol. 2003, 6, 427–430. [Google Scholar] [CrossRef]
- Foroumadi, A.; Mansouri, S.; Kiani, Z.; Rahmani, A. Synthesis and in vitro antibacterial evaluation of N-[5-(5-nitro-2-thienyl)-1,3,4-thiadiazole-2-yl] piperazinyl quinolones. Eur. J. Med. Chem. 2003, 38, 851–854. [Google Scholar] [CrossRef]
- Foroumadi, A.; Emami, S.; Hassanzadeh, A.; Rajaee, M.; Sokhanvar, K.; Moshafi, M.H.; Shafiee, A. Synthesis and antibacterial activity of N-(5-benzylthio-1,3,4-thiadiazol-2-yl) and N-(5-benzylsulfonyl-1,3,4-thiadiazol-2-yl)piperazinyl quinolone derivatives. Bioorganic Med. Chem. Lett. 2005, 15, 4488–4492. [Google Scholar] [CrossRef]
- Foroumadi, A.; Soltani, F.; Moshafi, M.H.; Ashraf-Askari, R. Synthesis and in vitro antibacterial activity of some N-(5-aryl-1,3,4-thiadiazole-2-yl)piperazinyl quinolone derivatives. Farmaco 2003, 58, 1023–1028. [Google Scholar] [CrossRef]
- Foroumadi, A.; Emami, S.; Mehni, M.; Moshafi, M.H.; Shafiee, A. Synthesis and antibacterial activity of N-[2-(5-bromothiophen-2-yl)-2-oxoethyl] and N-[(2–5-bromothiophen-2-yl)-2-oximinoethyl] derivatives of piperazinyl quinolones. Bioorganic Med. Chem. Lett. 2005, 15, 4536–4539. [Google Scholar] [CrossRef]
- Foroumadi, A.; Oboudiat, M.; Emami, S.; Karimollah, A.; Saghaee, L.; Moshafi, M.; Shafiee, A. Synthesis and antibacterial activity of N-[2-[5-(methylthio)thiophen-2-yl]-2-oxoethyl] and N-[2-[5-(methylthio)thiophen-2-yl]-2-(oxyimino)ethyl]piperazinylquinolone derivatives. Bioorganic Med. Chem. 2006, 14, 3421–3427. [Google Scholar] [CrossRef]
- Alovero, F.; Nieto, M.; Mazzieri, M.; Then, R.; Manzo, R. Mode of action of sulfanilyl fluoroquinolones. Antimicrob. Agents Chemother. 1998, 42, 1495–1498. [Google Scholar]
- Allemandi, D.A.; Alovero, F.L.; Manzo, R.H. In-vitro activity of new sulphanilil fluoroquinolones against Staphylococcus aureus. J. Antimicrob. Chemother. 1994, 34, 261–265. [Google Scholar] [CrossRef]
- Nieto, M.; Alovero, F.; Manzo, R.; Mazzieri, M. Benzenecarboxamide analogs of Fluoroquinolones (BCFQs). Antibacterial activity and Sar studies. Lett. Drug Des. Discov. 2006, 3, 108–111. [Google Scholar] [CrossRef]
- Beger, R.D.; Freeman, J.P.; Lay, J.O.; Wilkes, J.G.; Miller, D.W. Use of 13C-NMR spectrometric data to produce a predictive model of estrogen receptor binding activity. J. Chem. Inf. Comput. Sci. 2000, 41, 219–224. [Google Scholar]
- Takac, M.J. Effects of substituents on the NMR features of basic bicyclic ring systems of fluoroquinolone antibiotics and the relationships between NMR chemical shifts, molecular descriptors and drug-likeness parameters. Acta Pharm. 2010, 60, 237–254. [Google Scholar] [CrossRef]
- Zieba, A.; Maslankiewicz, A.; Sitkowski, J. Spectral assignments and reference data. Magn. Reson. Chem. 2004, 42, 903–904. [Google Scholar] [CrossRef]
- Mhaidat, N.; Qandil, A.; Al-Balas, Q.; Hassan, M.; Jaradat, S.; Matalkah, A.; Thorne, R. Methoxyphenylcipro induces antitumor activity in human cancer cells. Arch. Pharm. Res. 2013, 36, 1023–1028. [Google Scholar] [CrossRef]
- Vazquez, J.L.; Merino, S.; Domenech, O.; Berlanga, M.; Vinas, M.; Montero, M.T.; Hernandez-Borrell, J. Determination of the partition coefficients of a homologous series of ciprofloxacin: Influence of the N-4 piperazinyl alkylation on the antimicrobial activity. Int. J. Pharm. 2001, 220, 53–62. [Google Scholar] [CrossRef]
- Takenouchi, T.; Tabata, F.; Iwata, Y.; Hanzawa, H.; Sugawara, M.; Ohya, S. Hydrophilicity of quinolones is not an exclusive factor for decreased activity in efflux-mediated resistant mutants of Staphylococcus aureus. Antimicrob. Agents Chemother. 1996, 40, 1835–1842. [Google Scholar]
- Csizmadia, F.; Tsantili-Kakoulidou, A.; Panderi, I.; Darvas, F. Prediction of distribution coefficient from structure. 1. Estimation method. J. Pharm. Sci. 1997, 86, 865–871. [Google Scholar] [CrossRef]
- Bartzatt, R.; Cirillo, S.L.G.; Cirillo, J.D. Design of ciprofloxacin derivatives that inhibit growth of methicillin resistant Staphylococcus aureus (MRSA) and methicillin susceptible Staphylococcus aureus (MSSA). Med. Chem. 2010, 6, 51–56. [Google Scholar] [CrossRef]
- Piddock, L.J.V.; Jin, Y.-F.; Ricci, V.; Asuquo, A.E. Quinolone accumulation by Pseudomonas aeruginosa, Staphylococcus aureus and Escherichia coli. J. Antimicrob. Chemother. 1999, 43, 61–70. [Google Scholar]
- Nguyen, H.A.; Grellet, J.; Dubois, V.; Saux, M.-C.; Quentin, C. Factors compromising the activity of moxifloxacin against intracellular Staphylococcus aureus. J. Antimicrob. Chemother. 2007, 59, 755–758. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Qandil, A.M.; Al-Zoubi, L.O.; Al-Bakri, A.G.; Amawi, H.A.; Al-Balas, Q.A.; Alkatheri, A.M.; Albekairy, A.M. Synthesis, Antibacterial Evaluation and QSAR of α-Substituted-N4-Acetamides of Ciprofloxacin and Norfloxacin. Antibiotics 2014, 3, 244-269. https://doi.org/10.3390/antibiotics3030244
Qandil AM, Al-Zoubi LO, Al-Bakri AG, Amawi HA, Al-Balas QA, Alkatheri AM, Albekairy AM. Synthesis, Antibacterial Evaluation and QSAR of α-Substituted-N4-Acetamides of Ciprofloxacin and Norfloxacin. Antibiotics. 2014; 3(3):244-269. https://doi.org/10.3390/antibiotics3030244
Chicago/Turabian StyleQandil, Amjad M., Lorca O. Al-Zoubi, Amal G. Al-Bakri, Haneen A. Amawi, Qosay A. Al-Balas, Abdulmalik M. Alkatheri, and Abdulkareem M. Albekairy. 2014. "Synthesis, Antibacterial Evaluation and QSAR of α-Substituted-N4-Acetamides of Ciprofloxacin and Norfloxacin" Antibiotics 3, no. 3: 244-269. https://doi.org/10.3390/antibiotics3030244