
Choline Binding Proteins from Streptococcus pneumoniae: A Dual Role as Enzybiotics and Targets for the Design of New Antimicrobials


Abstract
:1. Introduction
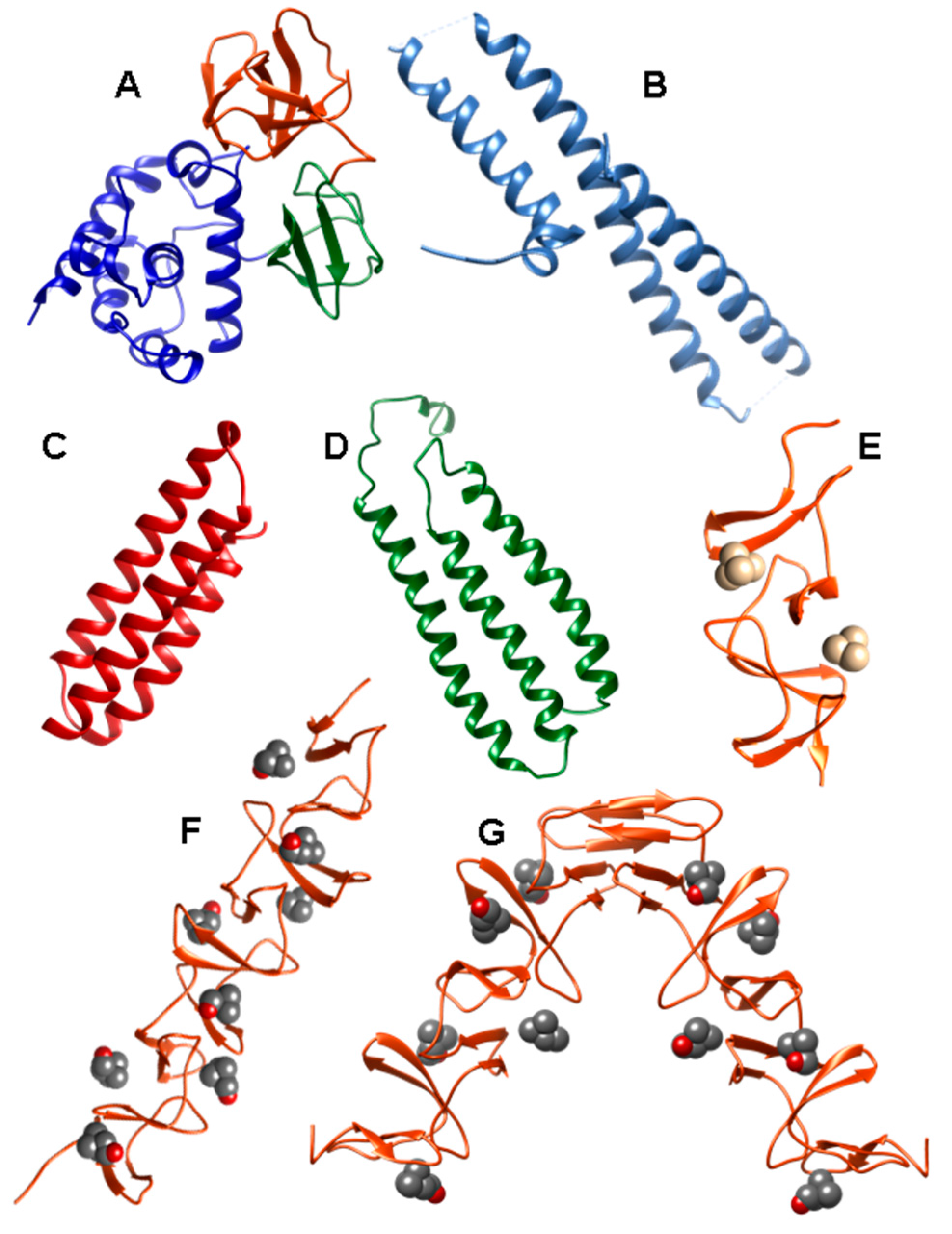
2. Pneumococcal Host-Encoded CBPs
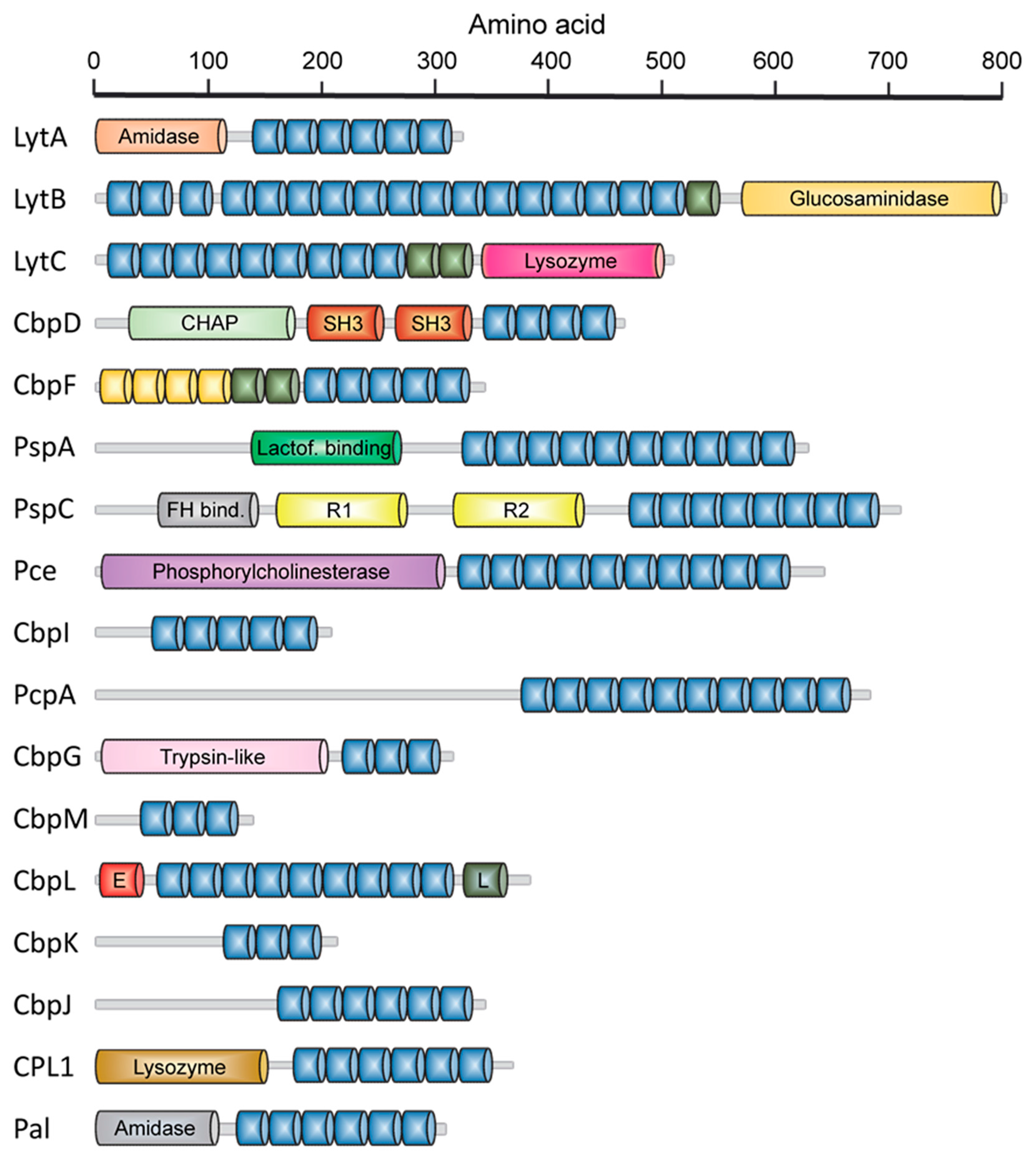
2.1. LytA Amidase
2.2. LytB N-Acetylglucosaminidase
2.3. LytC Lysozyme
2.4. CbpD Amidase/Endopeptidase
2.5. Choline-Binding Protein CbpF
2.6. Pneumococcal Surface Protein A (PspA)
2.7. Pneumococcal Surface Protein C (PspC)
2.8. Phosphorylcholine Esterase (Pce)
2.9. Other Choline Binding Proteins
3. Phage-Encoded CBPs
4. Actions to Control Pneumococcal Infections Based on CBPs
4.1. Enzybiotics
4.2. Inhibition of CBPs
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- O’Brien, K.L.; Wolfson, L.J.; Watt, J.P.; Henkle, E.; Deloria-Knoll, M.; McCall, N.; Lee, E.; Mulholland, K.; Levine, O.S.; Cherian, T.; et al. Burden of disease caused by Streptococcus pneumoniae in children younger than 5 years: Global estimates. Lancet 2009, 374, 893–902. [Google Scholar] [CrossRef]
- Blasi, F.; Mantero, M.; Santus, P.; Tarsia, P. Understanding the burden of pneumococcal disease in adults. Clin. Microbiol. Infect. 2012, 18 (Suppl. 5), 7–14. [Google Scholar] [CrossRef] [PubMed]
- UNICEF. Pneumonia: The Forgotten Killer Of Children; United Nations Children’s Emergency Fund: New York, NY, USA, 2006. [Google Scholar]
- UNICEF. Antimicrobial Resistance: Global Report on Surveillance; World Health Organization: Geneva, Switzerland, 2014. [Google Scholar]
- WHO. Weekly Epidemiological Record; World Health Organization: Geneva, Switzerland, 2007; pp. 93–104. [Google Scholar]
- Revai, K.; McCormick, D.P.; Patel, J.; Grady, J.J.; Saeed, K.; Chonmaitree, T. Effect of pneumococcal conjugate vaccine on nasopharyngeal bacterial colonization during acute otitis media. Pediatrics 2006, 117, 1823–1829. [Google Scholar] [CrossRef] [PubMed]
- Rodrigo, C.; Lim, W.S. The relevance of pneumococcal serotypes. Curr. Infect. Dis Rep. 2014, 16, 403. [Google Scholar] [CrossRef] [PubMed]
- Rose, M.; Zielen, S. Impact of infant immunization programs with pneumococcal conjugate vaccine in europe. Expert Rev. Vaccines 2009, 8, 1351–1364. [Google Scholar] [CrossRef] [PubMed]
- Whitney, C.G.; Farley, M.M.; Hadler, J.; Harrison, L.H.; Bennett, N.M.; Lynfield, R.; Reingold, A.; Cieslak, P.R.; Pilishvili, T.; Jackson, D.; et al. Decline in invasive pneumococcal disease after the introduction of protein-polysaccharide conjugate vaccine. N. Engl. J. Med. 2003, 348, 1737–1746. [Google Scholar] [CrossRef] [PubMed]
- Centers for disease control and prevention. Direct and indirect effects of routine vaccination of children with 7-valent pneumococcal conjugate vaccine on incidence of invasive pneumococcal disease-united states, 1998–2003. MMWR Morb. Mortal. Wkly. Rep. 2005, 54, 893–897. [Google Scholar]
- Paradiso, P.R. Advances in pneumococcal disease prevention: 13-Valent pneumococcal conjugate vaccine for infants and children. Clin. Infect. Dis. 2011, 52, 1241–1247. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Use of 13-valent pneumococcal conjugate vaccine and 23-valent pneumococcal polysaccharide vaccine for adults with immunocompromising conditions: Recommendations of the advisory committee on immunization practices (ACIP). MMWR Morb. Mortal. Wkly. Rep. 2012, 61, 816–819. [Google Scholar]
- Afonso, E.T.; Minamisava, R.; Bierrenbach, A.L.; Escalante, J.J.; Alencar, A.P.; Domingues, C.M.; Morais-Neto, O.L.; Toscano, C.M.; Andrade, A.L. Effect of 10-valent pneumococcal vaccine on pneumonia among children, Brazil. Emerg. Infect. Dis. 2013, 19, 589–597. [Google Scholar] [CrossRef] [PubMed]
- van der Linden, M.; Falkenhorst, G.; Perniciaro, S.; Imohl, M. Effects of infant pneumococcal conjugate vaccination on serotype distribution in invasive pneumococcal disease among children and adults in Germany. PLoS ONE 2015, 10, e0131494. [Google Scholar]
- Weil-Olivier, C.; van der Linden, M.; de Schutter, I.; Dagan, R.; Mantovani, L. Prevention of pneumococcal diseases in the post-seven valent vaccine era: A European perspective. BMC Infect. Dis. 2012, 12, 207. [Google Scholar] [CrossRef] [PubMed]
- Alexandre, C.; Dubos, F.; Courouble, C.; Pruvost, I.; Varon, E.; Hospital Network for Evaluating the Management of Common Childhood Diseases; Martinot, A. Rebound in the incidence of pneumococcal meningitis in Northern France: Effect of serotype replacement. Acta Paediatr. 2010, 99, 1686–1690. [Google Scholar] [CrossRef] [PubMed]
- Johnson, H.L.; Deloria-Knoll, M.; Levine, O.S.; Stoszek, S.K.; Freimanis Hance, L.; Reithinger, R.; Muenz, L.R.; O’Brien, K.L. Systematic evaluation of serotypes causing invasive pneumococcal disease among children under five: The pneumococcal global serotype project. PLoS Med. 2010, 7. [Google Scholar] [CrossRef] [PubMed]
- Coates, A.R.; Halls, G.; Hu, Y. Novel classes of antibiotics or more of the same? Br. J. Pharmacol. 2011, 163, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Albrich, W.C.; Monnet, D.L.; Harbarth, S. Antibiotic selection pressure and resistance in Streptococcus pneumoniae and Streptococcus pyogenes. Emerg. Infect. Dis. 2004, 10, 514–517. [Google Scholar] [CrossRef] [PubMed]
- Henriques-Normark, B. Molecular epidemiology and mechanisms for antibiotic resistance in Streptococcus pneumoniae. In Molecular Biology of Streptococci; Hakenbeck, R., Chhatwal, G.S., Eds.; Horizon Bioscience: Norfolk, UK, 2007; pp. 269–290. [Google Scholar]
- Todorova, K.; Maurer, P.; Rieger, M.; Becker, T.; Bui, N.K.; Gray, J.; Vollmer, W.; Hakenbeck, R. Transfer of penicillin resistance from Streptococcus oralis to Streptococcus pneumoniae identifies mure as resistance determinant. Mol. Microbiol. 2015, 97, 866–880. [Google Scholar] [CrossRef] [PubMed]
- Felmingham, D.; Canton, R.; Jenkins, S.G. Regional trends in beta-lactam, macrolide, fluoroquinolone and telithromycin resistance among streptococcus pneumoniae isolates 2001–2004. J. Infect. 2007, 55, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Lovering, A.L.; Safadi, S.S.; Strynadka, N.C. Structural perspective of peptidoglycan biosynthesis and assembly. Annu. Rev. Biochem. 2012, 81, 451–478. [Google Scholar] [CrossRef] [PubMed]
- Skov Sorensen, U.B.; Blom, J.; Birch-Andersen, A.; Henrichsen, J. Ultrastructural localization of capsules, cell wall polysaccharide, cell wall proteins, and F antigen in pneumococci. Infect. Immun. 1988, 56, 1890–1896. [Google Scholar] [PubMed]
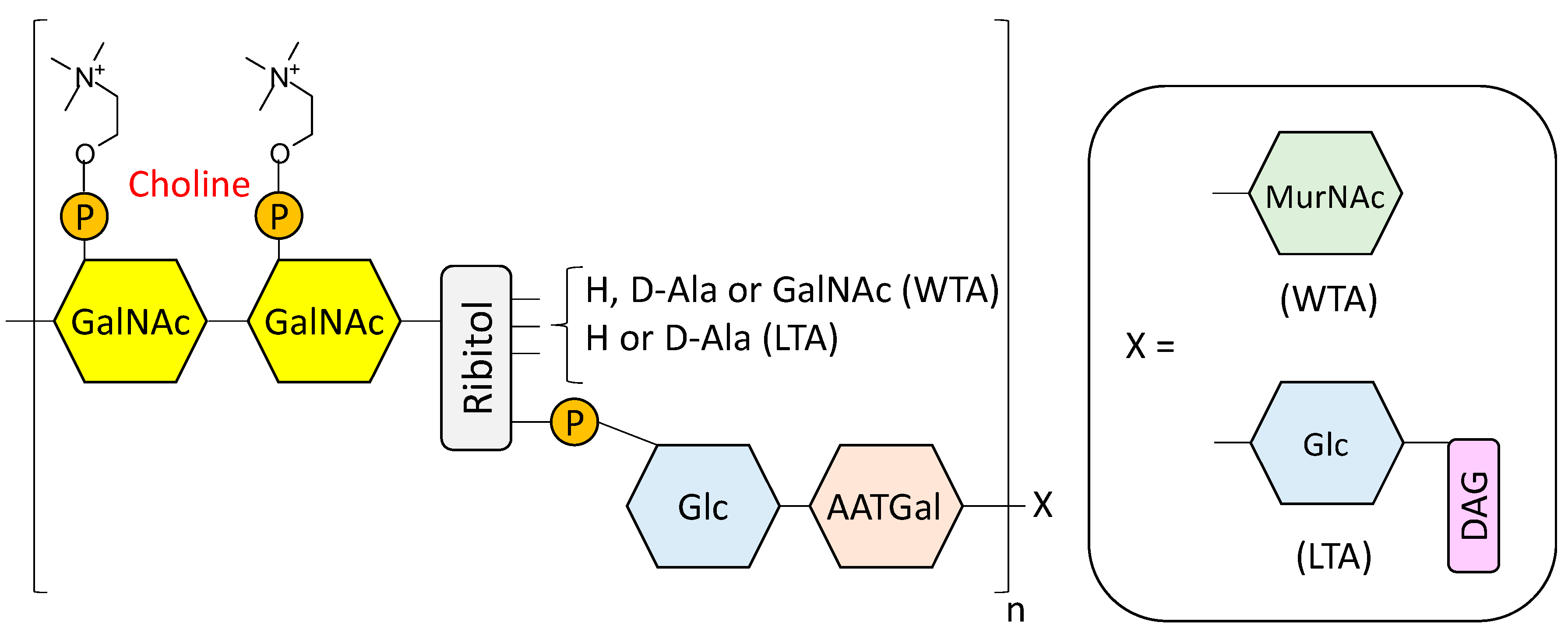
- Fischer, W. Lipoteichoic Acids and Lipoglycans. In Bacterial cell wall; Ghuysen, J.M., Hakenbeck, R., Eds.; Elsevier Science B.V: Amsterdam, The Netherlands, 1994; pp. 199–215. [Google Scholar]
- Seo, H.S.; Cartee, R.T.; Pritchard, D.G.; Nahm, M.H. A new model of pneumococcal lipoteichoic acid structure resolves biochemical, biosynthetic, and serologic inconsistencies of the current model. J. Bacteriol. 2008, 190, 2379–2387. [Google Scholar] [CrossRef] [PubMed]
- Gisch, N.; Kohler, T.; Ulmer, A.J.; Muthing, J.; Pribyl, T.; Fischer, K.; Lindner, B.; Hammerschmidt, S.; Zahringer, U. Structural reevaluation of Streptococcus pneumoniae lipoteichoic acid and new insights into its immunostimulatory potency. J. Biol. Chem. 2013, 288, 15654–15667. [Google Scholar] [CrossRef] [PubMed]
- Denapaite, D.; Bruckner, R.; Hakenbeck, R.; Vollmer, W. Biosynthesis of teichoic acids in Streptococcus pneumoniae and closely related species: Lessons from genomes. Microb. Drug Resist. 2012, 18, 344–358. [Google Scholar] [CrossRef] [PubMed]
- Gisch, N.; Peters, K.; Zähringer, U.; Vollmer, W. The pneumococcal cell wall. In Streptococcus Pneumoniae: Molecular Mechanisms of Host-Pathogen Interactions, 1st ed.; Brown, J.M., Hammerschmidt, S., Orihuela, C., Eds.; Academic Press: Cambridge, MA, USA, 2015; pp. 145–167. [Google Scholar]
- Fischer, H.; Tomasz, A. Peptidoglycan cross-linking and teichoic acid attachment in Streptococcus pneumoniae. J. Bacteriol. 1985, 163, 46–54. [Google Scholar] [PubMed]
- Fischer, W.; Behr, T.; Hartmann, R.; Peter-Katalinic, J.; Egge, H. Teichoic acid and lipoteichoic acid of Streptococcus pneumoniae possess identical chain structures. Eur. J. Biochem. 1993, 215, 851–857. [Google Scholar] [CrossRef] [PubMed]
- Behr, T.; Fischer, W.; Peter-Katalinic, J.; Egge, H. The structure of pneumococcal lipoteichoic acid. Improved preparation, chemical and mass spectrometric studies. Eur. J. Biochem. 1992, 207, 1063–1075. [Google Scholar] [CrossRef] [PubMed]
- Jennings, H.J.; Lugowski, C.; Young, N.M. Structure of the complex polysaccharide c-substance from Streptococcus pneumoniae type 1. Biochemistry 1980, 19, 4712–4719. [Google Scholar] [CrossRef] [PubMed]
- Vialle, S.; Sepulcri, P.; Dubayle, J.; Talaga, P. The teichoic acid (C-polysaccharide) synthesized by Streptococcus pneumoniae serotype 5 has a specific structure. Carbohydr. Res. 2005, 340, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Brundish, D.E.; Baddiley, J. Pneumococcal C-substance, a ribitol teichoic acid containing choline phosphate. Biochem. J. 1968, 110, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, C.; Jansson, P.E.; Skov Sorensen, U.B. The pneumococcal common antigen C-polysaccharide occurs in different forms. Mono-substituted or di-substituted with phosphocholine. Eur. J. Biochem. 1999, 265, 1091–1097. [Google Scholar] [CrossRef] [PubMed]
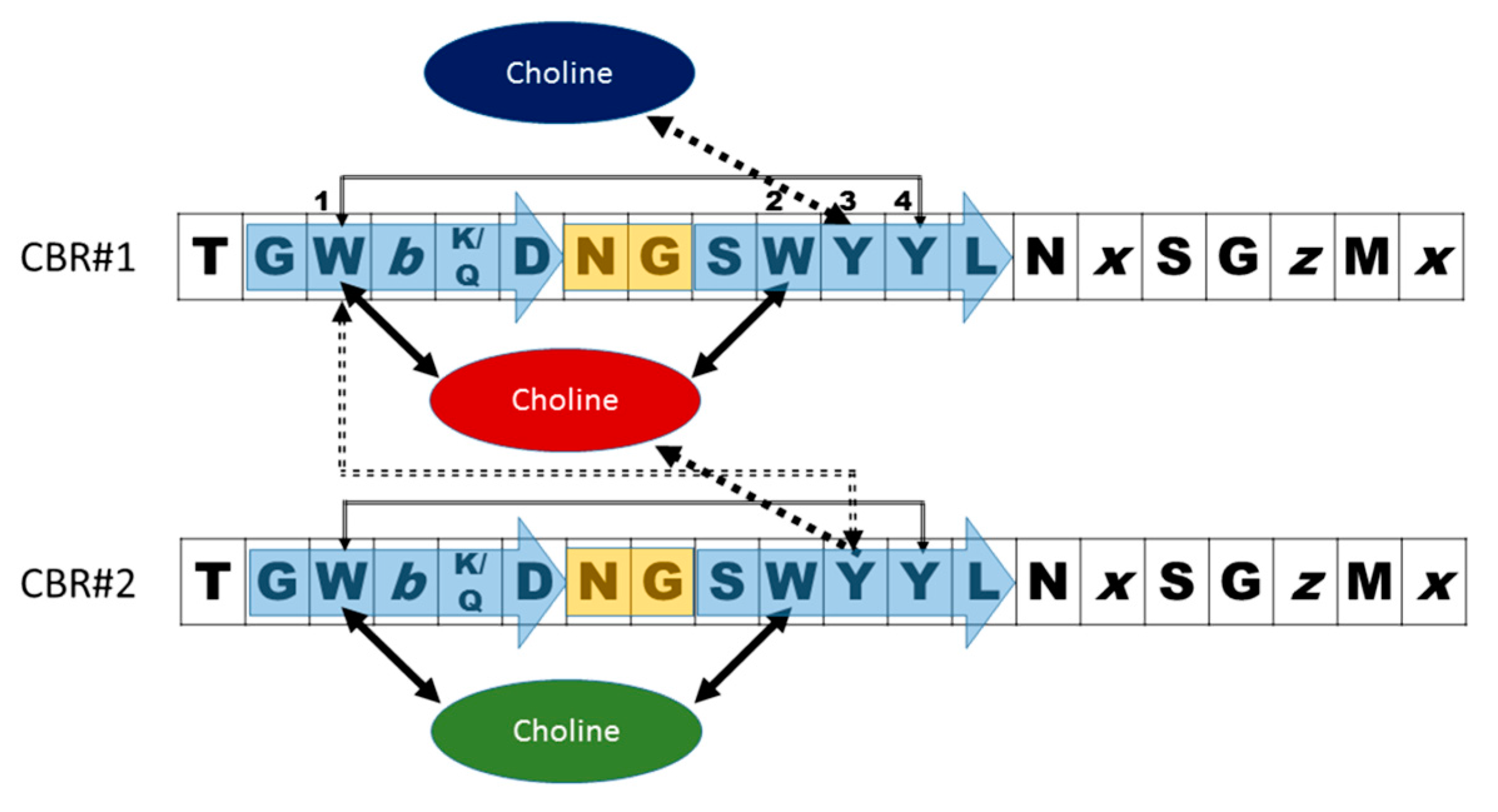
- Garcia, J.L.; Sanchez-Beato, A.R.; Medrano, F.J.; Lopez, R. Versatility of choline-binding domain. Microb. Drug Resist. 1998, 4, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Galán-Bartual, S.P.-D., I; García, P.; Hermoso, J.A. Structure and function of choline-binding proteins. In Streptococcus Pneumoniae: Molecular Mechanisms of Host-Pathogen Interactions, 1st ed.; Brown, J.M., Hammerschmidt, S., Orihuela, C., Eds.; Academic Press: Cambridge, MA, USA, 2015; pp. 207–226. [Google Scholar]
- Weiser, J.N.; Goldberg, J.B.; Pan, N.; Wilson, L.; Virji, M. The phosphorylcholine epitope undergoes phase variation on a 43-kilodalton protein in Pseudomonas aeruginosa and on pili of Neisseria meningitidis and Neisseria gonorrhoeae. Infect. Immun. 1998, 66, 4263–4267. [Google Scholar] [PubMed]
- Weiser, J.N.; Pan, N.; McGowan, K.L.; Musher, D.; Martin, A.; Richards, J. Phosphorylcholine on the lipopolysaccharide of Haemophilus influenzae contributes to persistence in the respiratory tract and sensitivity to serum killing mediated by C-reactive protein. J. Exp. Med. 1998, 187, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Gould, J.M.; Weiser, J.N. The inhibitory effect of C-reactive protein on bacterial phosphorylcholine platelet-activating factor receptor-mediated adherence is blocked by surfactant. J. Infect. Dis. 2002, 186, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Hakenbeck, R.; Madhour, A.; Denapaite, D.; Bruckner, R. Versatility of choline metabolism and choline-binding proteins in Streptococcus pneumoniae and commensal streptococci. FEMS Microbiol. Rev. 2009, 33, 572–586. [Google Scholar] [CrossRef] [PubMed]
- Fischer, W. Phosphocholine of pneumococcal teichoic acids: Role in bacterial physiology and pneumococcal infection. Res. Microbiol. 2000, 151, 421–427. [Google Scholar] [CrossRef]
- Volanakis, J.E.; Kaplan, M.H. Specificity of c-reactive protein for choline phosphate residues of pneumococcal C-polysaccharide. Proc. Soc. Exp. Biol. Med. 1971, 136, 612–614. [Google Scholar] [CrossRef] [PubMed]
- Vassal-Stermann, E.; Lacroix, M.; Gout, E.; Laffly, E.; Pedersen, C.M.; Martin, L.; Amoroso, A.; Schmidt, R.R.; Zahringer, U.; Gaboriaud, C.; et al. Human L-ficolin recognizes phosphocholine moieties of pneumococcal teichoic acid. J. Immunol. 2014, 193, 5699–5708. [Google Scholar] [CrossRef] [PubMed]
- Cundell, D.R.; Gerard, N.P.; Gerard, C.; Idanpaan-Heikkila, I.; Tuomanen, E.I. Streptococcus pneumoniae anchor to activated human cells by the receptor for platelet-activating factor. Nature 1995, 377, 435–438. [Google Scholar] [CrossRef] [PubMed]
- Kharat, A.S.; Tomasz, A. Drastic reduction in the virulence of Streptococcus pneumoniae expressing type 2 capsular polysaccharide but lacking choline residues in the cell wall. Mol. Microbiol. 2006, 60, 93–107. [Google Scholar] [CrossRef] [PubMed]
- Lopez, R.; Garcia, E. Recent trends on the molecular biology of pneumococcal capsules, lytic enzymes, and bacteriophage. FEMS Microbiol. Rev. 2004, 28, 553–580. [Google Scholar] [CrossRef] [PubMed]
- Rane, L.; Subbarow, Y. Nutritional requirements of the pneumococcus: I. Growth factors for types I, II, V, VII, VIII. J. Bacteriol. 1940, 40, 695–704. [Google Scholar] [PubMed]
- Badger, E. The structural specificity of choline for the growth of type iii pneumococcus. J. Biol. Chem. 1944, 153, 183–191. [Google Scholar]
- Yother, J.; Leopold, K.; White, J.; Fischer, W. Generation and properties of a streptococcus pneumoniae mutant which does not require choline or analogs for growth. J. Bacteriol. 1998, 180, 2093–2101. [Google Scholar] [PubMed]
- Severin, A.; Horne, D.; Tomasz, A. Autolysis and cell wall degradation in a choline-independent strain of streptococcus pneumoniae. Microb. Drug Resist. 1997, 3, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Damjanovic, M.; Kharat, A.S.; Eberhardt, A.; Tomasz, A.; Vollmer, W. The essential tacF gene is responsible for the choline-dependent growth phenotype of Streptococcus pneumoniae. J. Bacteriol. 2007, 189, 7105–7111. [Google Scholar] [CrossRef] [PubMed]
- Tomasz, A. Biological consequences of the replacement of choline by ethanolamine in the cell wall of pneumococcus: Chanin formation, loss of transformability, and loss of autolysis. Proc. Natl. Acad. Sci. USA 1968, 59, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Ware, D.; Watt, J.; Swiatlo, E. Utilization of putrescine by Streptococcus pneumoniae during growth in choline-limited medium. J. Microbiol. 2005, 43, 398–405. [Google Scholar] [PubMed]
- Perez-Dorado, I.; Galan-Bartual, S.; Hermoso, J.A. Pneumococcal surface proteins: When the whole is greater than the sum of its parts. Mol. Oral Microbiol. 2012, 27, 221–245. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, S.; Hammerschmidt, S. Versatility of pneumococcal surface proteins. Microbiology 2006, 152, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Frolet, C.; Beniazza, M.; Roux, L.; Gallet, B.; Noirclerc-Savoye, M.; Vernet, T.; Di Guilmi, A.M. New adhesin functions of surface-exposed pneumococcal proteins. BMC Microbiol. 2010, 10, 190. [Google Scholar] [CrossRef] [PubMed]
- Garcia, E.; Garcia, J.L.; Garcia, P.; Arraras, A.; Sanchez-Puelles, J.M.; Lopez, R. Molecular evolution of lytic enzymes of Streptococcus pneumoniae and its bacteriophages. Proc. Natl. Acad. Sci. USA 1988, 85, 914–918. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Puelles, J.M.; Sanz, J.M.; Garcia, J.L.; Garcia, E. Cloning and expression of gene fragments encoding the choline-binding domain of pneumococcal murein hydrolases. Gene 1990, 89, 69–75. [Google Scholar] [CrossRef]
- Garcia, P.; Garcia, J.L.; Garcia, E.; Sanchez-Puelles, J.M.; Lopez, R. Modular organization of the lytic enzymes of Streptococcus pneumoniae and its bacteriophages. Gene 1990, 86, 81–88. [Google Scholar] [CrossRef]
- Sanz, J.M.; Diaz, E.; Garcia, J.L. Studies on the structure and function of the N-terminal domain of the pneumococcal murein hydrolases. Mol. Microbiol. 1992, 6, 921–931. [Google Scholar] [CrossRef] [PubMed]
- Yother, J.; White, J.M. Novel surface attachment mechanism of the Streptococcus pneumoniae protein PspA. J. Bacteriol. 1994, 176, 2976–2985. [Google Scholar] [PubMed]
- Brooks-Walter, A.; Briles, D.E.; Hollingshead, S.K. The PspC gene of Streptococcus pneumoniae encodes a polymorphic protein, PspC, which elicits cross-reactive antibodies to PspA and provides immunity to pneumococcal bacteremia. Infect. Immun. 1999, 67, 6533–6542. [Google Scholar] [PubMed]
- Pfam Database. Available online: http://pfam.xfam.org/family/PF01473 (accessed on 1 June 2016).
- Diaz, E.; Lopez, R.; Garcia, J.L. Chimeric phage-bacterial enzymes: A clue to the modular evolution of genes. Proc. Natl. Acad. Sci. USA 1990, 87, 8125–8129. [Google Scholar] [CrossRef] [PubMed]
- Diaz, E.; Lopez, R.; Garcia, J.L. Chimeric pneumococcal cell wall lytic enzymes reveal important physiological and evolutionary traits. J. Biol. Chem. 1991, 266, 5464–5471. [Google Scholar] [PubMed]
- Croux, C.; Ronda, C.; Lopez, R.; Garcia, J.L. Interchange of functional domains switches enzyme specificity: Construction of a chimeric pneumococcal-clostridial cell wall lytic enzyme. Mol. Microbiol. 1993, 9, 1019–1025. [Google Scholar] [CrossRef] [PubMed]
- Lopez, R.; Garcia, E.; Garcia, P.; Garcia, J.L. The pneumococcal cell wall degrading enzymes: A modular design to create new lysins? Microb. Drug Resist. 1997, 3, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Doolittle, R.F. The multiplicity of domains in proteins. Annu. Rev. Biochem. 1995, 64, 287–314. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.; Clamp, M.; Barton, G.J. Jalview version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed]
- Jalview. Available online: http://www.jalview.org/ (accessed on 1 June 2016).
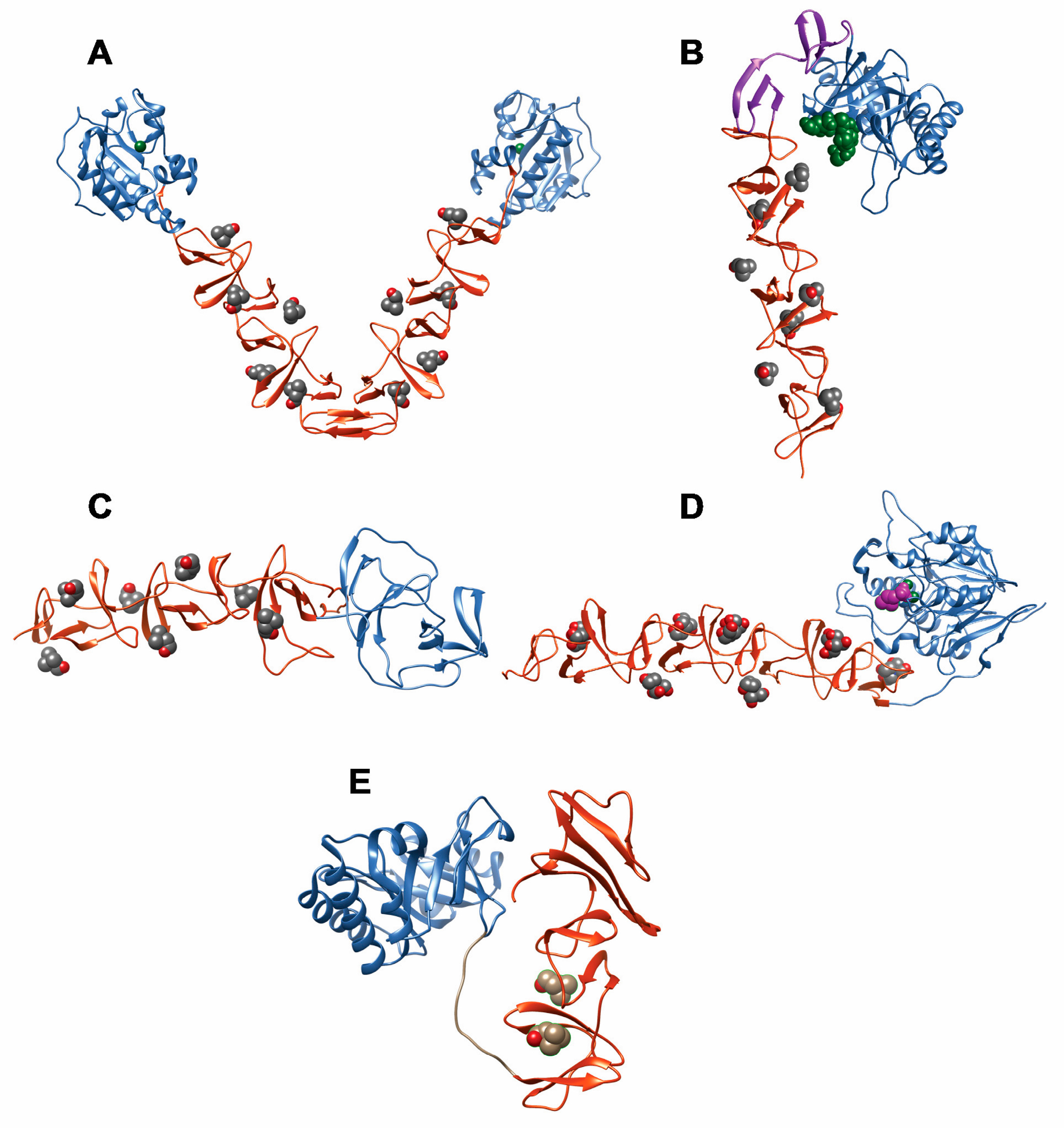
- Fernandez-Tornero, C.; Garcia, E.; Lopez, R.; Gimenez-Gallego, G.; Romero, A. Two new crystal forms of the choline-binding domain of the major pneumococcal autolysin: Insights into the dynamics of the active homodimer. J. Mol. Biol. 2002, 321, 163–173. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Quiñonero, D.; Frontera, A.; Deyà, P.M.; Alkorta, I.; Elguero, J. Interaction of positively and negatively charged aromatic hydrocarbons with benzene and triphenylene: Towards a model of pure organic insulators. Chem. Phys. Lett 2008, 460, 406–410. [Google Scholar] [CrossRef]
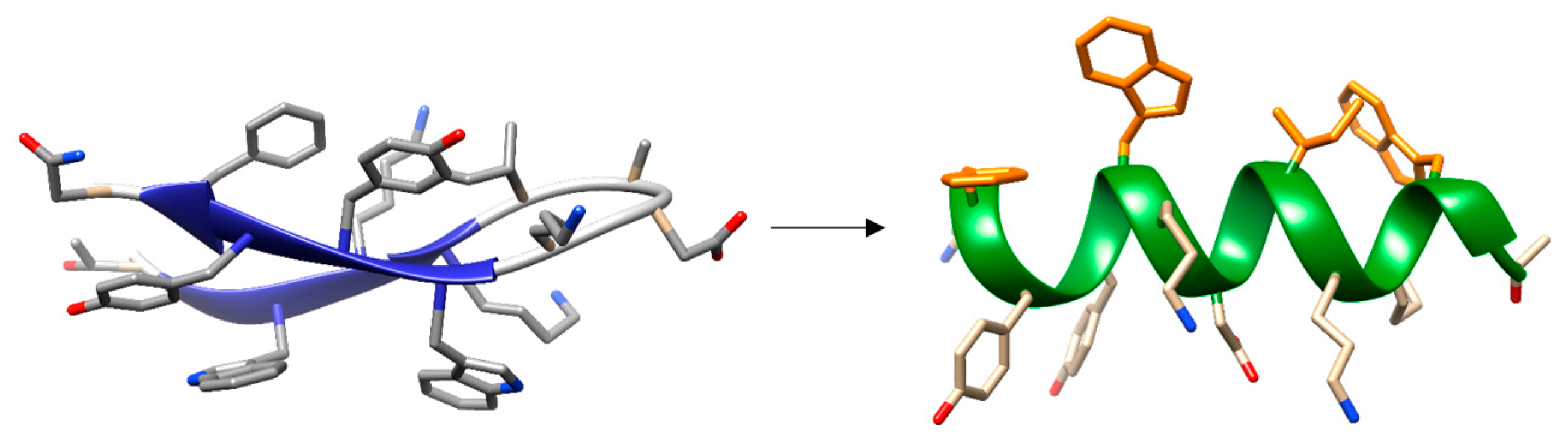
- Maestro, B.; Santiveri, C.M.; Jimenez, M.A.; Sanz, J.M. Structural autonomy of a beta-hairpin peptide derived from the pneumococcal choline-binding protein LytA. Protein Eng. Des. Sel. 2011, 24, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Zamora-Carreras, H.; Maestro, B.; Strandberg, E.; Ulrich, A.S.; Sanz, J.M.; Jimenez, M.A. Micelle-triggered beta-hairpin to alpha-helix transition in a 14-residue peptide from a choline-binding repeat of the pneumococcal autolysin LytA. Chemistry 2015, 21, 8076–8089. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.L.; Diaz, E.; Romero, A.; Garcia, P. Carboxy-terminal deletion analysis of the major pneumococcal autolysin. J. Bacteriol. 1994, 176, 4066–4072. [Google Scholar] [PubMed]
- Varea, J.; Saiz, J.L.; Lopez-Zumel, C.; Monterroso, B.; Medrano, F.J.; Arrondo, J.L.; Iloro, I.; Laynez, J.; Garcia, J.L.; Menendez, M. Do sequence repeats play an equivalent role in the choline-binding module of pneumococcal LytA amidase? J. Biol. Chem. 2000, 275, 26842–26855. [Google Scholar] [CrossRef] [PubMed]
- Perez-Dorado, I.; Gonzalez, A.; Morales, M.; Sanles, R.; Striker, W.; Vollmer, W.; Mobashery, S.; Garcia, J.L.; Martinez-Ripoll, M.; Garcia, P.; et al. Insights into pneumococcal fratricide from the crystal structures of the modular killing factor LytC. Nat. Struct. Mol. Biol. 2010, 17, 576–581. [Google Scholar] [CrossRef] [PubMed]
- Hermoso, J.A.; Monterroso, B.; Albert, A.; Galan, B.; Ahrazem, O.; Garcia, P.; Martinez-Ripoll, M.; Garcia, J.L.; Menendez, M. Structural basis for selective recognition of pneumococcal cell wall by modular endolysin from phage Cp-1. Structure 2003, 11, 1239–1249. [Google Scholar] [CrossRef] [PubMed]
- Monterroso, B.; Saiz, J.L.; Garcia, P.; Garcia, J.L.; Menendez, M. Insights into the structure-function relationships of pneumococcal cell wall lysozymes, LytC and Cpl-1. J. Biol. Chem. 2008, 283, 28618–28628. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Cheng, W.; Morlot, C.; Bai, X.H.; Jiang, Y.L.; Wang, W.; Roper, D.I.; Vernet, T.; Dong, Y.H.; Chen, Y.; et al. Full-length structure of the major autolysin LytA. Acta Crystallogr. D Biol. Crystallogr. 2015, 71, 1373–1381. [Google Scholar] [CrossRef] [PubMed]
- Medrano, F.J.; Gasset, M.; Lopez-Zumel, C.; Usobiaga, P.; Garcia, J.L.; Menendez, M. Structural characterization of the unligated and choline-bound forms of the major pneumococcal autolysin LytA amidase. Conformational transitions induced by temperature. J. Biol. Chem. 1996, 271, 29152–29161. [Google Scholar] [PubMed]
- Wizemann, T.M.; Heinrichs, J.H.; Adamou, J.E.; Erwin, A.L.; Kunsch, C.; Choi, G.H.; Barash, S.C.; Rosen, C.A.; Masure, H.R.; Tuomanen, E.; et al. Use of a whole genome approach to identify vaccine molecules affording protection against Streptococcus pneumoniae infection. Infect. Immun. 2001, 69, 1593–1598. [Google Scholar] [CrossRef] [PubMed]
- Briles, D.E.; Hollingshead, S.; Brooks-Walter, A.; Nabors, G.S.; Ferguson, L.; Schilling, M.; Gravenstein, S.; Braun, P.; King, J.; Swift, A. The potential to use PspA and other pneumococcal proteins to elicit protection against pneumococcal infection. Vaccine 2000, 18, 1707–1711. [Google Scholar] [CrossRef]
- Briles, D.E.; Hollingshead, S.K.; Swiatlo, E.; Brooks-Walter, A.; Szalai, A.; Virolainen, A.; McDaniel, L.S.; Benton, K.A.; White, P.; Prellner, K.; et al. PspA and PspC: Their potential for use as pneumococcal vaccines. Microb. Drug Resist. 1997, 3, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Nabors, G.S.; Braun, P.A.; Herrmann, D.J.; Heise, M.L.; Pyle, D.J.; Gravenstein, S.; Schilling, M.; Ferguson, L.M.; Hollingshead, S.K.; Briles, D.E.; et al. Immunization of healthy adults with a single recombinant pneumococcal surface protein A (PspA) variant stimulates broadly cross-reactive antibodies to heterologous PspA molecules. Vaccine 2000, 18, 1743–1754. [Google Scholar] [CrossRef]
- Ochs, M.M.; Bartlett, W.; Briles, D.E.; Hicks, B.; Jurkuvenas, A.; Lau, P.; Ren, B.; Millar, A. Vaccine-induced human antibodies to PspA augment complement c3 deposition on Streptococcus pneumoniae. Microb. Pathog. 2008, 44, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Howard, L.V.; Gooder, H. Specificity of the autolysin of Streptococcus (diplococcus) pneumoniae. J. Bacteriol. 1974, 117, 796–804. [Google Scholar] [PubMed]
- Gillespie, S.H.; McHugh, T.D.; Ayres, H.; Dickens, A.; Efstratiou, A.; Whiting, G.C. Allelic variation in Streptococcus pneumoniae autolysin (N-acetyl muramoyl-l-alanine amidase). Infect. Immun. 1997, 65, 3936–3938. [Google Scholar] [PubMed]
- Llull, D.; Lopez, R.; Garcia, E. Characteristic signatures of the LytA gene provide a basis for rapid and reliable diagnosis of Streptococcus pneumoniae infections. J. Clin. Microbiol. 2006, 44, 1250–1256. [Google Scholar] [CrossRef] [PubMed]
- Martner, A.; Dahlgren, C.; Paton, J.C.; Wold, A.E. Pneumolysin released during Streptococcus pneumoniae autolysis is a potent activator of intracellular oxygen radical production in neutrophils. Infect. Immun. 2008, 76, 4079–4087. [Google Scholar] [CrossRef] [PubMed]
- Eldholm, V.; Johnsborg, O.; Haugen, K.; Ohnstad, H.S.; Havarstein, L.S. Fratricide in Streptococcus pneumoniae: Contributions and role of the cell wall hydrolases CbpD, LytA and LytC. Microbiology 2009, 155, 2223–2234. [Google Scholar] [CrossRef] [PubMed]
- Martner, A.; Skovbjerg, S.; Paton, J.C.; Wold, A.E. Streptococcus pneumoniae autolysis prevents phagocytosis and production of phagocyte-activating cytokines. Infect. Immun. 2009, 77, 3826–3837. [Google Scholar] [CrossRef] [PubMed]
- Tomasz, A.; Albino, A.; Zanati, E. Multiple antibiotic resistance in a bacterium with suppressed autolytic system. Nature 1970, 227, 138–140. [Google Scholar] [CrossRef] [PubMed]
- Tomasz, A.; Waks, S. Mechanism of action of penicillin: Triggering of the pneumococcal autolytic enzyme by inhibitors of cell wall synthesis. Proc. Natl. Acad. Sci. USA 1975, 72, 4162–4166. [Google Scholar] [CrossRef] [PubMed]
- Moreillon, P.; Tomasz, A. Penicillin resistance and defective lysis in clinical isolates of pneumococci: Evidence for two kinds of antibiotic pressure operating in the clinical environment. J. Infect. Dis. 1988, 157, 1150–1157. [Google Scholar] [CrossRef] [PubMed]
- Moscoso, M.; Domenech, M.; Garcia, E. Vancomycin tolerance in clinical and laboratory Streptococcus pneumoniae isolates depends on reduced enzyme activity of the major LytA autolysin or cooperation between CiahH histidine kinase and capsular polysaccharide. Mol. Microbiol. 2010, 77, 1052–1064. [Google Scholar] [PubMed]
- Kietzman, C.C.; Gao, G.; Mann, B.; Myers, L.; Tuomanen, E.I. Dynamic capsule restructuring by the main pneumococcal autolysin LytA in response to the epithelium. Nat. Commun. 2016, 7, 10859. [Google Scholar] [CrossRef] [PubMed]
- Mellroth, P.; Daniels, R.; Eberhardt, A.; Ronnlund, D.; Blom, H.; Widengren, J.; Normark, S.; Henriques-Normark, B. LytA, major autolysin of Streptococcus pneumoniae, requires access to nascent peptidoglycan. J. Biol. Chem. 2012, 287, 11018–11029. [Google Scholar] [CrossRef] [PubMed]
- Mellroth, P.; Sandalova, T.; Kikhney, A.; Vilaplana, F.; Hesek, D.; Lee, M.; Mobashery, S.; Normark, S.; Svergun, D.; Henriques-Normark, B.; et al. Structural and functional insights into peptidoglycan access for the lytic amidase Lyta of Streptococcus pneumoniae. MBio 2014, 5, e01120–e01113. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Tornero, C.; Lopez, R.; Garcia, E.; Gimenez-Gallego, G.; Romero, A. A novel solenoid fold in the cell wall anchoring domain of the pneumococcal virulence factor LytA. Nat. Struct. Biol. 2001, 8, 1020–1024. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Tornero, C.; Garcia, E.; de Pascual-Teresa, B.; Lopez, R.; Gimenez-Gallego, G.; Romero, A. Ofloxacin-like antibiotics inhibit pneumococcal cell wall-degrading virulence factors. J. Biol. Chem. 2005, 280, 19948–19957. [Google Scholar] [CrossRef] [PubMed]
- Usobiaga, P.; Medrano, F.J.; Gasset, M.; Garcia, J.L.; Saiz, J.L.; Rivas, G.; Laynez, J.; Menendez, M. Structural organization of the major autolysin from Streptococcus pneumoniae. J. Biol. Chem. 1996, 271, 6832–6838. [Google Scholar] [PubMed]
- Maestro, B.; Sanz, J.M. Accumulation of partly folded states in the equilibrium unfolding of the pneumococcal choline-binding module C-LytA. Biochem. J. 2005, 387, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.; Lopez, R.; Garcia, E. Key role of amino acid residues in the dimerization and catalytic activation of the autolysin LytA, an important virulence factor in Streptococcus pneumoniae. J. Biol. Chem. 2007, 282, 17729–17737. [Google Scholar] [CrossRef] [PubMed]
- Maestro, B.; Sanz, J.M. Extensive unfolding of the C-LytA choline-binding module by submicellar concentrations of sodium dodecyl sulphate. FEBS Lett. 2007, 581, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Tomasz, A.; Westphal, M. Abnormal autolytic enzyme in a pneumococus with altered teichoic acid composition. Proc. Natl. Acad. Sci. USA 1971, 68, 2627–2630. [Google Scholar] [CrossRef] [PubMed]
- De Las Rivas, B.; Garcia, J.L.; Lopez, R.; Garcia, P. Purification and polar localization of pneumococcal LytB, a putative endo-beta-N-acetylglucosaminidase: The chain-dispersing murein hydrolase. J. Bacteriol. 2002, 184, 4988–5000. [Google Scholar] [CrossRef] [PubMed]
- Garcia, P.; Gonzalez, M.P.; Garcia, E.; Lopez, R.; Garcia, J.L. LytB, a novel pneumococcal murein hydrolase essential for cell separation. Mol. Microbiol. 1999, 31, 1275–1281. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Puelles, J.M.; Ronda, C.; Garcia, J.L.; Garcia, P.; Lopez, R.; Garcia, E. Searching for autolysin functions. Characterization of a pneumococcal mutant deleted in the LytA gene. Eur. J. Biochem. 1986, 158, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Rico-Lastres, P.; Diez-Martinez, R.; Iglesias-Bexiga, M.; Bustamante, N.; Aldridge, C.; Hesek, D.; Lee, M.; Mobashery, S.; Gray, J.; Vollmer, W.; et al. Substrate recognition and catalysis by LytB, a pneumococcal peptidoglycan hydrolase involved in virulence. Sci. Rep. 2015, 5, 16198. [Google Scholar] [CrossRef] [PubMed]
- Moscoso, M.; Garcia, E.; Lopez, R. Biofilm formation by Streptococcus pneumoniae: Role of choline, extracellular DNA, and capsular polysaccharide in microbial accretion. J. Bacteriol. 2006, 188, 7785–7795. [Google Scholar] [CrossRef] [PubMed]
- Gosink, K.K.; Mann, E.R.; Guglielmo, C.; Tuomanen, E.I.; Masure, H.R. Role of novel choline binding proteins in virulence of Streptococcus pneumoniae. Infect. Immun. 2000, 68, 5690–5695. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Sevillano, E.; Moscoso, M.; Garcia, P.; Garcia, E.; Yuste, J. Nasopharyngeal colonization and invasive disease are enhanced by the cell wall hydrolases lytb and lytc of Streptococcus pneumoniae. PLoS ONE 2011, 6, e23626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moscoso, M.; Obregon, V.; Lopez, R.; Garcia, J.L.; Garcia, E. Allelic variation of polymorphic locus LytB, encoding a choline-binding protein, from streptococci of the mitis group. Appl. Environ. Microbiol. 2005, 71, 8706–8713. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.H.; Chen, H.J.; Jiang, Y.L.; Wen, Z.; Huang, Y.; Cheng, W.; Li, Q.; Qi, L.; Zhang, J.R.; Chen, Y.; et al. Structure of pneumococcal peptidoglycan hydrolase LytB reveals insights into the bacterial cell wall remodeling and pathogenesis. J. Biol. Chem. 2014, 289, 23403–23416. [Google Scholar] [CrossRef] [PubMed]
- Garcia, P.; Paz Gonzalez, M.; Garcia, E.; Garcia, J.L.; Lopez, R. The molecular characterization of the first autolytic lysozyme of Streptococcus pneumoniae reveals evolutionary mobile domains. Mol. Microbiol. 1999, 33, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Claverys, J.P.; Havarstein, L.S. Cannibalism and fratricide: Mechanisms and raisons d’etre. Nat. Rev. Microbiol. 2007, 5, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Johnsborg, O.; Eldholm, V.; Bjornstad, M.L.; Havarstein, L.S. A predatory mechanism dramatically increases the efficiency of lateral gene transfer in Streptococcus pneumoniae and related commensal species. Mol. Microbiol. 2008, 69, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Chi, F.; Nolte, O.; Bergmann, C.; Ip, M.; Hakenbeck, R. Crossing the barrier: Evolution and spread of a major class of mosaic PBP2x in Streptococcus pneumoniae, S. mitis and S. oralis. Int. J. Med. Microbiol. 2007, 297, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Brueggemann, A.B.; Pai, R.; Crook, D.W.; Beall, B. Vaccine escape recombinants emerge after pneumococcal vaccination in the United States. PLoS Pathog. 2007, 3, e168. [Google Scholar] [CrossRef] [PubMed]
- Johnsborg, O.; Havarstein, L.S. Regulation of natural genetic transformation and acquisition of transforming DNA in Streptococcus pneumoniae. FEMS Microbiol. Rev. 2009, 33, 627–642. [Google Scholar] [CrossRef] [PubMed]
- Molina, R.; Gonzalez, A.; Stelter, M.; Perez-Dorado, I.; Kahn, R.; Morales, M.; Moscoso, M.; Campuzano, S.; Campillo, N.E.; Mobashery, S.; et al. Crystal structure of CbpF, a bifunctional choline-binding protein and autolysis regulator from Streptococcus pneumoniae. EMBO Rep. 2009, 10, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Monterroso, B.; Lopez-Zumel, C.; Garcia, J.L.; Saiz, J.L.; Garcia, P.; Campillo, N.E.; Menendez, M. Unravelling the structure of the pneumococcal autolytic lysozyme. Biochem. J. 2005, 391, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Eldholm, V.; Johnsborg, O.; Straume, D.; Ohnstad, H.S.; Berg, K.H.; Hermoso, J.A.; Havarstein, L.S. Pneumococcal CbpD is a murein hydrolase that requires a dual cell envelope binding specificity to kill target cells during fratricide. Mol. Microbiol. 2010, 76, 905–917. [Google Scholar] [CrossRef] [PubMed]
- Guiral, S.; Mitchell, T.J.; Martin, B.; Claverys, J.P. Competence-programmed predation of noncompetent cells in the human pathogen Streptococcus pneumoniae: Genetic requirements. Proc. Natl. Acad. Sci. USA 2005, 102, 8710–8715. [Google Scholar] [CrossRef] [PubMed]
- Kausmally, L.; Johnsborg, O.; Lunde, M.; Knutsen, E.; Havarstein, L.S. Choline-binding protein D (CbpD) in streptococcus pneumoniae is essential for competence-induced cell lysis. J. Bacteriol. 2005, 187, 4338–4345. [Google Scholar] [CrossRef] [PubMed]
- Bateman, A.; Rawlings, N.D. The CHAP domain: A large family of amidases including GSP amidase and peptidoglycan hydrolases. Trends Biochem. Sci. 2003, 28, 234–237. [Google Scholar] [CrossRef]
- Layec, S.; Gerard, J.; Legue, V.; Chapot-Chartier, M.P.; Courtin, P.; Borges, F.; Decaris, B.; Leblond-Bourget, N. The CHAP domain of Cse functions as an endopeptidase that acts at mature septa to promote Streptococcus thermophilus cell separation. Mol. Microbiol. 2009, 71, 1205–1217. [Google Scholar] [CrossRef] [PubMed]
- Molina, R.; Gonzalez, A.; Moscoso, M.; Garcia, P.; Stelter, M.; Kahn, R.; Hermoso, J.A. Crystallization and preliminary x-ray diffraction studies of choline-binding protein F from Streptococcus pneumoniae. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2007, 63, 742–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
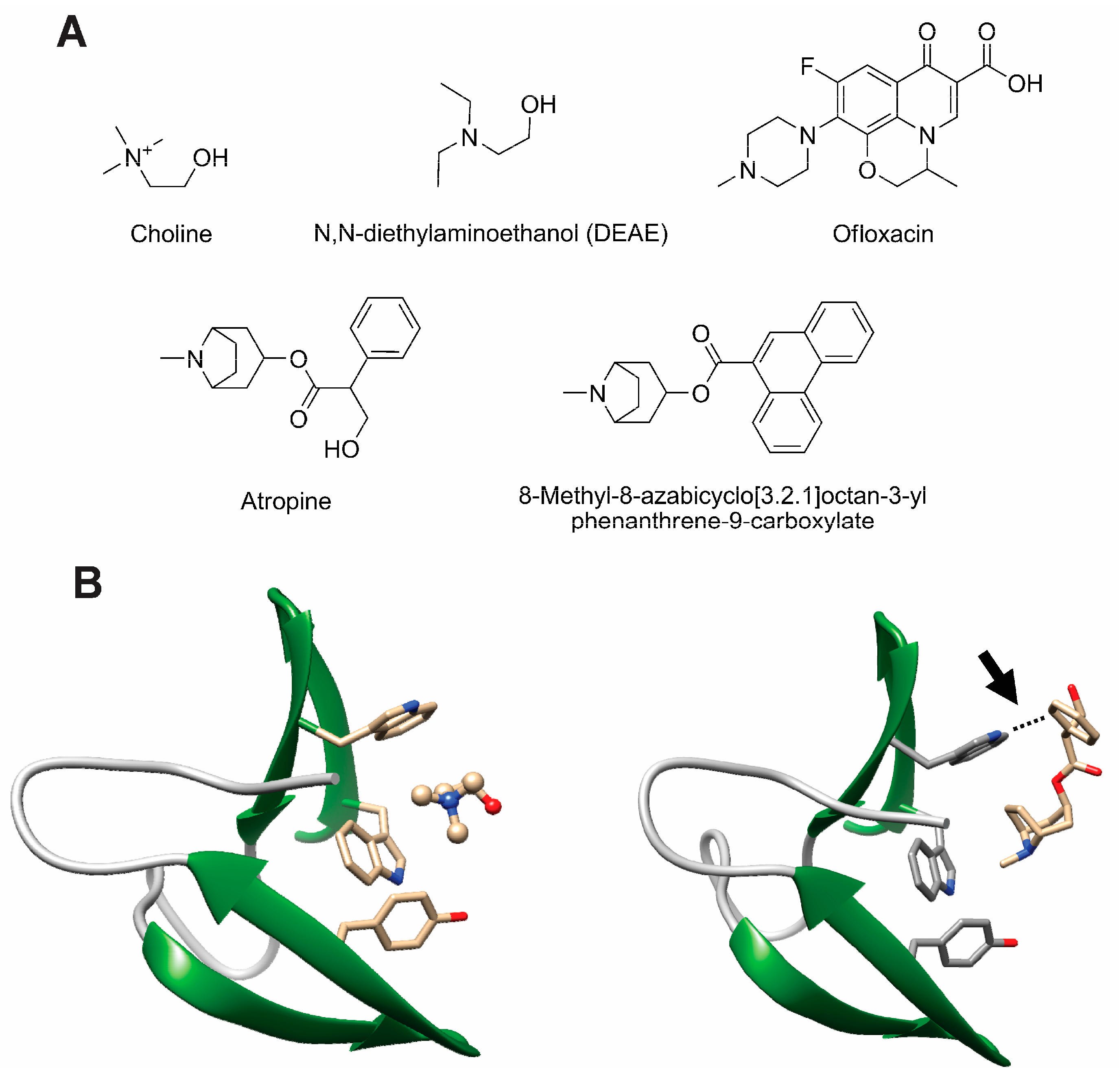
- Silva-Martin, N.; Retamosa, M.G.; Maestro, B.; Bartual, S.G.; Rodes, M.J.; Garcia, P.; Sanz, J.M.; Hermoso, J.A. Crystal structures of CbpF complexed with atropine and ipratropium reveal clues for the design of novel antimicrobials against Streptococcus pneumoniae. Biochim. Biophys. Acta 2014, 1840, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Wren, B.W. A family of clostridial and streptococcal ligand-binding proteins with conserved C-terminal repeat sequences. Mol. Microbiol. 1991, 5, 797–803. [Google Scholar] [CrossRef] [PubMed]
- Janecek, S.; Svensson, B.; Russell, R.R. Location of repeat elements in glucansucrases of Leuconostoc. and Streptococcus species. FEMS Microbiol. Lett. 2000, 192, 53–57. [Google Scholar] [CrossRef] [PubMed]
- McDaniel, L.S.; Scott, G.; Kearney, J.F.; Briles, D.E. Monoclonal antibodies against protease-sensitive pneumococcal antigens can protect mice from fatal infection with Streptococcus pneumoniae. J. Exp. Med. 1984, 160, 386–397. [Google Scholar] [CrossRef] [PubMed]
- McDaniel, L.S.; Scott, G.; Widenhofer, K.; Carroll, J.M.; Briles, D.E. Analysis of a surface protein of Streptococcus pneumoniae recognised by protective monoclonal antibodies. Microb. Pathog. 1986, 1, 519–531. [Google Scholar] [CrossRef]
- Waltman, W.D.; McDaniel, L.S.; Gray, B.M.; Briles, D.E. Variation in the molecular weight of PspA (pneumococcal surface protein A) among Streptococcus pneumoniae. Microb. Pathog. 1990, 8, 61–69. [Google Scholar] [CrossRef]
- Crain, M.J.; Waltman, W.D., 2nd; Turner, J.S.; Yother, J.; Talkington, D.F.; McDaniel, L.S.; Gray, B.M.; Briles, D.E. Pneumococcal surface protein A (PspA) is serologically highly variable and is expressed by all clinically important capsular serotypes of Streptococcus pneumoniae. Infect. Immun. 1990, 58, 3293–3299. [Google Scholar] [PubMed]
- Briles, D.E.; Yother, J.; McDaniel, L.S. Role of pneumococcal surface protein a in the virulence of Streptococcus pneumoniae. Rev. Infect. Dis. 1988, 10, S372–S374. [Google Scholar] [CrossRef] [PubMed]
- McDaniel, L.S.; Yother, J.; Vijayakumar, M.; McGarry, L.; Guild, W.R.; Briles, D.E. Use of insertional inactivation to facilitate studies of biological properties of pneumococcal surface protein A (PspA). J. Exp. Med. 1987, 165, 381–394. [Google Scholar] [CrossRef] [PubMed]
- Tu, A.H.; Fulgham, R.L.; McCrory, M.A.; Briles, D.E.; Szalai, A.J. Pneumococcal surface protein a inhibits complement activation by Streptococcus pneumoniae. Infect. Immun. 1999, 67, 4720–4724. [Google Scholar] [PubMed]
- Yuste, J.; Botto, M.; Paton, J.C.; Holden, D.W.; Brown, J.S. Additive inhibition of complement deposition by pneumolysin and PspA facilitates Streptococcus pneumoniae septicemia. J. Immunol. 2005, 175, 1813–1819. [Google Scholar] [CrossRef] [PubMed]
- Quin, L.R.; Moore, Q.C., 3rd; McDaniel, L.S. Pneumolysin, PspA, and PspC contribute to pneumococcal evasion of early innate immune responses during bacteremia in mice. Infect. Immun. 2007, 75, 2067–2070. [Google Scholar] [CrossRef] [PubMed]
- Hammerschmidt, S.; Bethe, G.; Remane, P.H.; Chhatwal, G.S. Identification of pneumococcal surface protein A as a lactoferrin-binding protein of Streptococcus pneumoniae. Infect. Immun. 1999, 67, 1683–1687. [Google Scholar] [PubMed]
- Shaper, M.; Hollingshead, S.K.; Benjamin, W.H., Jr.; Briles, D.E. PspA protects Streptococcus pneumoniae from killing by apolactoferrin, and antibody to PspA enhances killing of pneumococci by apolactoferrin. Infect. Immun. 2004, 72, 5031–5040. [Google Scholar] [CrossRef] [PubMed]
- Senkovich, O.; Cook, W.J.; Mirza, S.; Hollingshead, S.K.; Protasevich, II.; Briles, D.E.; Chattopadhyay, D. Structure of a complex of human lactoferrin N-lobe with pneumococcal surface protein A provides insight into microbial defense mechanism. J. Mol. Biol. 2007, 370, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Jedrzejas, M.J. Unveiling molecular mechanisms of pneumococcal surface protein A interactions with antibodies and lactoferrin. Clin. Chim. Acta 2006, 367, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Briles, D.E.; King, J.D.; Gray, M.A.; McDaniel, L.S.; Swiatlo, E.; Benton, K.A. PspA, a protection-eliciting pneumococcal protein: Immunogenicity of isolated native PspA in mice. Vaccine 1996, 14, 858–867. [Google Scholar] [CrossRef]
- Talkington, D.F.; Crimmins, D.L.; Voellinger, D.C.; Yother, J.; Briles, D.E. A 43-kilodalton pneumococcal surface protein, PspA: Isolation, protective abilities, and structural analysis of the amino-terminal sequence. Infect. Immun. 1991, 59, 1285–1289. [Google Scholar] [PubMed]
- McDaniel, L.S.; Sheffield, J.S.; Delucchi, P.; Briles, D.E. PspA, a surface protein of streptococcus pneumoniae, is capable of eliciting protection against pneumococci of more than one capsular type. Infect. Immun. 1991, 59, 222–228. [Google Scholar] [PubMed]
- Jedrzejas, M.J.; Lamani, E.; Becker, R.S. Characterization of selected strains of pneumococcal surface protein A. J. Biol. Chem. 2001, 276, 33121–33128. [Google Scholar] [CrossRef] [PubMed]
- Yother, J.; Briles, D.E. Structural properties and evolutionary relationships of PspA, a surface protein of streptococcus pneumoniae, as revealed by sequence analysis. J. Bacteriol. 1992, 174, 601–609. [Google Scholar] [PubMed]
- Jedrzejas, M.J.; Hollingshead, S.K.; Lebowitz, J.; Chantalat, L.; Briles, D.E.; Lamani, E. Production and characterization of the functional fragment of pneumococcal surface protein A. Arch. Biochem. Biophys. 2000, 373, 116–125. [Google Scholar] [CrossRef] [PubMed]
- McDaniel, L.S.; Ralph, B.A.; McDaniel, D.O.; Briles, D.E. Localization of protection-eliciting epitopes on PspA of streptococcus pneumoniae between amino acid residues 192 and 260. Microb. Pathog. 1994, 17, 323–337. [Google Scholar] [CrossRef] [PubMed]
- Hakansson, A.; Roche, H.; Mirza, S.; McDaniel, L.S.; Brooks-Walter, A.; Briles, D.E. Characterization of binding of human lactoferrin to pneumococcal surface protein A. Infect. Immun. 2001, 69, 3372–3381. [Google Scholar] [CrossRef] [PubMed]
- Rosenow, C.; Ryan, P.; Weiser, J.N.; Johnson, S.; Fontan, P.; Ortqvist, A.; Masure, H.R. Contribution of novel choline-binding proteins to adherence, colonization and immunogenicity of Streptococcus pneumoniae. Mol. Microbiol. 1997, 25, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Hammerschmidt, S.; Talay, S.R.; Brandtzaeg, P.; Chhatwal, G.S. SpsA, a novel pneumococcal surface protein with specific binding to secretory immunoglobulin a and secretory component. Mol. Microbiol. 1997, 25, 1113–1124. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, P.; Brooks-Walter, A.; Virolainen-Julkunen, A.; Hollingshead, S.K.; Briles, D.E. Role of pneumococcal surface protein C in nasopharyngeal carriage and pneumonia and its ability to elicit protection against carriage of Streptococcus pneumoniae. Infect. Immun. 2002, 70, 2526–2534. [Google Scholar] [CrossRef] [PubMed]
- Brock, S.C.; McGraw, P.A.; Wright, P.F.; Crowe, J.E., Jr. The human polymeric immunoglobulin receptor facilitates invasion of epithelial cells by Streptococcus pneumoniae in a strain-specific and cell type-specific manner. Infect. Immun. 2002, 70, 5091–5095. [Google Scholar] [CrossRef] [PubMed]
- Iannelli, F.; Chiavolini, D.; Ricci, S.; Oggioni, M.R.; Pozzi, G. Pneumococcal surface protein C contributes to sepsis caused by Streptococcus pneumoniae in mice. Infect. Immun. 2004, 72, 3077–3080. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.R.; Mostov, K.E.; Lamm, M.E.; Nanno, M.; Shimida, S.; Ohwaki, M.; Tuomanen, E. The polymeric immunoglobulin receptor translocates pneumococci across human nasopharyngeal epithelial cells. Cell 2000, 102, 827–837. [Google Scholar] [CrossRef]
- Hammerschmidt, S.; Tillig, M.P.; Wolff, S.; Vaerman, J.P.; Chhatwal, G.S. Species-specific binding of human secretory component to SpsA protein of Streptococcus pneumoniae via a hexapeptide motif. Mol. Microbiol. 2000, 36, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Orihuela, C.J.; Mahdavi, J.; Thornton, J.; Mann, B.; Wooldridge, K.G.; Abouseada, N.; Oldfield, N.J.; Self, T.; Ala’Aldeen, D.A.; Tuomanen, E.I. Laminin receptor initiates bacterial contact with the blood brain barrier in experimental meningitis models. J. Clin. Invest. 2009, 119, 1638–1646. [Google Scholar] [CrossRef] [PubMed]
- Mestecky, J.; Moro, I.; Underdown, B.J. Mucosal immunoglobulins. In Mucosal immunology, 2nd ed.; Ogra, P.L., Mestecky, J., Lamm, M.E., Strober, W., Bienenstock, J., McGhee, J.R., Eds.; Academic Press: San Diego, CA, USA, 1999; pp. 133–152. [Google Scholar]
- Zipfel, P.F.; Hallstrom, T.; Hammerschmidt, S.; Skerka, C. The complement fitness Factor H: Role in human diseases and for immune escape of pathogens, like pneumococci. Vaccine 2008, 26 (Suppl. 8), I67–I74. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Finkel, D.; Hostetter, M.K. Novel purification scheme and functions for a C3-binding protein from Streptococcus pneumoniae. Biochemistry 2000, 39, 5450–5457. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.L.; Hostetter, M.K. C3 as substrate for adhesion of Streptococcus pneumoniae. J. Infect. Dis 2000, 182, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Janulczyk, R.; Iannelli, F.; Sjoholm, A.G.; Pozzi, G.; Bjorck, L. Hic, a novel surface protein of Streptococcus pneumoniae that interferes with complement function. J. Biol. Chem. 2000, 275, 37257–37263. [Google Scholar] [CrossRef] [PubMed]
- Dave, S.; Brooks-Walter, A.; Pangburn, M.K.; McDaniel, L.S. PspC, a pneumococcal surface protein, binds human Factor H. Infect. Immun. 2001, 69, 3435–3437. [Google Scholar] [CrossRef] [PubMed]
- Dieudonne-Vatran, A.; Krentz, S.; Blom, A.M.; Meri, S.; Henriques-Normark, B.; Riesbeck, K.; Albiger, B. Clinical isolates of Streptococcus pneumoniae bind the complement inhibitor C4b-binding protein in a pspc allele-dependent fashion. J. Immunol. 2009, 182, 7865–7877. [Google Scholar] [CrossRef] [PubMed]
- Voss, S.; Hallstrom, T.; Saleh, M.; Burchhardt, G.; Pribyl, T.; Singh, B.; Riesbeck, K.; Zipfel, P.F.; Hammerschmidt, S. The choline-binding protein PspC of Streptococcus pneumoniae interacts with the C-terminal heparin-binding domain of vitronectin. J. Biol. Chem. 2013, 288, 15614–15627. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Glover, D.T.; Szalai, A.J.; Hollingshead, S.K.; Briles, D.E. PspA and PspC minimize immune adherence and transfer of pneumococci from erythrocytes to macrophages through their effects on complement activation. Infect. Immun. 2007, 75, 5877–5885. [Google Scholar] [CrossRef] [PubMed]
- Iannelli, F.; Oggioni, M.R.; Pozzi, G. Allelic variation in the highly polymorphic locus PspC of Streptococcus pneumoniae. Gene 2002, 284, 63–71. [Google Scholar] [CrossRef]
- Achila, D.; Liu, A.; Banerjee, R.; Li, Y.; Martinez-Hackert, E.; Zhang, J.R.; Yan, H. Structural determinants of host specificity of complement Factor H recruitment by Streptococcus pneumoniae. Biochem. J. 2015, 465, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Hammerschmidt, S.; Agarwal, V.; Kunert, A.; Haelbich, S.; Skerka, C.; Zipfel, P.F. The host immune regulator Factor H interacts via two contact sites with the PspC protein of Streptococcus pneumoniae and mediates adhesion to host epithelial cells. J. Immunol. 2007, 178, 5848–5858. [Google Scholar] [CrossRef] [PubMed]
- Dave, S.; Carmicle, S.; Hammerschmidt, S.; Pangburn, M.K.; McDaniel, L.S. Dual roles of PspC, a surface protein of Streptococcus pneumoniae, in binding human secretory IgA and Factor H. J. Immunol. 2004, 173, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Duthy, T.G.; Ormsby, R.J.; Giannakis, E.; Ogunniyi, A.D.; Stroeher, U.H.; Paton, J.C.; Gordon, D.L. The human complement regulator Factor H binds pneumococcal surface protein PspC via short consensus repeats 13 to 15. Infect. Immun. 2002, 70, 5604–5611. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Ma, Y.; Zhang, J.R. Streptococcus pneumoniae recruits complement Factor H through the amino terminus of CbpA. J. Biol. Chem. 2006, 281, 15464–15474. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Mann, B.; Lewis, W.S.; Rowe, A.; Heath, R.; Stewart, M.L.; Hamburger, A.E.; Sivakolundu, S.; Lacy, E.R.; Bjorkman, P.J.; et al. Solution structure of choline binding protein A, the major adhesin of Streptococcus pneumoniae. EMBO J. 2005, 24, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Navarre, W.W.; Schneewind, O. Surface proteins of gram-positive bacteria and mechanisms of their targeting to the cell wall envelope. Microbiol. Mol. Biol. Rev. 1999, 63, 174–229. [Google Scholar] [PubMed]
- Holtje, J.V.; Tomasz, A. Teichoic acid phosphorylcholine esterase. A novel enzyme activity in pneumococcus. J. Biol. Chem. 1974, 249, 7032–7034. [Google Scholar] [PubMed]
- Vollmer, W.; Tomasz, A. Identification of the teichoic acid phosphorylcholine esterase in Streptococcus pneumoniae. Mol. Microbiol. 2001, 39, 1610–1622. [Google Scholar] [CrossRef] [PubMed]
- de las Rivas, B.; Garcia, J.L.; Lopez, R.; Garcia, P. Molecular characterization of the pneumococcal teichoic acid phosphorylcholine esterase. Microb. Drug Resist. 2001, 7, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Hermoso, J.A.; Lagartera, L.; Gonzalez, A.; Stelter, M.; Garcia, P.; Martinez-Ripoll, M.; Garcia, J.L.; Menendez, M. Insights into pneumococcal pathogenesis from the crystal structure of the modular teichoic acid phosphorylcholine esterase Pce. Nat. Struct. Mol. Biol. 2005, 12, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Attali, C.; Frolet, C.; Durmort, C.; Offant, J.; Vernet, T.; Di Guilmi, A.M. Streptococcus pneumoniae choline-binding protein E interaction with plasminogen/plasmin stimulates migration across the extracellular matrix. Infect. Immun. 2008, 76, 466–476. [Google Scholar] [CrossRef] [PubMed]
- Buey, R.M.; Monterroso, B.; Menendez, M.; Diakun, G.; Chacon, P.; Hermoso, J.A.; Diaz, J.F. Insights into molecular plasticity of choline binding proteins (pneumococcal surface proteins) by SAXS. J. Mol. Biol. 2007, 365, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Garau, G.; Lemaire, D.; Vernet, T.; Dideberg, O.; Di Guilmi, A.M. Crystal structure of phosphorylcholine esterase domain of the virulence factor choline-binding protein E from Streptococcus pneumoniae: New structural features among the metallo-beta-lactamase superfamily. J. Biol. Chem. 2005, 280, 28591–28600. [Google Scholar] [CrossRef] [PubMed]
- Lagartera, L.; Gonzalez, A.; Hermoso, J.A.; Saiz, J.L.; Garcia, P.; Garcia, J.L.; Menendez, M. Pneumococcal phosphorylcholine esterase, Pce, contains a metal binuclear center that is essential for substrate binding and catalysis. Protein Sci. 2005, 14, 3013–3024. [Google Scholar] [CrossRef] [PubMed]
- Paterson, N.G.; Riboldi-Tunicliffe, A.; Mitchell, T.J.; Isaacs, N.W. Overexpression, purification and crystallization of a choline-binding protein CbpI from Streptococcus pneumoniae. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2006, 62, 672–675. [Google Scholar] [CrossRef] [PubMed]
- Tettelin, H.; Nelson, K.E.; Paulsen, I.T.; Eisen, J.A.; Read, T.D.; Peterson, S.; Heidelberg, J.; DeBoy, R.T.; Haft, D.H.; Dodson, R.J.; et al. Complete genome sequence of a virulent isolate of Streptococcus pneumoniae. Science 2001, 293, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Beato, A.R.; Lopez, R.; Garcia, J.L. Molecular characterization of PcpA: A novel choline-binding protein of Streptococcus pneumoniae. FEMS Microbiol. Lett. 1998, 164, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Glover, D.T.; Hollingshead, S.K.; Briles, D.E. Streptococcus pneumoniae surface protein PcpA elicits protection against lung infection and fatal sepsis. Infect. Immun. 2008, 76, 2767–2776. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.W.; Briles, D.E.; Myers, L.E.; Hollingshead, S.K. Mn2+-dependent regulation of multiple genes in Streptococcus pneumoniae through PsaR and the resultant impact on virulence. Infect. Immun. 2006, 74, 1171–1180. [Google Scholar] [CrossRef] [PubMed]
- Kloosterman, T.G.; Witwicki, R.M.; van der Kooi-Pol, M.M.; Bijlsma, J.J.; Kuipers, O.P. Opposite effects of Mn2+ and Zn2+ on PsaR-mediated expression of the virulence genes pcpA, prtA, and psaBCA of Streptococcus pneumoniae. J. Bacteriol. 2008, 190, 5382–5393. [Google Scholar] [CrossRef] [PubMed]
- Manzoor, I.; Shafeeq, S.; Kloosterman, T.G.; Kuipers, O.P. Co2+-dependent gene expression in streptococcus pneumoniae: Opposite effect of Mn2+ and Co2+ on the expression of the virulence genes psaBCA, pcpA, and prtA. Front. Microbiol. 2015, 6, 748. [Google Scholar] [CrossRef] [PubMed]
- Manzoor, I.; Shafeeq, S.; Kuipers, O.P. Ni2+-dependent and PsaR-mediated regulation of the virulence genes pcpA, psaBCA, and prtA in Streptococcus pneumoniae. PLoS ONE 2015, 10, e0142839. [Google Scholar] [CrossRef] [PubMed]
- Hava, D.L.; Camilli, A. Large-scale identification of serotype 4 Streptococcus pneumoniae virulence factors. Mol. Microbiol. 2002, 45, 1389–1406. [Google Scholar] [PubMed]
- Mann, B.; Orihuela, C.; Antikainen, J.; Gao, G.; Sublett, J.; Korhonen, T.K.; Tuomanen, E. Multifunctional role of choline binding protein G in pneumococcal pathogenesis. Infect. Immun. 2006, 74, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, W.; Frolet, C.; Bao, R.; di Guilmi, A.M.; Vernet, T.; Chen, Y. Structure of the choline-binding domain of Spr1274 in Streptococcus pneumoniae. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2009, 65, 757–761. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Fernandez, J.B.; Bartual, S.G.; Hermoso, J.A. Crystal Structure of the Choline-Binding Domain of CbpL from Streptococcus Pneumoniae. Protein Data Bank (PDB) Entry 4CNL. Available online: http://www.rcsb.org/pdb/explore.do?structureId=4CNL (accessed on 9 June 2016).
- Muñoz-Elias, E.J.; Marcano, J.; Camilli, A. Isolation of Streptococcus pneumoniae biofilm mutants and their characterization during nasopharyngeal colonization. Infect. Immun. 2008, 76, 5049–5061. [Google Scholar] [CrossRef] [PubMed]
- Bruckner, R.; Nuhn, M.; Reichmann, P.; Weber, B.; Hakenbeck, R. Mosaic genes and mosaic chromosomes-genomic variation in Streptococcus pneumoniae. Int. J. Med. Microbiol. 2004, 294, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Fischetti, V.A. Bacteriophage endolysins: A novel anti-infective to control gram-positive pathogens. Int. J. Med. Microbiol. 2010, 300, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, M.M.; Garcia, J.L.; Lopez, R.; Garcia, P. The lytic enzyme of the pneumococcal phage Dp-1: A chimeric lysin of intergeneric origin. Mol. Microbiol. 1997, 25, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Perez-Dorado, I.; Campillo, N.E.; Monterroso, B.; Hesek, D.; Lee, M.; Paez, J.A.; Garcia, P.; Martinez-Ripoll, M.; Garcia, J.L.; Mobashery, S.; et al. Elucidation of the molecular recognition of bacterial cell wall by modular pneumococcal phage endolysin Cpl-1. J. Biol. Chem. 2007, 282, 24990–24999. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.; Loomis, L.; Fischetti, V.A. Prevention and elimination of upper respiratory colonization of mice by group a streptococci by using a bacteriophage lytic enzyme. Proc. Natl. Acad. Sci. USA 2001, 98, 4107–4112. [Google Scholar] [CrossRef] [PubMed]
- Hermoso, J.A.; Garcia, J.L.; Garcia, P. Taking aim on bacterial pathogens: From phage therapy to enzybiotics. Curr. Opin. Microbiol. 2007, 10, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Borysowski, J.; Gorksi, A. Enzybiotics and Their Potential Applications in Medicine. In Enzybiotics: Antibiotic enzymes as drugs and therapeutics; Villa, T.G., Veiga-Crespo, P., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2010; pp. 1–26. [Google Scholar]
- Fenton, M.; Ross, P.; McAuliffe, O.; O’Mahony, J.; Coffey, A. Recombinant bacteriophage lysins as antibacterials. Bioeng. Bugs 2010, 1, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Pastagia, M.; Schuch, R.; Fischetti, V.A.; Huang, D.B. Lysins: The arrival of pathogen-directed anti-infectives. J. Med. Microbiol. 2013, 62, 1506–1516. [Google Scholar] [CrossRef] [PubMed]
- Veiga-Crespo, P.; Ageitos, J.M.; Poza, M.; Villa, T.G. Enzybiotics: A look to the future, recalling the past. J. Pharm. Sci. 2007, 96, 1917–1924. [Google Scholar] [CrossRef] [PubMed]
- Hojckova, K.; Stano, M.; Klucar, L. Phibiotics: Catalogue of therapeutic enzybiotics, relevant research studies and practical applications. BMC Microbiol. 2013, 13, 53. [Google Scholar] [CrossRef] [PubMed]
- Loeffler, J.M.; Nelson, D.; Fischetti, V.A. Rapid killing of Streptococcus pneumoniae with a bacteriophage cell wall hydrolase. Science 2001, 294, 2170–2172. [Google Scholar] [CrossRef] [PubMed]
- Loeffler, J.M.; Djurkovic, S.; Fischetti, V.A. Phage lytic enzyme Cpl-1 as a novel antimicrobial for pneumococcal bacteremia. Infect. Immun. 2003, 71, 6199–6204. [Google Scholar] [CrossRef] [PubMed]
- Jado, I.; Lopez, R.; Garcia, E.; Fenoll, A.; Casal, J.; García, P. Phage lytic enzymes as therapy for antibiotic-resistant Streptococcus pneumoniae infection in a murine sepsis model. J. Antimicrob. Chemother. 2003, 52, 967–973. [Google Scholar] [CrossRef] [PubMed]
- Entenza, J.M.; Loeffler, J.M.; Grandgirard, D.; Fischetti, V.A.; Moreillon, P. Therapeutic effects of bacteriophage Cpl-1 lysin against Streptococcus pneumoniae endocarditis in rats. Antimicrob. Agents Chemother. 2005, 49, 4789–4792. [Google Scholar] [CrossRef] [PubMed]
- McCullers, J.A.; Karlstrom, A.; Iverson, A.R.; Loeffler, J.M.; Fischetti, V.A. Novel strategy to prevent otitis media caused by colonizing Streptococcus pneumoniae. PLoS Pathog. 2007, 3, e28. [Google Scholar] [CrossRef] [PubMed]
- Grandgirard, D.; Loeffler, J.M.; Fischetti, V.A.; Leib, S.L. Phage lytic enzyme Cpl-1 for antibacterial therapy in experimental pneumococcal meningitis. J. Infect. Dis. 2008, 197, 1519–1522. [Google Scholar] [CrossRef] [PubMed]
- Witzenrath, M.; Schmeck, B.; Doehn, J.M.; Tschernig, T.; Zahlten, J.; Loeffler, J.M.; Zemlin, M.; Muller, H.; Gutbier, B.; Schutte, H.; et al. Systemic use of the endolysin Cpl-1 rescues mice with fatal pneumococcal pneumonia. Crit. Care Med. 2009, 37, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Loeffler, J.M.; Fischetti, V.A. Synergistic lethal effect of a combination of phage lytic enzymes with different activities on penicillin-sensitive and -resistant Streptococcus pneumoniae strains. Antimicrob. Agents Chemother. 2003, 47, 375–377. [Google Scholar] [CrossRef] [PubMed]
- Djurkovic, S.; Loeffler, J.M.; Fischetti, V.A. Synergistic killing of Streptococcus pneumoniae with the bacteriophage lytic enzyme Cpl-1 and penicillin or gentamicin depends on the level of penicillin resistance. Antimicrob. Agents Chemother. 2005, 49, 1225–1228. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Cerrato, V.; Garcia, P.; Del Prado, G.; Garcia, E.; Gracia, M.; Huelves, L.; Ponte, C.; Lopez, R.; Soriano, F. In vitro interactions of LytA, the major pneumococcal autolysin, with two bacteriophage lytic enzymes (Cpl-1 and Pal), cefotaxime and moxifloxacin against antibiotic-susceptible and -resistant streptococcus pneumoniae strains. J. Antimicrob. Chemother. 2007, 60, 1159–1162. [Google Scholar] [CrossRef] [PubMed]
- Vouillamoz, J.; Entenza, J.M.; Giddey, M.; Fischetti, V.A.; Moreillon, P.; Resch, G. Bactericidal synergism between daptomycin and the phage lysin Cpl-1 in a mouse model of pneumococcal bacteraemia. Int. J. Antimicrob. Agents 2013, 42, 416–421. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Cerrato, V.; Garcia, P.; Huelves, L.; Garcia, E.; Del Prado, G.; Gracia, M.; Ponte, C.; Lopez, R.; Soriano, F. Pneumococcal LytA autolysin, a potent therapeutic agent in experimental peritonitis-sepsis caused by highly beta-lactam-resistant Streptococcus pneumoniae. Antimicrob. Agents Chemother. 2007, 51, 3371–3373. [Google Scholar] [CrossRef] [PubMed]
- Garcia, P.; Lopez, R.; Ronda, C.; Garcia, E.; Tomasz, A. Mechanism of phage-induced lysis in pneumococci. J. Gen. Microbiol. 1983, 129, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Resch, G.; Moreillon, P.; Fischetti, V.A. A stable phage lysin (Cpl-1) dimer with increased antipneumococcal activity and decreased plasma clearance. Int. J. Antimicrob. Agents 2011, 38, 516–521. [Google Scholar] [CrossRef] [PubMed]
- Sanz, J.M.; Garcia, P.; Garcia, J.L. Construction of a multifunctional pneumococcal murein hydrolase by module assembly. Eur. J. Biochem. 1996, 235, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Berry, A.M.; Lock, R.A.; Hansman, D.; Paton, J.C. Contribution of autolysin to virulence of Streptococcus pneumoniae. Infect. Immun. 1989, 57, 2324–2330. [Google Scholar] [PubMed]
- Canvin, J.R.; Marvin, A.P.; Sivakumaran, M.; Paton, J.C.; Boulnois, G.J.; Andrew, P.W.; Mitchell, T.J. The role of pneumolysin and autolysin in the pathology of pneumonia and septicemia in mice infected with a type 2 pneumococcus. J. Infect. Dis. 1995, 172, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Hirst, R.A.; Gosai, B.; Rutman, A.; Guerin, C.J.; Nicotera, P.; Andrew, P.W.; O’Callaghan, C. Streptococcus pneumoniae deficient in pneumolysin or autolysin has reduced virulence in meningitis. J. Infect. Dis. 2008, 197, 744–751. [Google Scholar] [CrossRef] [PubMed]
- Holtje, J.V.; Tomasz, A. Lipoteichoic acid: A specific inhibitor of autolysin activity in pneumococcus. Proc. Natl. Acad. Sci. USA 1975, 72, 1690–1694. [Google Scholar] [CrossRef] [PubMed]
- Giudicelli, S.; Tomasz, A. Attachment of pneumococcal autolysin to wall teichoic acids, an essential step in enzymatic wall degradation. J. Bacteriol. 1984, 158, 1188–1190. [Google Scholar] [PubMed]
- Briese, T.; Hakenbeck, R. Interaction of the pneumococcal amidase with lipoteichoic acid and choline. Eur. J. Biochem. 1985, 146, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Dalia, A.B.; Weiser, J.N. Minimization of bacterial size allows for complement evasion and is overcome by the agglutinating effect of antibody. Cell. Host Microbe 2011, 10, 486–496. [Google Scholar] [CrossRef] [PubMed]
- Sanz, J.M.; Garcia, J.L.; Laynez, J.; Usobiaga, P.; Menendez, M. Thermal stability and cooperative domains of Cpl1 lysozyme and its NH2- and COOH-terminal modules. Dependence on choline binding. J. Biol. Chem. 1993, 268, 6125–6130. [Google Scholar] [PubMed]
- Sanz, J.M.; Lopez, R.; Garcia, J.L. Structural requirements of choline derivatives for “conversion” of pneumococcal amidase. A new single-step procedure for purification of this autolysin. FEBS Lett. 1988, 232, 308–312. [Google Scholar] [CrossRef]
- Maestro, B.; Gonzalez, A.; Garcia, P.; Sanz, J.M. Inhibition of pneumococcal choline-binding proteins and cell growth by esters of bicyclic amines. FEBS J. 2007, 274, 364–376. [Google Scholar] [CrossRef] [PubMed]
- de Gracia Retamosa, M.; Diez-Martinez, R.; Maestro, B.; Garcia-Fernandez, E.; de Waal, B.; Meijer, E.W.; Garcia, P.; Sanz, J.M. Aromatic esters of bicyclic amines as antimicrobials against Streptococcus pneumoniae. Angew. Chem. Int. Ed. Engl. 2015, 54, 13673–13677. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Puelles, J.M.; Sanz, J.M.; Garcia, J.L.; Garcia, E. Immobilization and single-step purification of fusion proteins using DEAE-cellulose. Eur. J. Biochem. 1992, 203, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Caubin, J.; Martin, H.; Roa, A.; Cosano, I.; Pozuelo, M.; de La Fuente, J.M.; Sanchez-Puelles, J.M.; Molina, M.; Nombela, C. Choline-binding domain as a novel affinity tag for purification of fusion proteins produced in Pichia pastoris. Biotechnol. Bioeng. 2001, 74, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Ortega, S.; Garcia, J.L.; Zazo, M.; Varela, J.; Munoz-Willery, I.; Cuevas, P.; Gimenez-Gallego, G. Single-step purification on DEAE-Sephacel of recombinant polypeptides produced in Escherichia coli. Biotechnology 1992, 10, 795–798. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Echevarria, M.J.; Gimenez-Gallego, G.; Sabariegos-Jareno, R.; Diaz-Orejas, R. Kid, a small protein of the ParD stability system of plasmid R1, is an inhibitor of DNA replication acting at the initiation of DNA synthesis. J. Mol. Biol. 1995, 247, 568–577. [Google Scholar] [CrossRef]
- Maestro, B.; Velasco, I.; Castillejo, I.; Arevalo-Rodriguez, M.; Cebolla, A.; Sanz, J.M. Affinity partitioning of proteins tagged with choline-binding modules in aqueous two-phase systems. J. Chromatogr. A 2008, 1208, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Cunha, B.A.; Schoch, P.E.; Bottone, E.J. Antibiotic Essentials; Jones and Bartlett Publishers LLC: Sudbury, MA, USA, 2009. [Google Scholar]
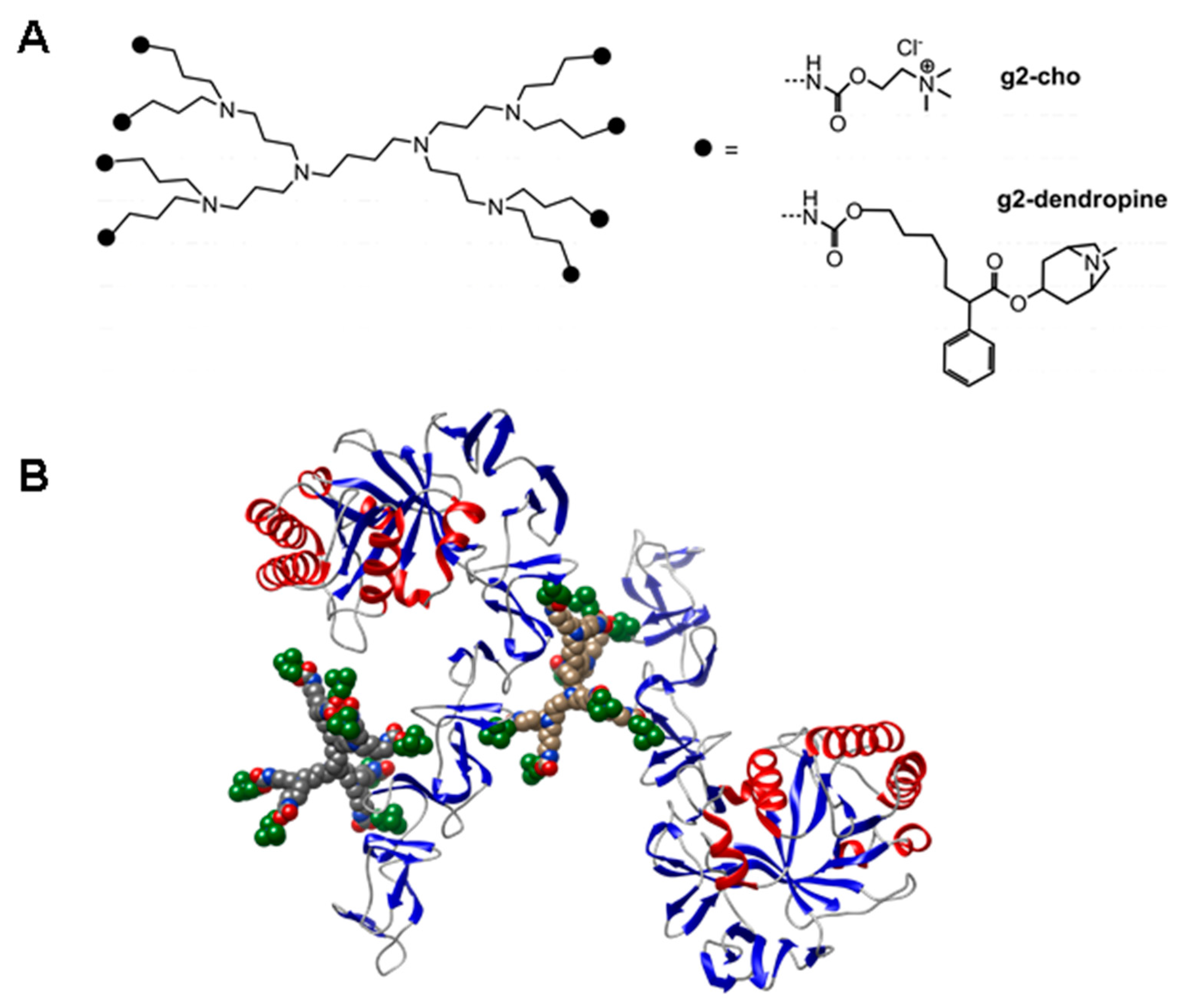
- Hernandez-Rocamora, V.M.; Maestro, B.; de Waal, B.; Morales, M.; Garcia, P.; Meijer, E.W.; Merkx, M.; Sanz, J.M. Multivalent choline dendrimers as potent inhibitors of pneumococcal cell-wall hydrolysis. Angew. Chem. Int. Ed. Engl. 2009, 48, 948–951. [Google Scholar] [CrossRef] [PubMed]
- Mammen, M.; Choi, S.-K.; Whitesides, G.M. Polyvalent interactions in biological systems: Implications for design and use of multivalent ligands and inhibitors. Angew. Chem. Int. Ed. 1998, 37, 2754–2794. [Google Scholar] [CrossRef]
- Ribes, S.; Riegelmann, J.; Redlich, S.; Maestro, B.; de Waal, B.; Meijer, E.W.; Sanz, J.M.; Nau, R. Multivalent choline dendrimers increase phagocytosis of Streptococcus pneumoniae R6 by microglial cells. Chemotherapy 2013, 59, 138–142. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CBP | Origin | Administration | Animal Model | Reference |
|---|---|---|---|---|
| CPL1 | Cp-1 phage | Intravenous | Murine bacteremia | [215] |
| Intravenous | Rat endocarditis | [217] | ||
| Intranasal | Mice nasal colonization | [218] | ||
| Intracisternal or intraparenteral | Rat meningitis | [219] | ||
| Intraperitoneal | Mice severe pneumonia | [220] | ||
| Oral | Mice nasal colonization | [207] | ||
| Pal | Dp-1 phage | Nasal and pharyngeal administration | Mice nasal colonization | [214] |
| CPL1 and Pal (separately or combined) | Cp-1 and Dp-1 phages | Intraparenteral | Murine sepsis | [216] |
| LytA | Streptococcus pneumoniae | Intraperitoneal | Mice peritonitis-sepsis | [225] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maestro, B.; Sanz, J.M. Choline Binding Proteins from Streptococcus pneumoniae: A Dual Role as Enzybiotics and Targets for the Design of New Antimicrobials. Antibiotics 2016, 5, 21. https://doi.org/10.3390/antibiotics5020021
Maestro B, Sanz JM. Choline Binding Proteins from Streptococcus pneumoniae: A Dual Role as Enzybiotics and Targets for the Design of New Antimicrobials. Antibiotics. 2016; 5(2):21. https://doi.org/10.3390/antibiotics5020021
Chicago/Turabian StyleMaestro, Beatriz, and Jesús M. Sanz. 2016. "Choline Binding Proteins from Streptococcus pneumoniae: A Dual Role as Enzybiotics and Targets for the Design of New Antimicrobials" Antibiotics 5, no. 2: 21. https://doi.org/10.3390/antibiotics5020021