
Ribosomal Antibiotics: Contemporary Challenges

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Main Findings
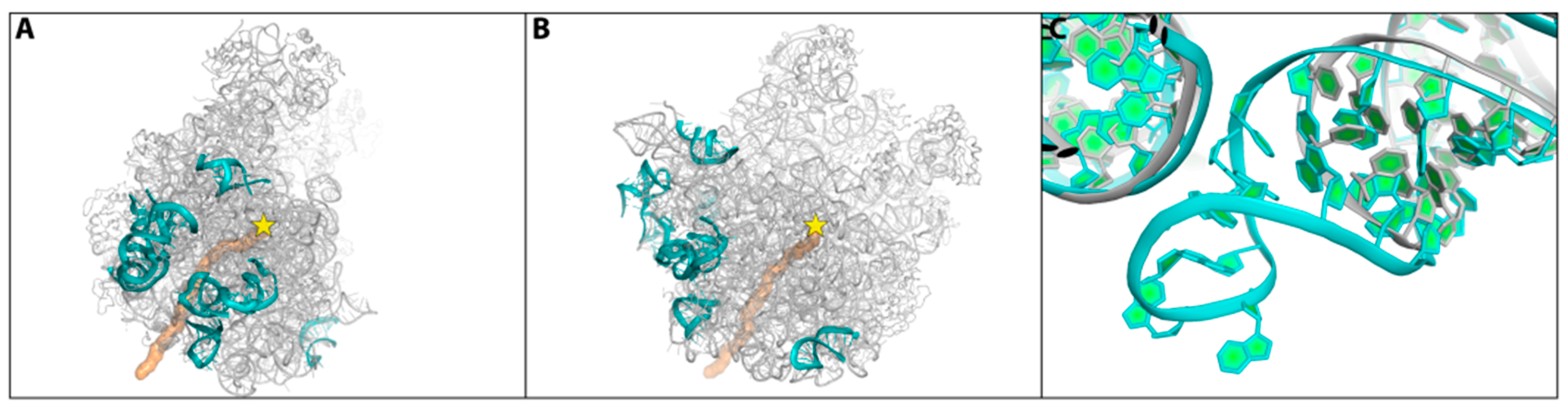
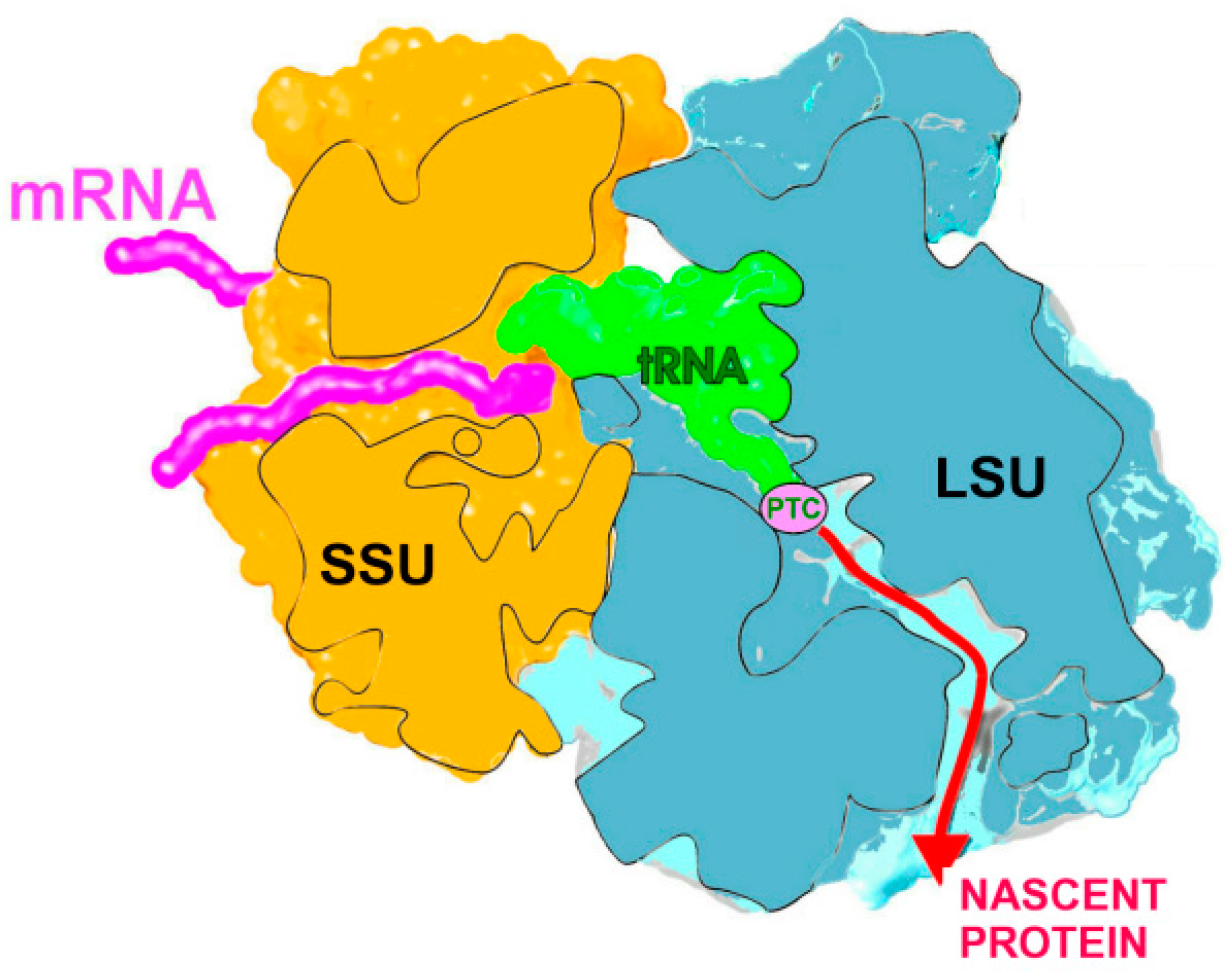
2.1. The Nascent Protein Exit Tunnel Seems to Be Invovled in Cellular Regulation
2.2. Inherent Flexity of Antibiotics Binding Pockets and of Their Surroundings
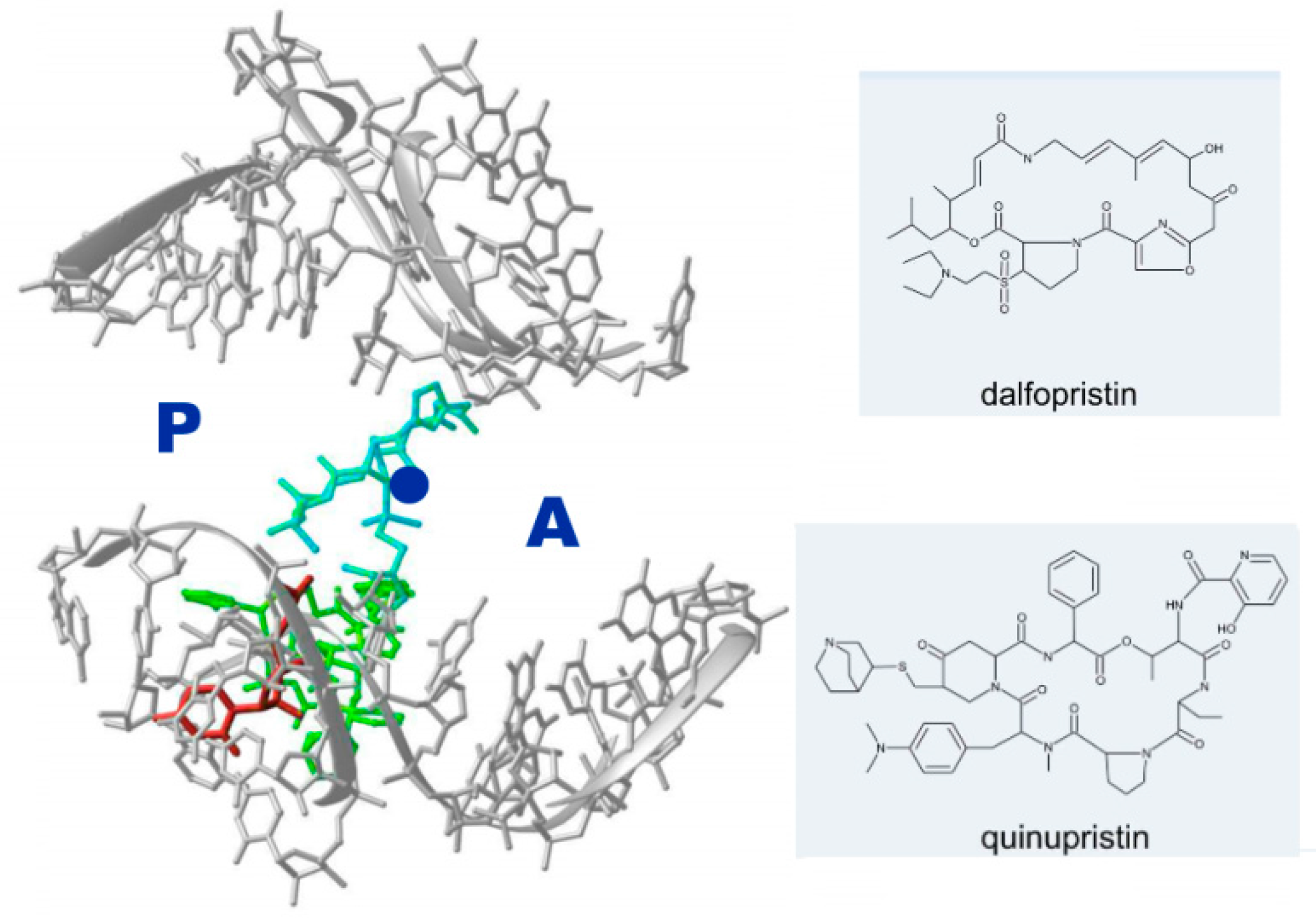
2.3. Antibiotic Pairs and Synergism
2.4. Species Specificity and Susceptibility to Antibiotics
2.5. Ecological Aspects: Degradable Antibiotics
3. Conclusions
- -
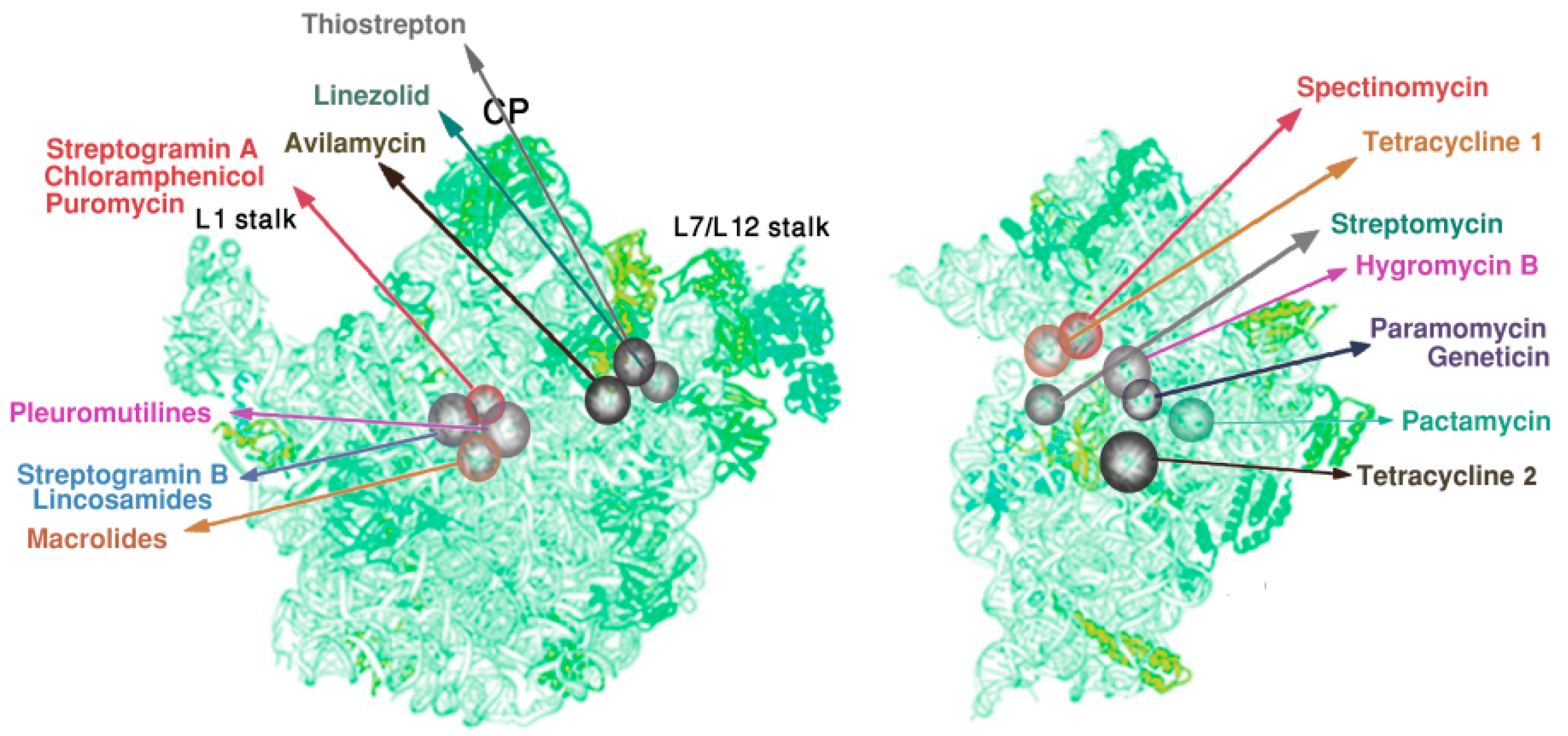
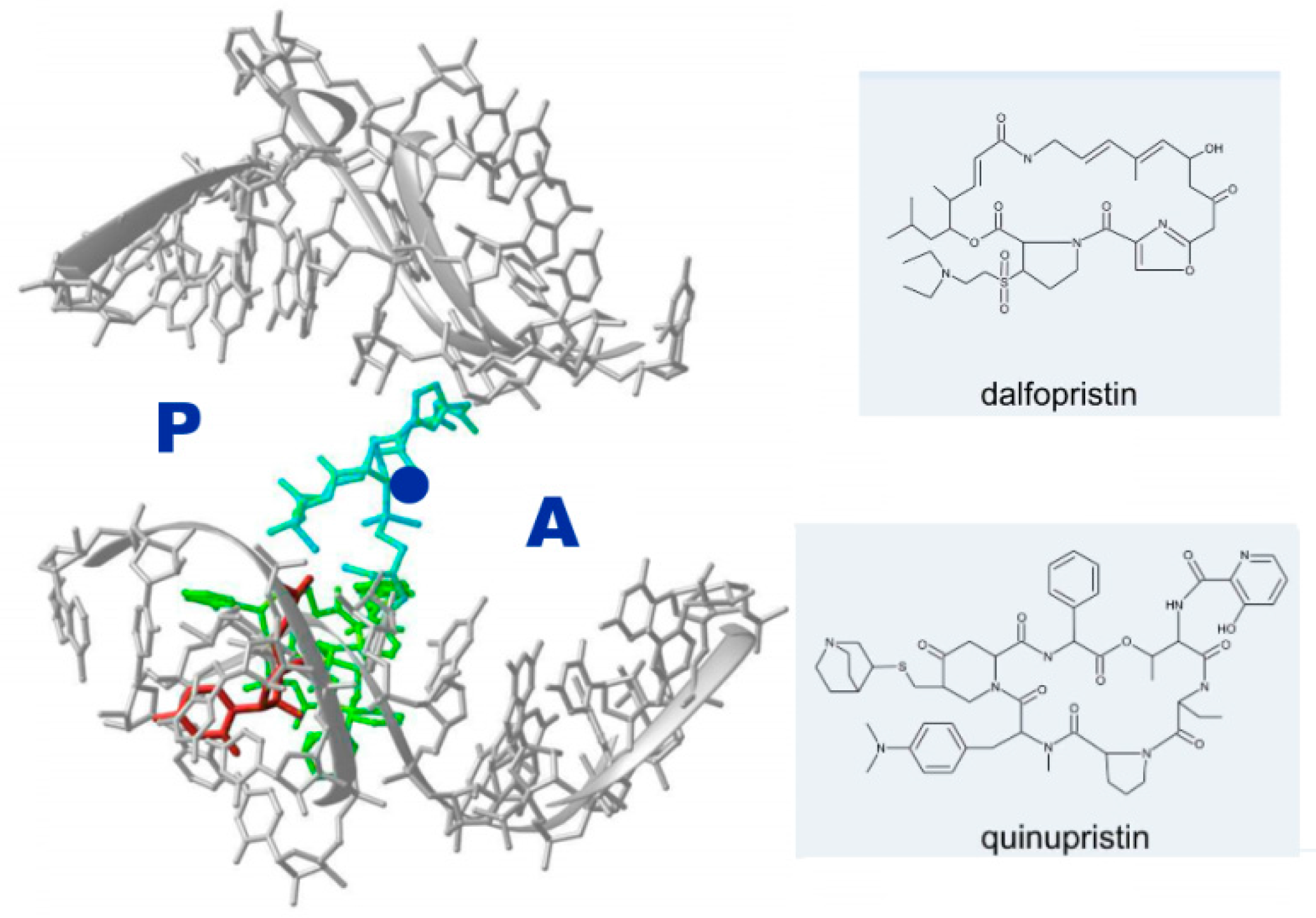
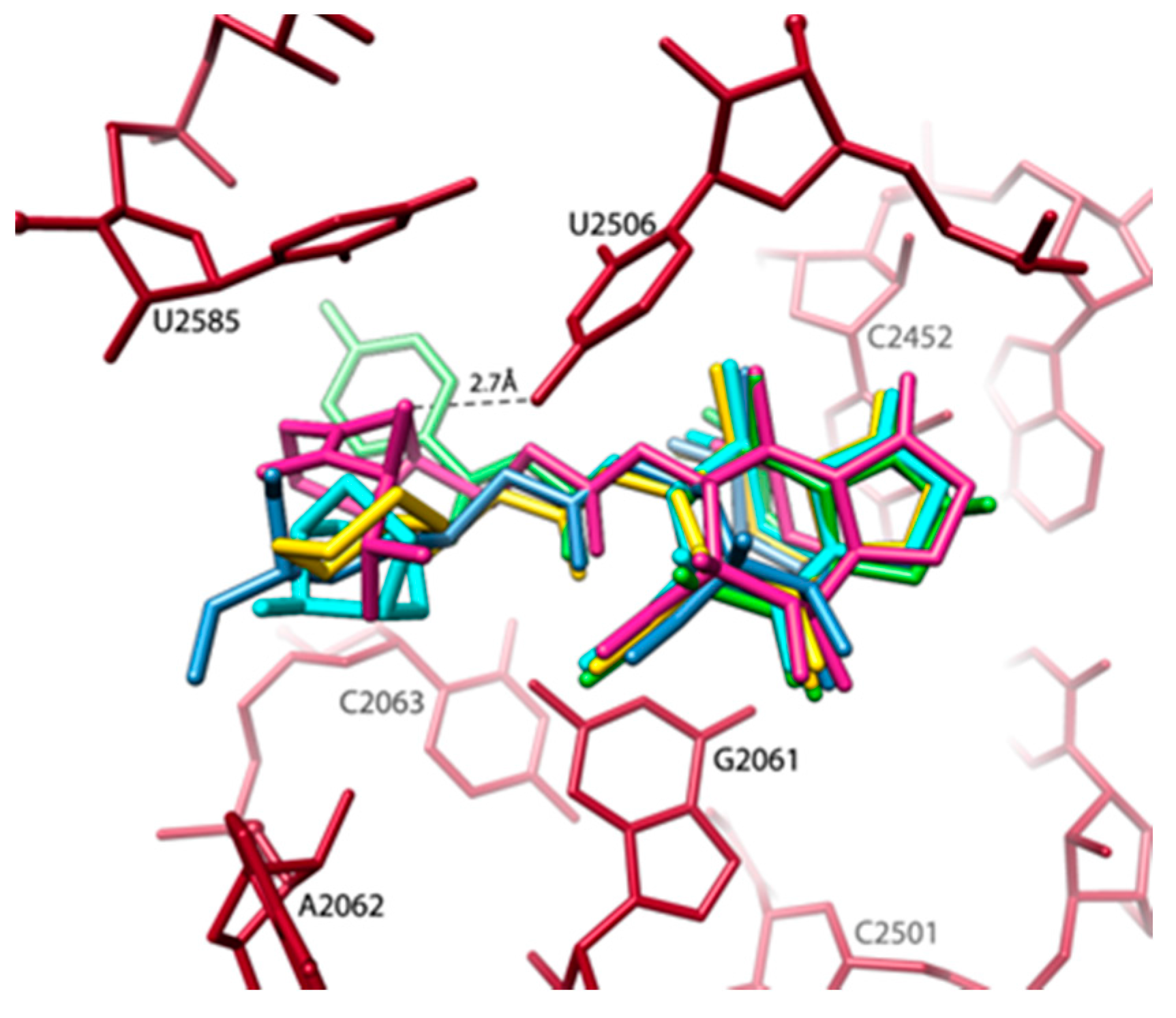
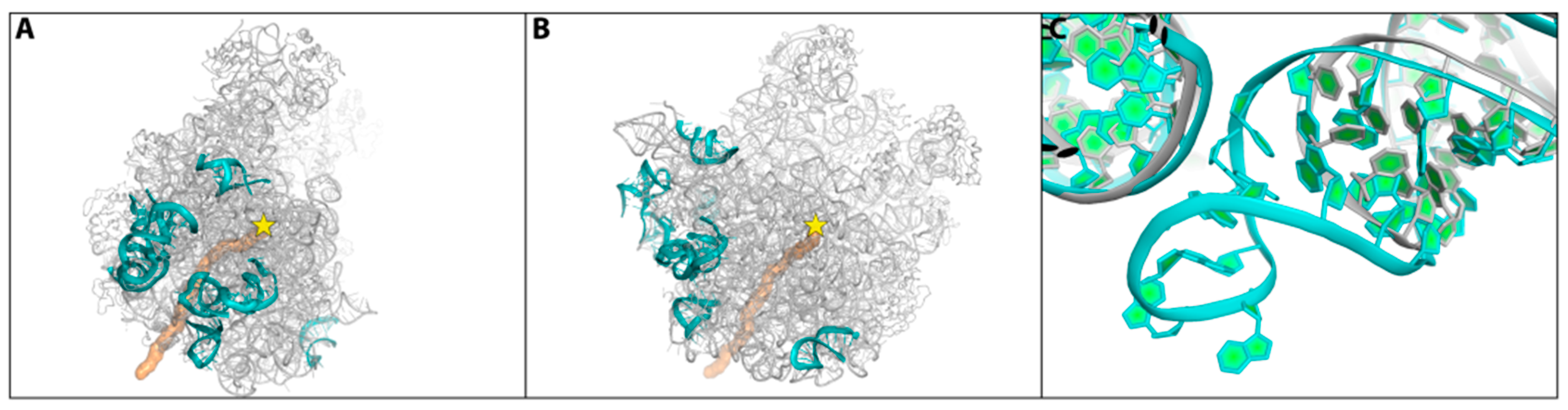
- Minimal inhibitory concentration (MIC) values of known antibiotics can be optimized. For example, the additional of a single hydrogen bond between an antibiotic and its pocket improves the MIC dramatically (Figure 3). Flexibility is a common property of antibiotics binding pockets. For example, in synergetic antibiotics, alterations of rRNA conformation proximal to the macrolide’s binding pockets can propagate towards the PTC. Additionally, it seems that there is an allosteric link between the tunnel and the catalytic center (PTC) of the ribosome.
- -
- It is suggested that species-specific structural motifs should be exploited for the creation of novel antibiotics with a better distinction between pathogens and useful bacterial species in the microbiome. In fact, the next generation antibiotics should be degradable and species-specific. Thus, the aim of immediate research should be to minimize resistance to antibiotics while preserving the microbiome as well as reducing the contamination of the environment.
- -
- The proposed design of “pathogen-specific antibiotics,” which is a revolution in the current concepts of antibiotics, is of immediate need. “Pathogen-specific antibiotics” means antibiotic drugs specific for each and every pathogen. This strategy requires the clinically fast identification of pathogens that is already being addressed [60].
- -
- The practical application of “pathogen-specific antibiotics” requires the swift determination of the structures of antibiotics targets (e.g., ribosomes) of all or most pathogens. For this aim, the recent exciting development of single particle 3D cryo-electron microscopy should be more suitable than X-ray crystallography, since it can be performed by the use of relatively small amounts and does not require crystals.
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fischbach, M.A.; Walsh, C.T. Antibiotics for emerging pathogens. Science 2009, 325, 1089–1093. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, A. Resistance to antibiotics found in isolated Amazonian tribe. Science AAAS 2015. [Google Scholar] [CrossRef]
- Schlunzen, F.; Zarivach, R.; Harms, J.; Bashan, A.; Tocilj, A.; Albrecht, R.; Yonath, A.; Franceschi, F. Structural basis for the interaction of antibiotics with the peptidyl transferase centre in eubacteria. Nature 2001, 413, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Auerbach, T.; Bashan, A.; Harms, J.; Schluenzen, F.; Zarivach, R.; Bartels, H.; Agmon, I.; Kessler, M.; Pioletti, M.; Franceschi, F.; et al. Antibiotics targeting ribosomes: Crystallographic studies. Curr. Drug Targets Infect. Disord. 2002, 2, 169–186. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.L.; Moore, P.B.; Steitz, T.A. Structures of five antibiotics bound at the peptidyl transferase center of the large ribosomal subunit. J. Mol. Biol. 2003, 330, 1061–1075. [Google Scholar] [CrossRef]
- Pfister, P.; Jenni, S.; Poehlsgaard, J.; Thomas, A.; Douthwaite, S.; Ban, N.; Bottger, E.C. The structural basis of macrolide-ribosome binding assessed using mutagenesis of 23S rRNA positions 2058 and 2059. J. Mol. Biol. 2004, 342, 1569–1581. [Google Scholar] [CrossRef] [PubMed]
- Schuwirth, B.S.; Borovinskaya, M.A.; Hau, C.W.; Zhang, W.; Vila-Sanjurjo, A.; Holton, J.M.; Cate, J.H. Structures of the bacterial ribosome at 3.5 A resolution. Science 2005, 310, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Dunkle, J.A.; Xiong, L.; Mankin, A.S.; Cate, J.H. Structures of the Escherichia coli ribosome with antibiotics bound near the peptidyl transferase center explain spectra of drug action. Proc. Natl. Acad. Sci. USA 2010, 107, 17152–17157. [Google Scholar] [CrossRef] [PubMed]
- Bulkley, D.; Innis, C.A.; Blaha, G.; Steitz, T.A. Revisiting the structures of several antibiotics bound to the bacterial ribosome. Proc. Natl. Acad. Sci. USA 2010, 107, 17158–17163. [Google Scholar] [CrossRef] [PubMed]
- Bulkley, D.; Johnson, F.; Steitz, T.A. The antibiotic thermorubin inhibits protein synthesis by binding to inter-subunit bridge B2a of the ribosome. J. Mol. Biol. 2012, 416, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Polikanov, Y.S.; Szal, T.; Jiang, F.; Gupta, P.; Matsuda, R.; Shiozuka, M.; Steitz, T.A.; Vazquez-Laslop, N.; Mankin, A.S. Negamycin interferes with decoding and translocation by simultaneous interaction with rRNA and tRNA. Mol. Cell. 2014, 56, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.N.; Lomakin, I.B.; Gagnon, M.G.; Steitz, T.A. The mechanism of inhibition of protein synthesis by the proline-rich peptide oncocin. Nat. Struct. Mol. Biol. 2015, 22, 466–469. [Google Scholar] [CrossRef] [PubMed]
- Amunts, A.; Fiedorczuk, K.; Truong, T.T.; Chandler, J.; Greenberg, E.P.; Ramakrishnan, V. Bactobolin A binds to a site on the 70S ribosome distinct from previously seen antibiotics. J. Mol. Biol. 2015, 427, 753–755. [Google Scholar] [CrossRef] [PubMed]
- Harms, J.M.; Bartels, H.; Schlunzen, F.; Yonath, A. Antibiotics acting on the translational machinery. J. Cell. Sci. 2003, 116, 1391–1393. [Google Scholar] [CrossRef] [PubMed]
- Noeske, J.; Wasserman, M.R.; Terry, D.S.; Altman, R.B.; Blanchard, S.C.; Cate, J.H. High-resolution structure of the Escherichia coli ribosome. Nat. Struct. Mol. Biol. 2015, 22, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Eyal, Z.; Matzov, D.; Krupkin, M.; Wekselman, I.; Paukner, S.; Zimmerman, E.; Rozenberg, H.; Bashan, A.; Yonath, A. Structural insights into species-specific features of the ribosome from the pathogen Staphylococcus aureus. Proc. Natl. Acad. Sci. USA 2015, 112, E5805–E5814. [Google Scholar] [CrossRef] [PubMed]
- Krupkin, M.; Wekselman, I.; Eyal, Z.; Matzov, D.; Rozenberg, H.; Diskin-Posner, Y.; Zimmerman, E.; Yonath, B.A.A. The orthosomycins avilamycin and evernimicin block IF2 and A-tRNA binding to the large ribosomal subunit. In Proceedings of the Ribosome Structure and Function EMBO Meeting, Strasbourg, France, 6–10 July 2016.
- Douthwaite, S. Designer drugs for discerning bugs. Proc. Natl. Acad. Sci. USA 2010, 107, 17065–17066. [Google Scholar] [CrossRef] [PubMed]
- Berisio, R.; Harms, J.; Schluenzen, F.; Zarivach, R.; Hansen, H.A.; Fucini, P.; Yonath, A. Structural insight into the antibiotic action of telithromycin against resistant mutants. J. Bacteriol. 2003, 185, 4276–4279. [Google Scholar] [CrossRef] [PubMed]
- Berisio, R.; Schluenzen, F.; Harms, J.; Bashan, A.; Auerbach, T.; Baram, D.; Yonath, A. Structural insight into the role of the ribosomal tunnel in cellular regulation. Nat. Struct. Biol. 2003, 10, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Schlunzen, F.; Harms, J.M.; Franceschi, F.; Hansen, H.A.; Bartels, H.; Zarivach, R.; Yonath, A. Structural basis for the antibiotic activity of ketolides and azalides. Structure 2003, 11, 329–338. [Google Scholar] [CrossRef]
- Llano-Sotelo, B.; Dunkle, J.; Klepacki, D.; Zhang, W.; Fernandes, P.; Cate, J.H.; Mankin, A.S. Binding and action of CEM-101, a new fluoroketolide antibiotic that inhibits protein synthesis. Antimicrob. Agents Chemother. 2010, 54, 4961–4970. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.; Chen, J.; Tsai, A.; Kornberg, G.; Puglisi, J.D. Sequence-dependent elongation dynamics on macrolide-bound ribosomes. Cell Rep. 2014, 7, 1534–1546. [Google Scholar] [CrossRef] [PubMed]
- Kannan, K.; Kanabar, P.; Schryer, D.; Florin, T.; Oh, E.; Bahroos, N.; Tenson, T.; Weissman, J.S.; Mankin, A.S. The general mode of translation inhibition by macrolide antibiotics. Proc. Natl. Acad. Sci. USA 2014, 111, 15958–15963. [Google Scholar] [CrossRef] [PubMed]
- Tu, D.; Blaha, G.; Moore, P.B.; Steitz, T.A. Structures of MLSBK antibiotics bound to mutated large ribosomal subunits provide a structural explanation for resistance. Cell 2005, 121, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Vester, B.; Douthwaite, S. Macrolide resistance conferred by base substitutions in 23S rRNA. Antimicrob. Agents Chemother. 2001, 45, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Vimberg, V.; Xiong, L.; Bailey, M.; Tenson, T.; Mankin, A. Peptide-mediated macrolide resistance reveals possible specific interactions in the nascent peptide exit tunnel. Mol. Microbiol. 2004, 54, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Blaha, G.; Gurel, G.; Schroeder, S.J.; Moore, P.B.; Steitz, T.A. Mutations outside the anisomycin-binding site can make ribosomes drug-resistant. J. Mol. Biol. 2008, 379, 505–519. [Google Scholar] [CrossRef] [PubMed]
- Nakatogawa, H.; Ito, K. The ribosomal exit tunnel functions as a discriminating gate. Cell 2002, 108, 629–636. [Google Scholar] [CrossRef]
- Gong, F.; Yanofsky, C. Rho’s role in transcription attenuation in the TNA operon of E. coli. Methods Enzymol. 2003, 371, 383–391. [Google Scholar] [PubMed]
- Woolhead, C.A.; Johnson, A.E.; Bernstein, H.D. Translation arrest requires two-way communication between a nascent polypeptide and the ribosome. Mol. Cell. 2006, 22, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Seidelt, B.; Innis, C.A.; Wilson, D.N.; Gartmann, M.; Armache, J.P.; Villa, E.; Trabuco, L.G.; Becker, T.; Mielke, T.; Schulten, K.; et al. Structural insight into nascent polypeptide chain-mediated translational stalling. Science 2009, 326, 1412–1415. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Chiba, S.; Pogliano, K. Divergent stalling sequences sense and control cellular physiology. Biochem. Biophys. Res. Commun. 2010, 393, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Chiba, S.; Ito, K. Multisite ribosomal stalling: A unique mode of regulatory nascent chain action revealed for MifM. Mol. Cell. 2012, 47, 863–872. [Google Scholar] [CrossRef] [PubMed]
- Ramu, H.; Vazquez-Laslop, N.; Klepacki, D.; Dai, Q.; Piccirilli, J.; Micura, R.; Mankin, A.S. Nascent peptide in the ribosome exit tunnel affects functional properties of the A-site of the peptidyl transferase center. Mol. Cell. 2011, 41, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Laslop, N.; Klepacki, D.; Mulhearn, D.C.; Ramu, H.; Krasnykh, O.; Franzblau, S.; Mankin, A.S. Role of antibiotic ligand in nascent peptide-dependent ribosome stalling. Proc. Natl. Acad. Sci. USA 2011, 108, 10496–10501. [Google Scholar] [CrossRef] [PubMed]
- Woolstenhulme, C.J.; Parajuli, S.; Healey, D.W.; Valverde, D.P.; Petersen, E.N.; Starosta, A.L.; Guydosh, N.R.; Johnson, W.E.; Wilson, D.N.; Buskirk, A.R. Nascent peptides that block protein synthesis in bacteria. Proc. Natl. Acad. Sci. USA 2013, 110, E878–E887. [Google Scholar] [CrossRef] [PubMed]
- Belousoff, M.J.; Shapira, T.; Bashan, A.; Zimmerman, E.; Rozenberg, H.; Arakawa, K.; Kinashi, H.; Yonath, A. Crystal structure of the synergistic antibiotic pair, lankamycin and lankacidin, in complex with the large ribosomal subunit. Proc. Natl. Acad. Sci. USA 2011, 108, 2717–2722. [Google Scholar] [CrossRef] [PubMed]
- Arenz, S.; Ramu, H.; Gupta, P.; Berninghausen, O.; Beckmann, R.; Vazquez-Laslop, N.; Mankin, A.S.; Wilson, D.N. Molecular basis for erythromycin-dependent ribosome stalling during translation of the ErmBL leader peptide. Nat. Commun. 2014, 5, 3501. [Google Scholar] [CrossRef] [PubMed]
- Berisio, R.; Corti, N.; Pfister, P.; Yonath, A.; Bottger, E.C. 23S rRNA 2058A-->G alteration mediates ketolide resistance in combination with deletion in L22. Antimicrob. Agents Chemother. 2006, 50, 3816–3823. [Google Scholar] [CrossRef] [PubMed]
- Wekselman, I.; Zimmerman, E.; Rozenberg, H.; Bashan, A.; Yonath, A. The structural basis for the mutated ribosomal protein L22 mediated resistance to erythromycin demonstrates the dynamics of the nascent protein exit tunnel. In Proceedings of the 7th FISEB Meeting, Eilat, Israel, 10–13 February 2014.
- Gregory, S.T.; Dahlberg, A.E. Erythromycin resistance mutations in ribosomal proteins L22 and L4 perturb the higher order structure of 23S ribosomal RNA. J. Mol. Biol. 1999, 289, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Sothiselvam, S.; Liu, B.; Han, W.; Ramu, H.; Klepacki, D.; Atkinson, G.C.; Brauer, A.; Remm, M.; Tenson, T.; Schulten, K.; et al. Macrolide antibiotics allosterically predispose the ribosome for translation arrest. Proc. Natl. Acad. Sci. USA 2014, 111, 9804–9809. [Google Scholar] [CrossRef] [PubMed]
- Pyetan, E.; Baram, D.; Auerbach-Nevo, T.; Yonath, A. Chemical parameters influencing fine-tuning in the binding of macrolide antibiotics to the ribosomal tunnel. Pure Appl. Chem. 2007, 79, 955–968. [Google Scholar] [CrossRef]
- Poulsen, S.M.; Karlsson, M.; Johansson, L.B.; Vester, B. The pleuromutilin drugs tiamulin and valnemulin bind to the RNA at the peptidyl transferase centre on the ribosome. Mol. Microbiol. 2001, 41, 1091–1099. [Google Scholar] [CrossRef] [PubMed]
- Auerbach, T.; Mermershtain, I.; Bashan, A.; Davidovich, C.; Rosenberg, H.; Sherman, D.H.; Yonath, A. Structural basis for the antibacterial activity of the 12-membered-ring mono-sugar macrolide methymycin. Biotechnology 2009, 84, 24–35. [Google Scholar]
- Toh, S.M.; Mankin, A.S. An indigenous posttranscriptional modification in the ribosomal peptidyl transferase center confers resistance to an array of protein synthesis inhibitors. J. Mol. Biol. 2008, 380, 593–597. [Google Scholar] [CrossRef] [PubMed]
- Schlunzen, F.; Pyetan, E.; Fucini, P.; Yonath, A.; Harms, J.M. Inhibition of peptide bond formation by pleuromutilins: The structure of the 50S ribosomal subunit from Deinococcus radiodurans in complex with tiamulin. Mol. Microbiol. 2004, 54, 1287–1294. [Google Scholar] [CrossRef] [PubMed]
- Davidovich, C.; Bashan, A.; Auerbach-Nevo, T.; Yaggie, R.D.; Gontarek, R.R.; Yonath, A. Induced-fit tightens pleuromutilins binding to ribosomes and remote interactions enable their selectivity. Proc. Natl. Acad. Sci. USA 2007, 104, 4291–4296. [Google Scholar] [CrossRef] [PubMed]
- Davidovich, C.; Bashan, A.; Yonath, A. Structural basis for cross-resistance to ribosomal PTC antibiotics. Proc. Natl. Acad. Sci. USA 2008, 105, 20665–20670. [Google Scholar] [CrossRef] [PubMed]
- Noeske, J.; Huang, J.; Olivier, N.B.; Giacobbe, R.A.; Zambrowski, M.; Cate, J.H. Synergy of streptogramin antibiotics occurs independently of their effects on translation. Antimicrob. Agents Chemother. 2014, 58, 5269–5279. [Google Scholar] [CrossRef] [PubMed]
- Harms, J.M.; Schlunzen, F.; Fucini, P.; Bartels, H.; Yonath, A. Alterations at the peptidyl transferase centre of the ribosome induced by the synergistic action of the streptogramins dalfopristin and quinupristin. BMC Biol. 2004, 2, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auerbach, T.; Mermershtain, I.; Davidovich, C.; Bashan, A.; Belousoff, M.; Wekselman, I.; Zimmerman, E.; Xiong, L.; Klepacki, D.; Arakawa, K.; et al. The structure of ribosome-lankacidin complex reveals ribosomal sites for synergistic antibiotics. Proc. Natl. Acad. Sci. USA 2010, 107, 1983–1988. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.N. On the specificity of antibiotics targeting the large ribosomal subunit. Ann. N. Y. Acad. Sci. 2011, 1241, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Kannan, K.; Mankin, A.S. Macrolide antibiotics in the ribosome exit tunnel: Species-specific binding and action. Ann. N. Y. Acad. Sci. 2011, 1241, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Meskauskas, A.; Petrov, A.N.; Dinman, J.D. Identification of functionally important amino acids of ribosomal protein L3 by saturation mutagenesis. Mol. Cell Biol. 2005, 25, 10863–10874. [Google Scholar] [CrossRef] [PubMed]
- Pringle, M.; Poehlsgaard, J.; Vester, B.; Long, K.S. Mutations in ribosomal protein L3 and 23S ribosomal RNA at the peptidyl transferase centre are associated with reduced susceptibility to tiamulin in Brachyspira spp. Isolates. Mol. Microbiol. 2004, 54, 1295–1306. [Google Scholar] [CrossRef] [PubMed]
- Weller, J.; Hill, W.E. Probing the initiation complex formation on E. coli ribosomes using short complementary DNA oligomers. Biochimie 1991, 73, 971–981. [Google Scholar] [CrossRef]
- Seefeldt, A.C.; Graf, M.; Perebaskine, N.; Nguyen, F.; Arenz, S.; Mardirossian, M.; Scocchi, M.; Wilson, D.N.; Innis, C.A. Structure of the mammalian antimicrobial peptide Bac7(1–16) bound within the exit tunnel of a bacterial ribosome. Nucleic Acids Res. 2016, 44, 2429–2438. [Google Scholar] [CrossRef] [PubMed]
- Lam, B.; Das, J.; Holmes, R.D.; Live, L.; Sage, A.; Sargent, E.H.; Kelley, S.O. Solution-based circuits enable rapid and multiplexed pathogen detection. Nat. Commun. 2013, 4, 2001–2003. [Google Scholar] [CrossRef] [PubMed]


© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Auerbach-Nevo, T.; Baram, D.; Bashan, A.; Belousoff, M.; Breiner, E.; Davidovich, C.; Cimicata, G.; Eyal, Z.; Halfon, Y.; Krupkin, M.; et al. Ribosomal Antibiotics: Contemporary Challenges. Antibiotics 2016, 5, 24. https://doi.org/10.3390/antibiotics5030024
Auerbach-Nevo T, Baram D, Bashan A, Belousoff M, Breiner E, Davidovich C, Cimicata G, Eyal Z, Halfon Y, Krupkin M, et al. Ribosomal Antibiotics: Contemporary Challenges. Antibiotics. 2016; 5(3):24. https://doi.org/10.3390/antibiotics5030024
Chicago/Turabian StyleAuerbach-Nevo, Tamar, David Baram, Anat Bashan, Matthew Belousoff, Elinor Breiner, Chen Davidovich, Giuseppe Cimicata, Zohar Eyal, Yehuda Halfon, Miri Krupkin, and et al. 2016. "Ribosomal Antibiotics: Contemporary Challenges" Antibiotics 5, no. 3: 24. https://doi.org/10.3390/antibiotics5030024