
The Novel Aminomethylcycline Omadacycline Has High Specificity for the Primary Tetracycline-Binding Site on the Bacterial Ribosome


Abstract
:1. Introduction
2. Results
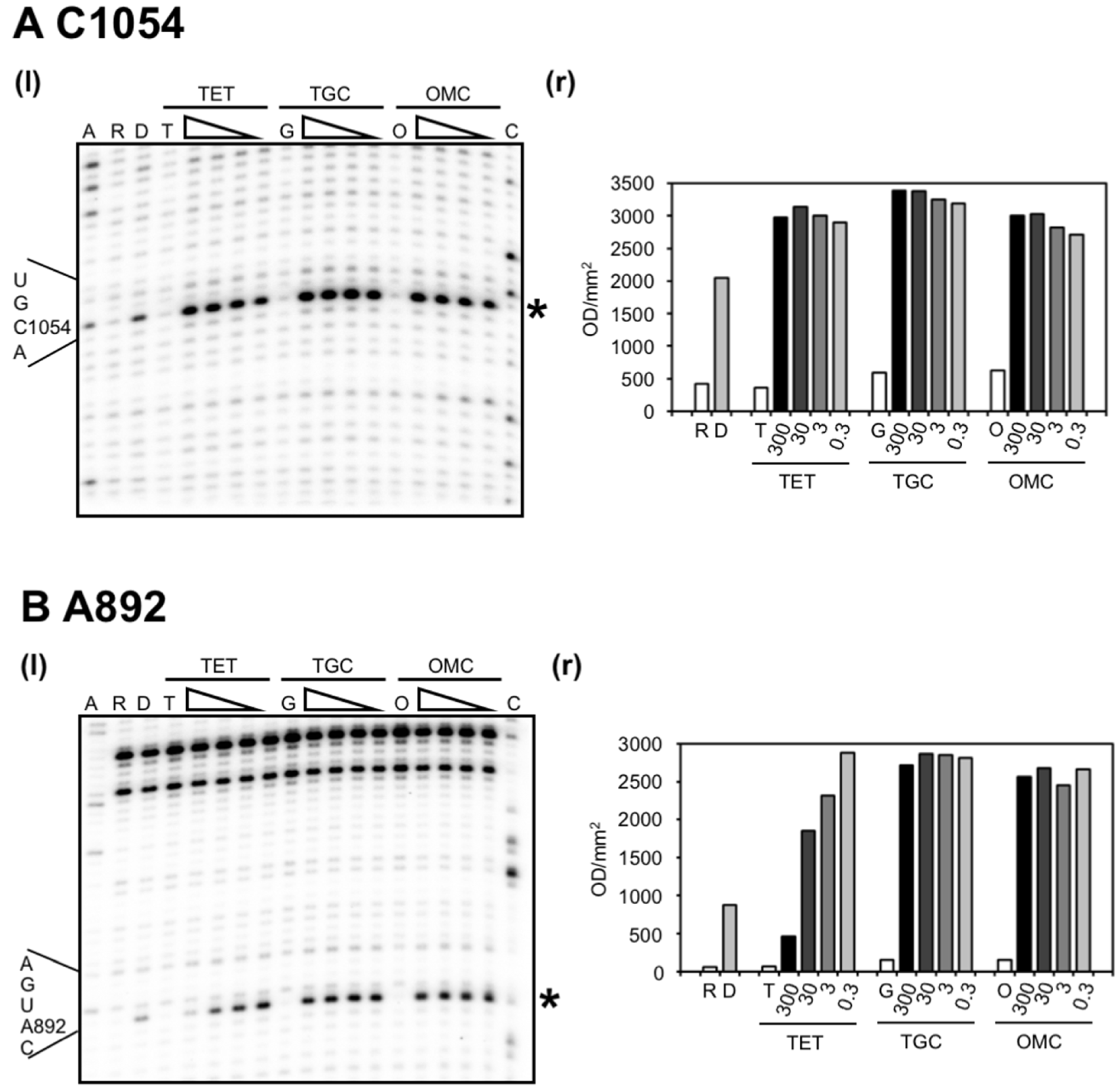
2.1. Omadacycline Is Susceptible to 16S rRNA Mutations Conferring TET Resistance
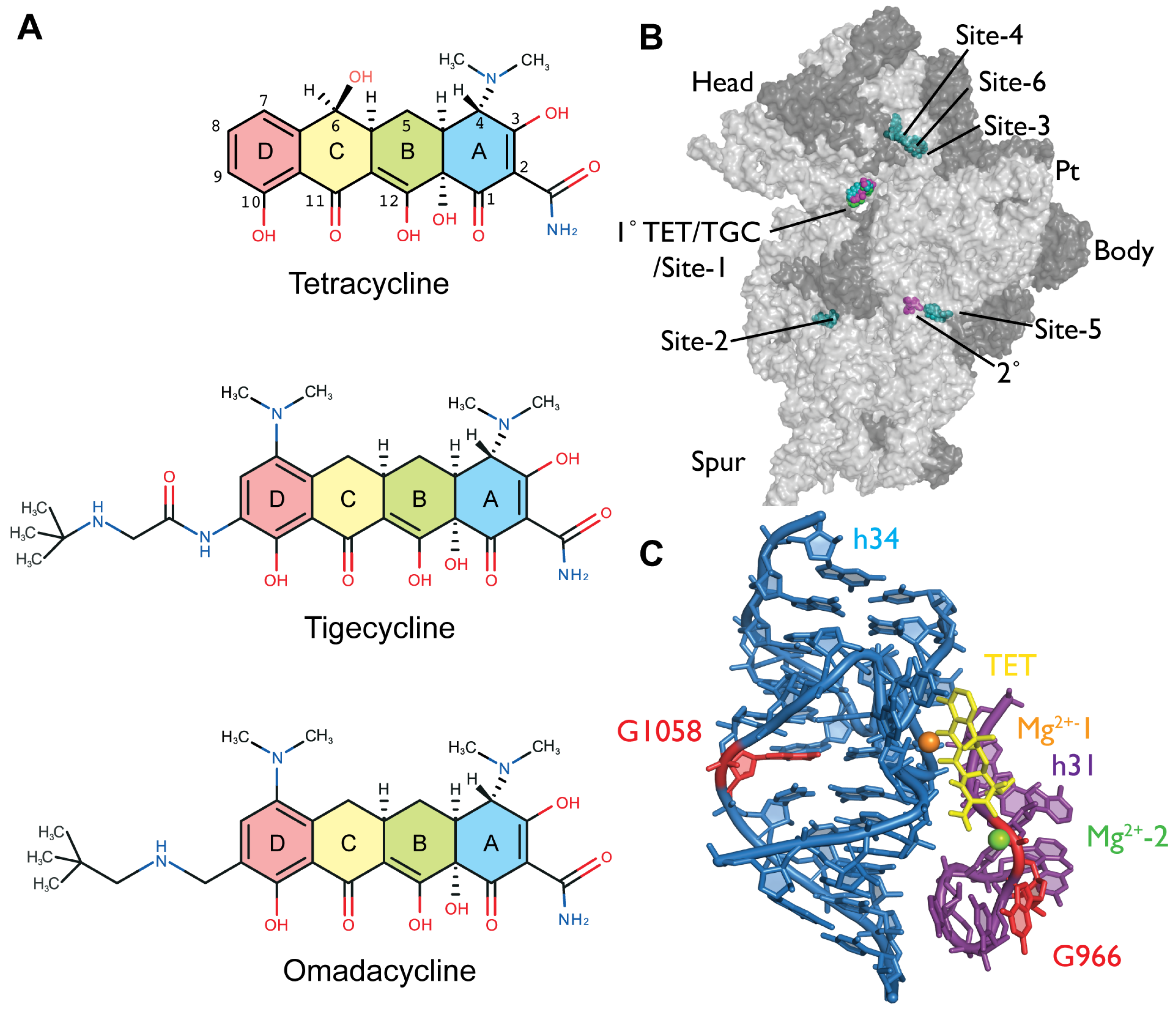
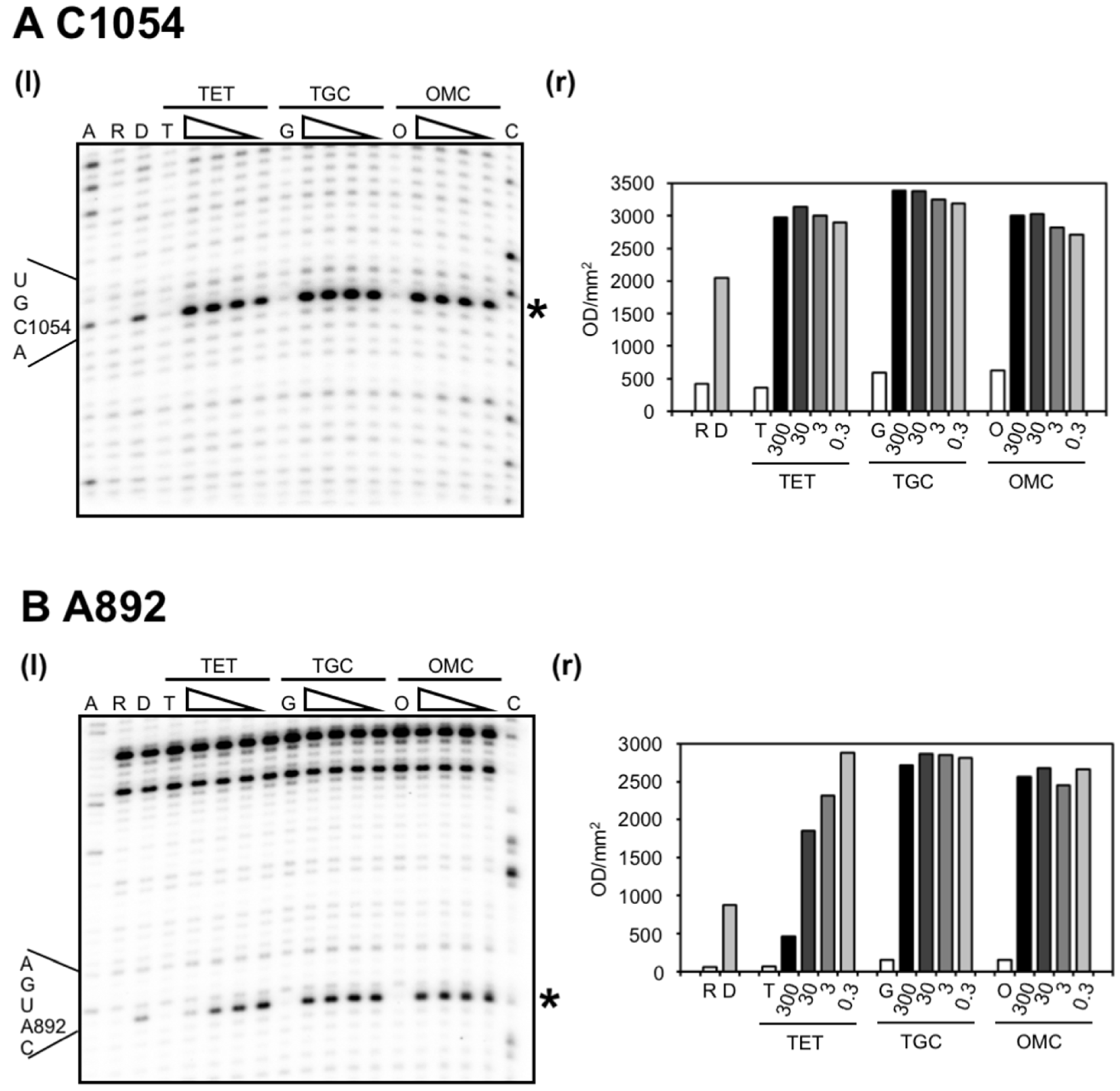
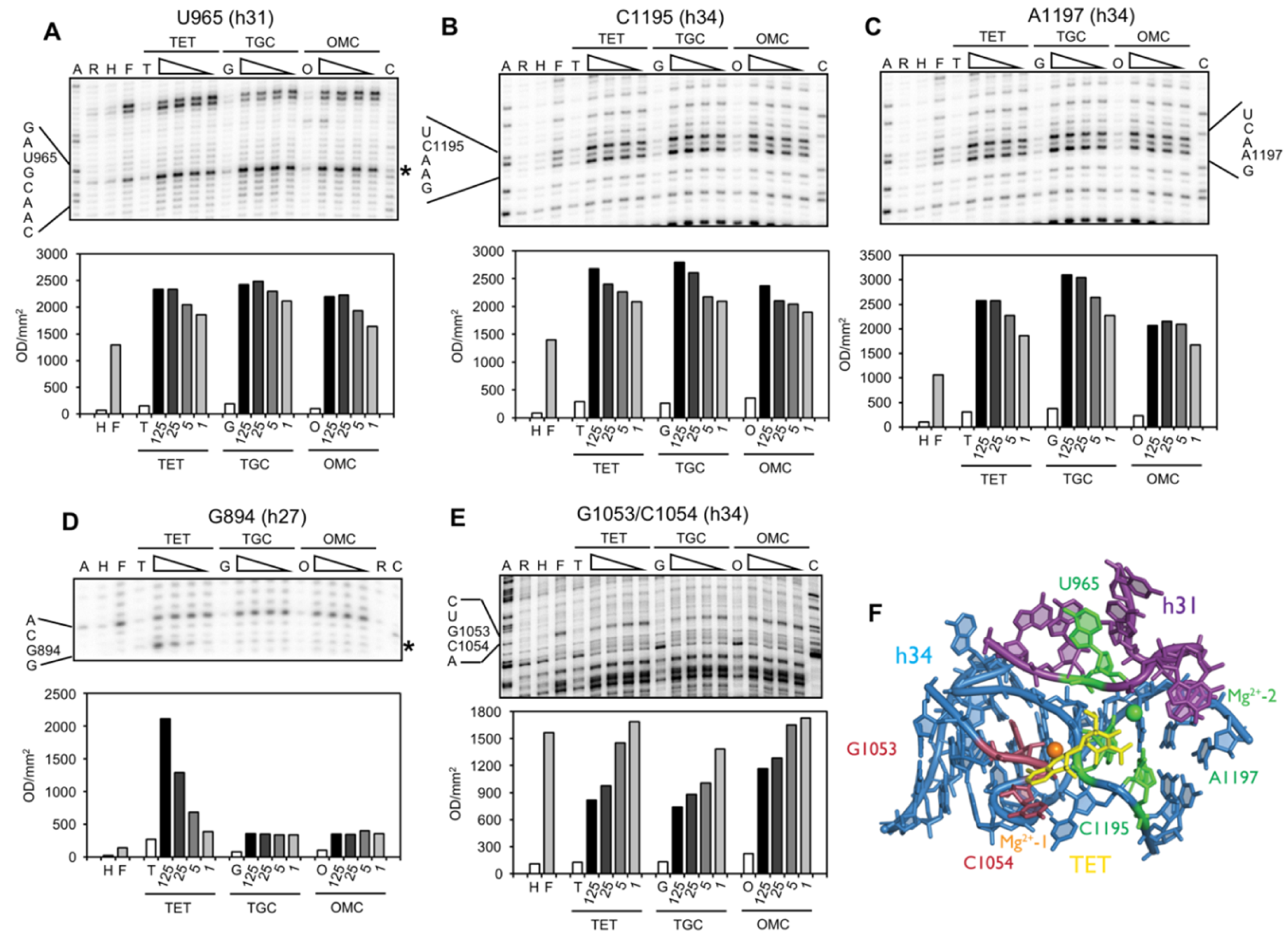
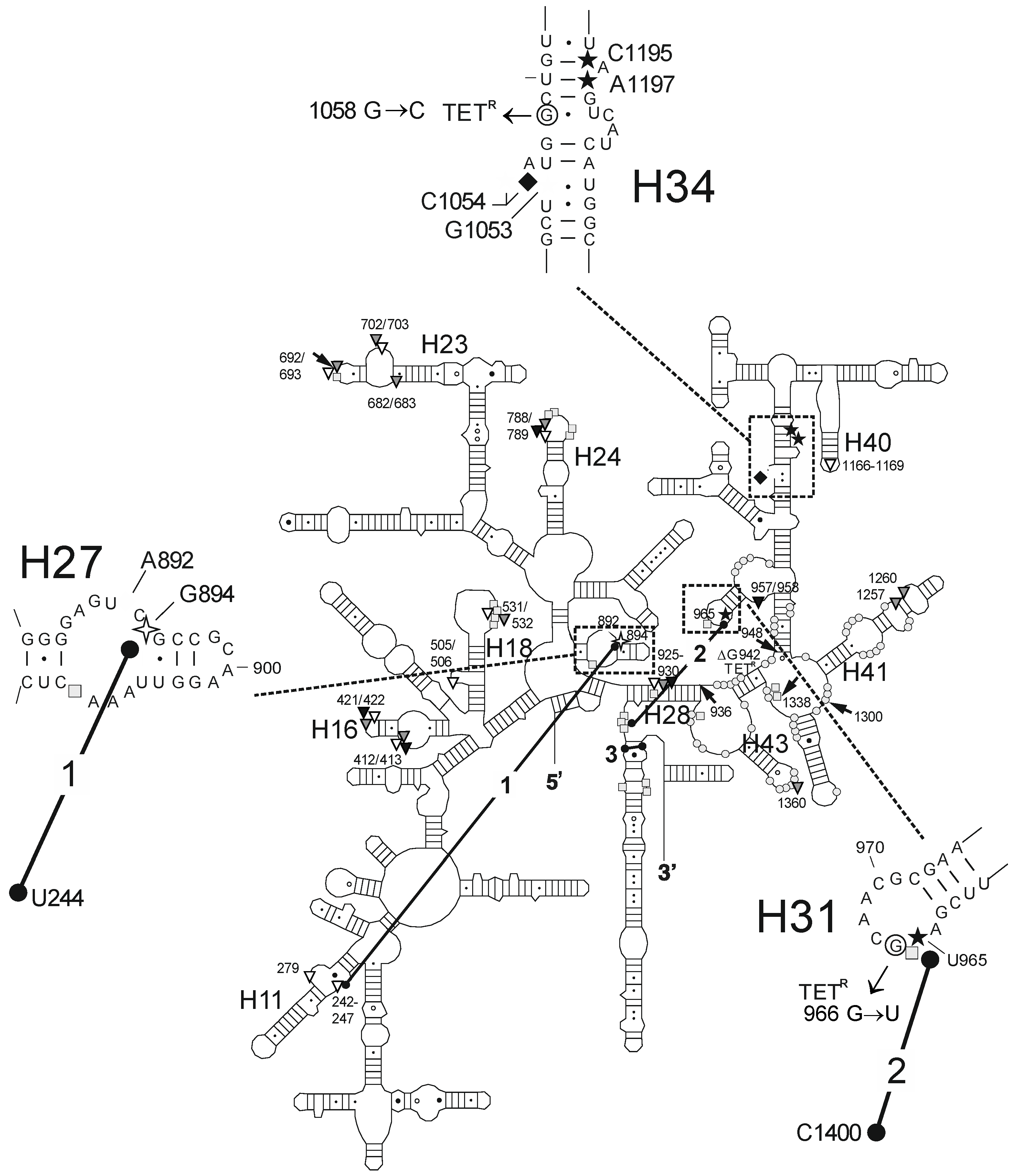
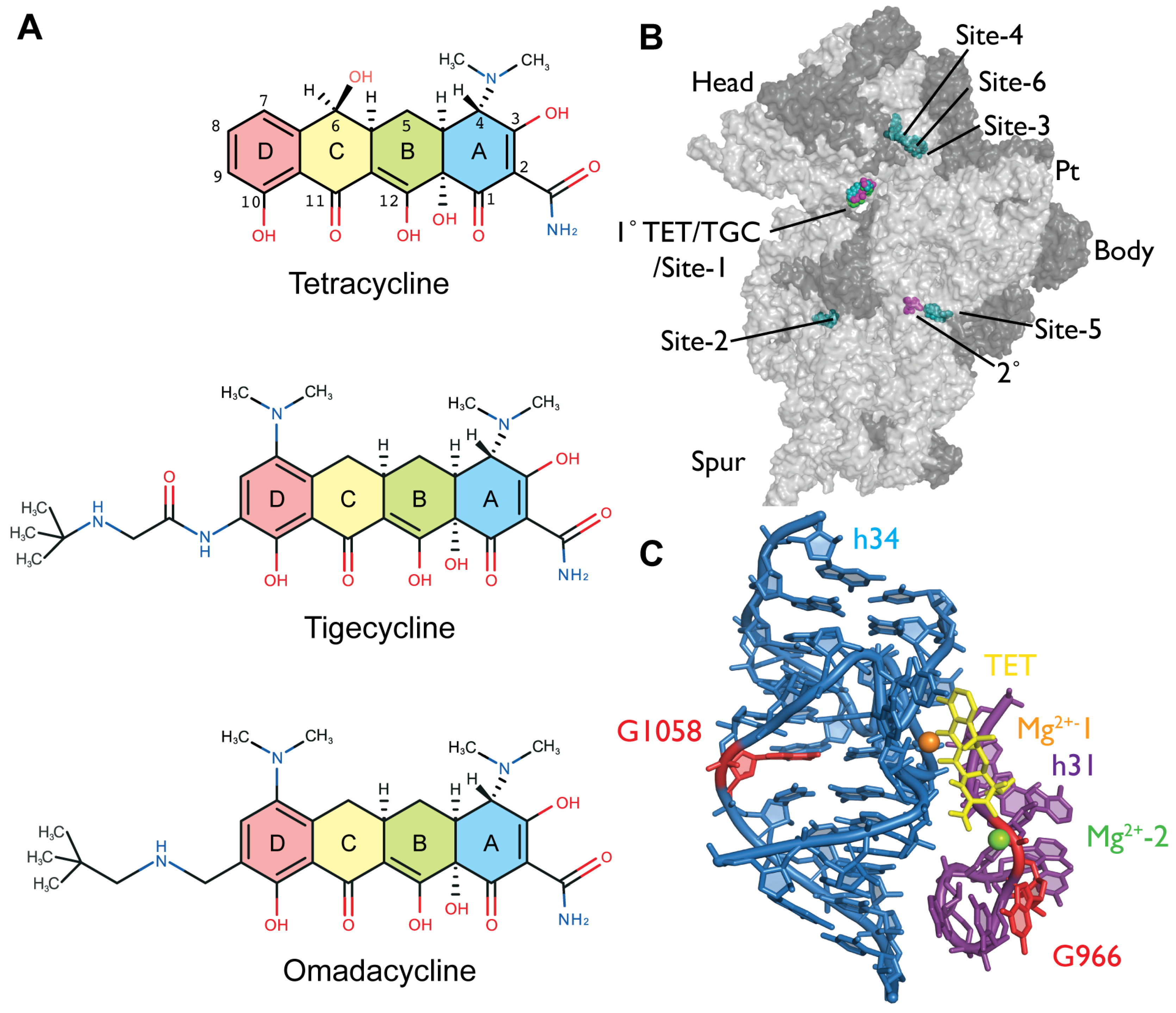
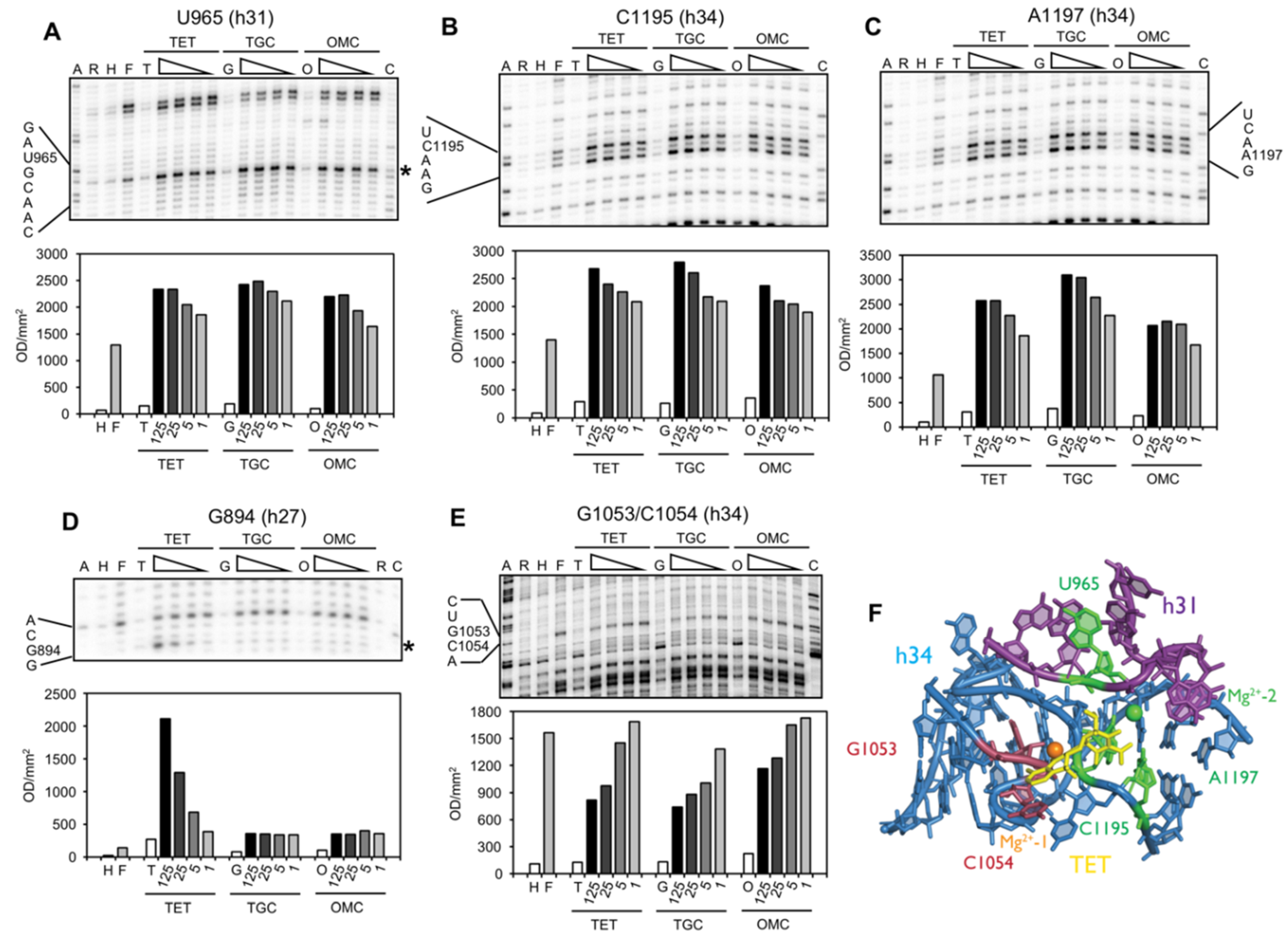
2.2. Chemical Probing Indicates That OMC Binds Specifically to the Primary TET Binding Site
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Strains and Plasmids
4.3. MIC Determinations
4.4. Isolation of 70S Ribosomes
4.5. Chemical Modification of 70S Ribosomes with DMS
4.6. Fenton-Mediated Hydroxyl Radical Cleavage Reactions
4.7. Extraction of rRNA
4.8. Primer Extension Reaction
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| TET | Tetracycline |
| TGC | Tigecycline |
| OMC | Omadacycline |
| aa | amino acid |
| tRNA | transfer RNA |
| rRNA | ribosomal RNA |
| MIC | minimal inhibitory concentration |
| CLSI | Clinical & Laboratory Standards Institute |
| DMS | Dimethyl sulfate |
| EDTA | Ethylenediaminetetraacetic acid |
| DEPC | Diethyl pyrocarbonate |
| cDNA | complementary DNA |
References
- Rasmussen, B.; Noller, H.F.; Daubresse, G.; Oliva, B.; Misulovin, Z.; Rothstein, D.M.; Ellestad, G.A.; Gluzman, Y.; Tally, F.P.; Chopra, I. Molecular basis of tetracycline action: Identification of analogs whose primary target is not the bacterial ribosome. Antimicrob. Agents Chemother. 1991, 35, 2306–2311. [Google Scholar] [CrossRef] [PubMed]
- Brodersen, D.E.; Clemons, W.M., Jr.; Carter, A.P.; Morgan-Warren, R.J.; Wimberly, B.T.; Ramakrishnan, V. The structural basis for the action of the antibiotics tetracycline, pactamycin, and hygromycin B on the 30S ribosomal subunit. Cell 2000, 103, 1143–1154. [Google Scholar] [CrossRef]
- Pioletti, M.; Schlünzen, F.; Harms, J.; Zarivach, R.; Glühmann, M.; Avila, H.; Bashan, A.; Bartels, H.; Auerbach, T.; Jacobi, C.; et al. Crystal structures of complexes of the small ribosomal subunit with tetracycline, edeine and IF3. EMBO J. 2001, 20, 1829–1839. [Google Scholar] [CrossRef] [PubMed]
- Jenner, L.; Starosta, A.L.; Terry, D.S.; Mikolajka, A.; Filonava, L.; Yusupov, M.; Blanchard, S.C.; Wilson, D.N.; Yusupova, G. Structural basis for potent inhibitory activity of the antibiotic tigecycline during protein synthesis. Proc. Natl. Acad. Sci. USA 2013, 110, 3812–3816. [Google Scholar] [CrossRef] [PubMed]
- Schedlbauer, A.; Kaminishi, T.; Ochoa-Lizarralde, B.; Dhimole, N.; Zhou, S.; López-Alonso, J.P.; Connell, S.R.; Fucini, P. Structural characterization of an alternative mode of tigecycline binding to the bacterial ribosome. Antimicrob. Agents Chemother. 2015, 59, 2849–2854. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, S.C.; Gonzalez, R.L.; Kim, H.D.; Chu, S.; Puglisi, J.D. tRNA selection and kinetic proofreading in translation. Nat. Struct. Mol. Biol. 2004, 11, 1008–1014. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.L.; Levy, S.B. The history of the tetracyclines. Ann. N. Y. Acad. Sci. 2011, 1241, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Chopra, I.; Roberts, M. Tetracycline antibiotics: Mode of action, applications, molecular biology, and epidemiology of bacterial resistance. Microbiol. Mol. Biol. Rev. 2001, 65, 232–260. [Google Scholar] [CrossRef] [PubMed]
- Marshall, B.M.; Levy, S.B. Food animals and antimicrobials: Impacts on human health. Clin. Microbiol. Rev. 2011, 24, 718–733. [Google Scholar] [CrossRef] [PubMed]
- Thaker, M.; Spanogiannopoulos, P.; Wright, G.D. The tetracycline resistome. Cell Mol. Life Sci. 2010, 67, 419–431. [Google Scholar] [CrossRef] [PubMed]
- Petersen, P.J.; Jacobus, N.V.; Weiss, W.J.; Sum, P.E.; Testa, R.T. In vitro and in vivo antibacterial activities of a novel glycylcycline, the 9-t-butylglycylamido derivative of minocycline (GAR-936). Antimicrob. Agents Chemother. 1999, 43, 738–744. [Google Scholar] [PubMed]
- Honeyman, L.; Ismail, M.; Nelson, M.L.; Bhatia, B.; Bowser, T.E.; Chen, J.; Mechiche, R.; Ohemeng, K.; Verma, A.K.; Cannon, E.P.; et al. Structure-activity relationship of the aminomethylcyclines and the discovery of omadacycline. Antimicrob. Agents Chemother. 2015, 59, 7044–7053. [Google Scholar] [CrossRef] [PubMed]
- Draper, M.P.; Weir, S.; Macone, A.; Donatelli, J.; Trieber, C.A.; Tanaka, S.K.; Levy, S.B. Mechanism of action of the novel aminomethylcycline antibiotic omadacycline. Antimicrob. Agents Chemother. 2014, 58, 1279–1283. [Google Scholar] [CrossRef] [PubMed]
- Macone, A.B.; Caruso, B.K.; Leahy, R.G.; Donatelli, J.; Weir, S.; Draper, M.P.; Tanaka, S.K.; Levy, S.B. In vitro and in vivo antibacterial activities of omadacycline, a novel aminomethylcycline. Antimicrob. Agents Chemother. 2014, 58, 1127–1135. [Google Scholar] [CrossRef] [PubMed]
- Bauer, G.; Berens, C.; Projan, S.J.; Hillen, W. Comparison of tetracycline and tigecycline binding to ribosomes mapped by dimethylsulphate and drug-directed Fe2+ cleavage of 16S rRNA. J. Antimicrob. Chemother. 2004, 53, 592–599. [Google Scholar] [CrossRef] [PubMed]
- Moore, I.F.; Hughes, D.W.; Wright, G.D. Tigecycline is modified by the flavin-dependent monooxygenase TetX. Biochemistry 2005, 44, 11829–11835. [Google Scholar] [CrossRef] [PubMed]
- Olson, M.W.; Ruzin, A.; Feyfant, E.; Rush, T.S., 3rd; O’Connell, J.; Bradford, P.A. Functional, biophysical, and structural bases for antibacterial activity of tigecycline. Antimicrob. Agents Chemother. 2006, 50, 2156–2166. [Google Scholar] [CrossRef] [PubMed]
- Berens, C. Interactions of tetracyclines with RNA. In Tetracyclines in Biology, Chemistry and Medicine; Nelson, M., Hillen, W., Greenwald, R.A., Eds.; Birkhäuser: Basel, Switzerland, 2002; pp. 177–196. [Google Scholar]
- Noel, G.J.; Draper, M.P.; Hait, H.; Tanaka, S.K.; Arbeit, R.D. A randomized, evaluator-blind, phase 2 study comparing the safety and efficacy of omadacycline to those of linezolid for treatment of complicated skin and skin structure infections. Antimicrob. Agents Chemother. 2012, 56, 5650–5654. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.I.; Eady, E.A.; Cove, J.H.; Cunliffe, W.J. 16S rRNA mutation associated with tetracycline resistance in a Gram-positive bacterium. Antimicrob. Agents Chemother. 1998, 42, 1702–1705. [Google Scholar] [PubMed]
- Nonaka, L.; Connell, S.R.; Taylor, D.E. 16S rRNA mutations that confer tetracycline resistance in Helicobacter pylori decrease drug binding in Escherichia coli ribosomes. J. Bacteriol. 2005, 187, 3708–3712. [Google Scholar] [CrossRef] [PubMed]
- Pringle, M.; Fellström, C.; Johansson, K.-E. Decreased susceptibility to doxycycline associated with a 16S rRNA gene mutation in Brachyspira hyodysenteriae. Vet. Microbiol. 2007, 123, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Dégrange, S.; Renaudin, H.; Charron, A.; Pereyre, S.; Bébéar, C.; Bébéar, C.M. Reduced susceptibility to tetracyclines is associated in vitro with the presence of 16S rRNA mutations in Mycoplasma hominis and Mycoplasma pneumoniae. J. Antimicrob. Chemother. 2008, 61, 1390–1392. [Google Scholar] [CrossRef] [PubMed]
- Amram, E.; Mikula, I.; Schnee, C.; Ayling, R.D.; Nicholas, R.A.; Rosales, R.S.; Harrus, S.; Lysnyansky, I. 16S rRNA gene mutations associated with decreased susceptibility to tetracycline in Mycoplasma bovis. Antimicrob. Agents Chemother. 2015, 59, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Moazed, D.; Noller, H.F. Interaction of antibiotics with functional sites in 16S ribosomal RNA. Nature 1987, 327, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Asai, T.; Zaporojets, D.; Squires, C.; Squires, C.L. An Escherichia coli strain with all chromosomal rRNA operons inactivated: Complete exchange of rRNA genes between bacteria. Proc. Natl. Acad. Sci. USA 1999, 96, 1971–1976. [Google Scholar] [CrossRef] [PubMed]
- Brosius, J.; Ullrich, A.; Raker, M.A.; Gray, A.; Dull, T.J.; Gutell, R.R.; Noller, H.F. Construction and fine mapping of recombinant plasmids containing the rrnB ribosomal RNA operon of E. coli. Plasmid 1981, 6, 112–118. [Google Scholar] [CrossRef]
- Palm, G.J.; Lederer, T.; Orth, P.; Saenger, W.; Takahashi, M.; Hillen, W.; Hinrichs, W. Specific binding of divalent metal ions to tetracycline and to the Tet repressor/tetracycline complex. J. Biol. Inorg. Chem. 2008, 13, 1097–1110. [Google Scholar] [CrossRef] [PubMed]
- Ettner, N.; Metzger, J.W.; Lederer, T.; Hulmes, J.D.; Kisker, C.; Hinrichs, W.; Ellestad, G.A.; Hillen, W. Proximity mapping of the Tet repressor-tetracycline-Fe2+ complex by hydrogen peroxide mediated protein cleavage. Biochemistry 1995, 34, 22–31. [Google Scholar] [CrossRef] [PubMed]
- McMurry, L.M.; Aldema-Ramos, M.L.; Levy, S.B. Fe2+-tetracycline-mediated cleavage of the Tn10 tetracycline efflux protein TetA reveals a substrate binding site near glutamine 225 in transmembrane helix 7. J. Bacteriol. 2002, 184, 5113–5120. [Google Scholar] [CrossRef] [PubMed]
- Heidrich, C.G.; Berens, C. Probing RNA structure and ligand binding sites on RNA by Fenton cleavage. In Handbook of RNA Biochemistry, 2nd ed.; Westhof, E., Bindereif, A., Schön, A., Hartmann, R.K., Eds.; WILEY-VCH: Weinheim, Germany, 2014; pp. 301–318. [Google Scholar]
- Hertweck, M.; Hiller, R.; Mueller, M.W. Inhibition of nuclear pre-mRNA splicing by antibiotics in vitro. Eur. J. Biochem. 2002, 269, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Oehler, R.; Polacek, N.; Steiner, G.; Barta, A. Interaction of tetracycline with RNA: Photoincorporation into ribosomal RNA of Escherichia coli. Nucleic Acids Res. 1997, 25, 1219–1224. [Google Scholar] [CrossRef] [PubMed]
- Moazed, D.; Noller, H.F. Transfer RNA shields specific nucleotides in 16S ribosomal RNA from attack by chemical probes. Cell 1986, 47, 985–994. [Google Scholar] [CrossRef]
- Noah, J.W.; Dolan, M.A.; Babin, P.; Wollenzien, P. Effects of tetracycline and spectinomycin on the tertiary structure of ribosomal RNA in the Escherichia coli 30S ribosomal subunit. J. Biol. Chem. 1999, 274, 16576–16581. [Google Scholar] [CrossRef] [PubMed]
- Powers, T.; Changchien, L.-M.; Craven, G.R.; Noller, H.F. Probing the assembly of the 3’ major domain of 16 S ribosomal RNA. Quaternary interactions involving ribosomal proteins S7, S9 and S19. J. Mol. Biol. 1988, 200, 309–319. [Google Scholar] [CrossRef]
- Trieber, C.A.; Taylor, D.E. Mutations in the 16S rRNA genes of Helicobacter pylori mediate resistance to tetracycline. J. Bacteriol. 2002, 184, 2131–2140. [Google Scholar] [CrossRef] [PubMed]
- Tuckman, M.; Petersen, P.J.; Projan, S.J. Mutations in the interdomain loop region of the tetA(A) tetracycline resistance gene increase efflux of minocycline and glycylcyclines. Microb. Drug Resist. 2000, 6, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Peleg, A.Y.; Adams, J.; Paterson, D.L. Tigecycline efflux as a mechanism for nonsusceptibility in Acinetobacter baumannii. Antimicrob. Agents Chemother. 2007, 51, 2065–2069. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, T.; Presedo, J.; Khan, A.A. The tetA gene decreases tigecycline sensitivity of Salmonella enterica isolates. Int. J. Antimicrob. Agents 2013, 42, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Deng, M.; Zhu, M.-H.; Li, J.-J.; Bi, S.; Sheng, Z.-K.; Hu, F.-S.; Zhang, J.-J.; Chen, W.; Xue, X.-W.; Sheng, J.-F.; et al. Molecular epidemiology and mechanisms of tigecycline resistance in clinical isolates of Acinetobacter baumannii from a chinese university hospital. Antimicrob. Agents Chemother. 2014, 58, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Villa, L.; Feudi, C.; Fortini, D.; García-Fernández, A.; Carattoli, A. Genomics of KPC-producing Klebsiella pneumoniae sequence type 512 clone highlights the role of RamR and ribosomal S10 protein mutations in conferring tigecycline resistance. Antimicrob. Agents Chemother. 2014, 58, 1707–1712. [Google Scholar] [CrossRef] [PubMed]
- Lambs, L.; Venturini, M.; Decock-Le Révérend, B.; Kozlowski, H.; Berthon, G. Metal ion-tetracycline interactions in biological fluids. Part 8. Potentiometric and spectroscopic studies on the formation of Ca(II) and Mg(II) complexes with 4-dedimethylamino-tetracycline and 6-desoxy-6-demethyl-tetracycline. J. Inorg. Biochem. 1988, 33, 193–210. [Google Scholar] [CrossRef]
- Gutell, R.R. Collection of small subunit (16S- and 16S-like) ribosomal RNA structures: 1994. Nucleic Acids Res. 1994, 22, 3502–3507. [Google Scholar] [CrossRef] [PubMed]
- Anokhina, M.M.; Barta, A.; Nierhaus, K.H.; Spiridonova, V.A.; Kopylov, A.M. Mapping of the second tetracycline binding site on the ribosomal small subunit of E. coli. Nucleic Acids Res. 2004, 32, 2594–2597. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. DH5α competent cells. Focus 1986, 8, 9. [Google Scholar]
- Deutscher, M.P.; Marlor, C.W.; Zaniewski, R. Ribonuclease T: New exoribonuclease possibly involved in end-turnover of tRNA. Proc. Natl. Acad. Sci. USA 1984, 81, 4290–4293. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.F.; Zuo, Y.; Li, Z.; Rudd, K.E.; Deutscher, M.P. The vacb gene required for virulence in Shigella flexneri and Escherichia coli encodes the exoribonuclease RNase R. J. Biol. Chem. 1998, 273, 14077–14080. [Google Scholar] [CrossRef] [PubMed]
- Brodersen, D.E.; Clemons, W.M., Jr.; Carter, A.P.; Wimberly, B.T.; Ramakrishnan, V. Crystal structure of the 30S ribosomal subunit from Thermus thermophilus: Structure of the proteins and their interactions with 16S RNA. J. Mol. Biol. 2002, 316, 725–768. [Google Scholar] [CrossRef] [PubMed]
- Blaha, G.; Stelzl, U.; Spahn, C.M.; Agrawal, R.K.; Frank, J.; Nierhaus, K.-H. Preparation of functional ribosomal complexes and effect of buffer conditions on tRNA positions observed by cryoelectron microscopy. Methods Enzymol. 2000, 317, 292–309. [Google Scholar] [PubMed]
- Berens, C.; Streicher, B.; Schroeder, R.; Hillen, W. Visualizing metal-ion-binding sites in group I introns by iron(II)-mediated Fenton reactions. Chem. Biol. 1998, 5, 163–175. [Google Scholar] [CrossRef]
- Moazed, D.; Stern, S.; Noller, H.F. Rapid chemical probing of conformation in 16S ribosomal RNA and 30S ribosomal subunits using primer extension. J. Mol. Biol. 1986, 187, 399–416. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Test Compound | MIC Parameter | Value (µg/mL) against Each Bacterial Strain | |||
|---|---|---|---|---|---|
| ATCC-25922 | TA527 (wt) | TA527/1058C | TA527/966U | ||
| Tetracycline | MIC (fold increase) | 1 | 2 | 8 (4×) | 16 (8×) |
| Tigecycline | MIC (fold increase) | 0.06 | 0.25 | 1 (4×) | 1 (4×) |
| Omadacycline | MIC (fold increase) | nd | 2 | 8 (4×) | 16 (8×) |
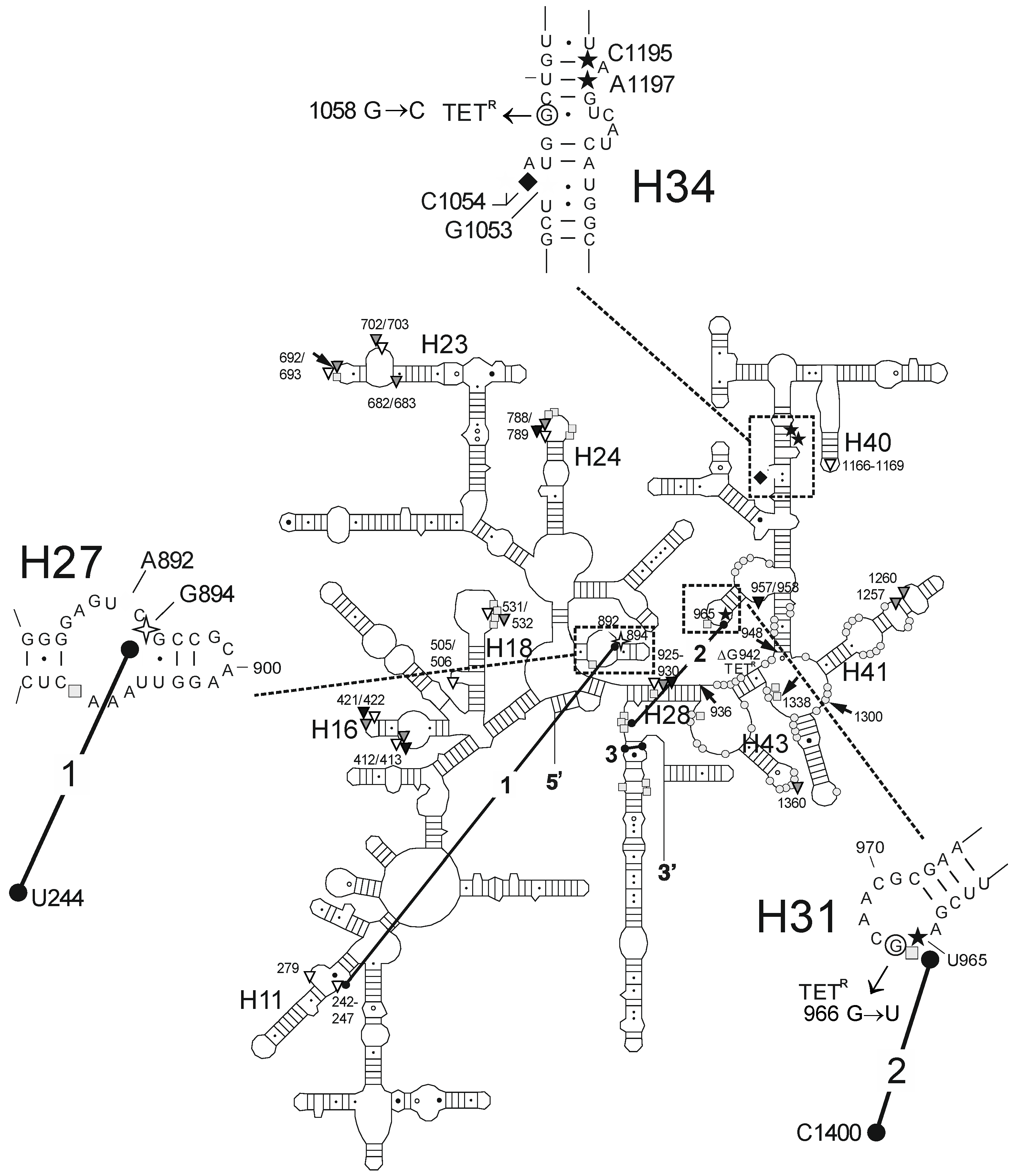
| Fe2+ Cleavage Sites a | Specific/Non-Specific b | TET-/TGC-Site c | Biochemical and Genetic Data a |
|---|---|---|---|
| G242-G247 (h11) | ns (T) | site-2/site-5 | protection against methylation by DMS at A892 (TET) [25] and A909 (tRNA) [34]; TET-inhibited crosslink U244 × G894 [35] |
| A279 (h11) | ns (T) | site-2/site-5 | protection against methylation by DMS at A892 (TET) [25] and A909 (tRNA) [34]; TET-inhibited crosslink U244 × G894 [35] |
| A412/G413 (h16) | ns (TGO) | ||
| U421/C422 (h16) | ns (TGO) | ||
| G505/G506 (h18) | ns (TG) | ||
| U531/A532 | ns (TG) | protection against methylation by DMS at G529-G532 (tRNA) [34] | |
| G682/G683 (h23) | ns (T) | protection against methylation by DMS at G693 (tRNA) [34] | |
| U692/G693 (h23) | ns (TG) | protection against methylation by DMS at G693 (tRNA) [34] | |
| A702/G703 (h23) | ns (TG) | protection against methylation by DMS at G693 (tRNA) [34] | |
| U788/U789 (h24) | ns (TGO) | protection against methylation by DMS at A790/G791/A794/C795 (tRNA) [34] | |
| G894 (h27) | s (T) | site-2/site-5 | protection against methylation by DMS at A892 (TET) [25] and A909 (tRNA) [34]; TET-inhibited crosslink U244 × G894 [35] |
| G925-C930 (h28) | ns (TGO) | protection against methylation by DMS at G928 (tRNA) [34] | |
| U957/A958 (h30/h31) | ns (O) | enhanced methylation by DMS at G954 and A977-C980, protection against methylation by DMS at A983 (S7) [36] | |
| U965 (h31) | s (TGO) | site-1 | inhibition of crosslink A967 × C1400 [35]; A965U/G966U/A967C mutation: TET resistance in H. pylori [37] |
| G1053/C1054 (h34) | s (TGO) | site-1 | enhanced methylation by DMS at C1054 (TET, TGC) [15,25]; G1058C mutation: TET resistance in P. acnes [20] and B. hyodysenteriae [22] |
| G1166-A1169 (h40) | ns (T) | site-3 | |
| C1195/A1197 (h34) | s (TGO) | site-1 | enhanced methylation by DMS at C1054 (TET, TGC) [15,25]; G1058C mutation: TET resistance in P. acnes [20] and B. hyodysenteriae [22] |
| A1257 (h41) | ns (G) | protection against methylation by DMS at A1256 (S7) [36] | |
| G1260 (h41) | ns (G) | enhanced methylation by DMS at A1261 (S7 + S9) [36] | |
| A1360 (h43) | ns (G) | protection against methylation by DMS at A1360 (S7) [36] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heidrich, C.G.; Mitova, S.; Schedlbauer, A.; Connell, S.R.; Fucini, P.; Steenbergen, J.N.; Berens, C. The Novel Aminomethylcycline Omadacycline Has High Specificity for the Primary Tetracycline-Binding Site on the Bacterial Ribosome. Antibiotics 2016, 5, 32. https://doi.org/10.3390/antibiotics5040032
Heidrich CG, Mitova S, Schedlbauer A, Connell SR, Fucini P, Steenbergen JN, Berens C. The Novel Aminomethylcycline Omadacycline Has High Specificity for the Primary Tetracycline-Binding Site on the Bacterial Ribosome. Antibiotics. 2016; 5(4):32. https://doi.org/10.3390/antibiotics5040032
Chicago/Turabian StyleHeidrich, Corina G., Sanya Mitova, Andreas Schedlbauer, Sean R. Connell, Paola Fucini, Judith N. Steenbergen, and Christian Berens. 2016. "The Novel Aminomethylcycline Omadacycline Has High Specificity for the Primary Tetracycline-Binding Site on the Bacterial Ribosome" Antibiotics 5, no. 4: 32. https://doi.org/10.3390/antibiotics5040032