
Nitrogen Trapping Ability of Hydrogen-Induced Vacancy and the Effect on the Formation of AlN in Aluminum


1
Division of Computational Physics, Institute for Computational Science, Ton Duc Thang University, Ho Chi Minh City 700000, Vietnam
2
Faculty of Electrical & Electronics Engineering, Ton Duc Thang University, Ho Chi Minh City 700000, Vietnam
3
The Center of Nanostructured Materials and Nanotechnologies, Belgorod State University, Belgorod 308034, Russia
4
Division of Computational Mathematics and Engineering, Institute for Computational Science, Ton Duc Thang University, Ho Chi Minh City 700000, Vietnam
5
Faculty of Civil Engineering, Ton Duc Thang University, Ho Chi Minh City 700000, Vietnam
*
Author to whom correspondence should be addressed.
Coatings 2017, 7(6), 79; https://doi.org/10.3390/coatings7060079
Submission received: 24 April 2017
/
Revised: 6 June 2017
/
Accepted: 6 June 2017
/
Published: 9 June 2017
(This article belongs to the Special Issue Modelling and Simulation of Coating)
Abstract
:This paper presents the ternary interaction of N, H, and vacancy point defects and the nitrogen trapping ability of aluminum vacancies induced by hydrogen by means of DFT methods employed in VASP (Vienna Ab initio Simulation Package) and Abinit packages. The obtained vacancy formation energy of 0.65 eV is close to experimental values. Although the N–vacancy complex is unstable with the negative binding energy of −0.51 eV, the stability of H–vacancy–N is proved by the positive binding energy of 0.59 eV and the appearance of the orbital hybridization in the density of state (DOS) of atoms connecting to this complex. Moreover, Al vacancies can trap more than 4 N atoms, which prevents the formation of aluminum nitride and subsequently affects not only the hardness of the Al surface but also many practical applications of AlN coating.
1. Introduction
Aluminum and its alloys are widely used in life and technique. Especially, the aluminum coated with an AlN layer can be used to improve LED performance [1,2], or it can be used as electronic packaging material [3,4]. In the optical industry, AlN can be used as protective coatings [5,6]. At the same time, the formation of AlN is often affected by point defects such as vacancies and light impurities. There have been many studies on the effect of separate impurities on the vacancy formation and nucleation in Al [7,8,9]. In the experimental studies of Chung et al. [10], they found that the hardness of the Al sample seems to hardly increase during nitrogen implantation at the fluence of 2 × 1018 ions/cm2. Ghoranneviss et al. [11] suggested that over-implantation of nitrogen causes excessive defects and vacancies. In other words, there should be sort of interactions between vacancy and nitrogen. However, theoretical study shows that the possibility of a vacancy bound to a nitrogen impurity was ruled out because of the repulsive interaction between aluminum vacancies and nitrogen [12]. In a previous study, we showed that an Al vacancy induced by H impurities can interact with N [13]. This indicates that Al–nitride may not be formed because of the forming of the H–N–vacancy complex. However, the stability of H–N–vacancy complex was not estimated, and the vacancy formation energy of 0.55 eV in [13] was far lower than the experimental value, which ranged from 0.63 to 0.67 eV [14,15]. The reason comes from the fact that, for the Al system, the vacancy formation enthalpy decreases when the temperature is lower [16]. It is shown in previous DFT studies that a mono-vacancy in the fcc lattice and in the hpc lattice can trap four H atoms [17]. Besides a temperature effect, the ability of the vacancy–H complex to capture an amount of N atoms is also necessary for studying the H–N–vacancy interaction. In this paper, we study the capability of H-induced vacancy in Al to trap nitrogen atoms using a combination of DFT methods in VASP and Abinit packages. We also perform the calculation for the same values obtained by applying the Abinit package in the previous study [13] to estimate the effect of electronic temperature on the accuracy of the theoretical results.
2. Materials and Methods
Both Abinit and VASP implement density functional theory, using plane-wave basic set and support projected augmented wave (PAW) [18]. The atoms were described by using PAW potentials constructed within the generalized gradient approximation (GGA) in the Perdew–Burke–Ernzerhof (PBE) form [19]. The VASP [20] package is employed, using the direct optimization methods and the conjugate-gradient algorithm [21] to study the models of 2 × 2 × 2 fcc Al. The convergence of the total energy regarding the cut off energy Ecut and the k-point meshes was tested. It is observed that the cut-off energy of 400 eV and the k-point mesh of (4 × 4 × 4) give the convergence in total energy with 1 meV/atom difference. The geometry optimizations are performed until the net force on each atom meets a criterion of 5 meV/A. The obtained lattice constant of 4.1 Å is close to the experimental value of 4.05 Å [22].
In the case of impurity–vacancy interaction, VASP requires a good predicted configuration, so we firstly performed relaxation with Abinit package with the following steps:
The hydrogen atom is placed at the center of the vacancy space, then the Nose-Hoover [23] molecular dynamics was performed with a temperature of 300 K to activate atoms out of or across local minima; then, atomic movements were cooled down by setting the temperature to 100 K. The obtained configuration with a good beginning position of the hydrogen atom in the vacancy space was optimized using the Verlet algorithm [24] along with BFGS relaxation. In the optimization process, firstly the positions of ions are relaxed without changing the volume and unit cell parameters, and the full relaxation of both position and the volume is then performed (optcell = 2).
The obtained configuration containing hydrogen and vacancy point defects is used to add first the nitrogen atom into the center of the vacancy space. The relaxation and optimization processes are performed in the same way hydrogen is added into the vacancy space.
The obtained configuration containing hydrogen, a vacancy, and nitrogen point defects is used to add the second nitrogen atom into the center of the vacancy space. Then, the relaxation and optimization processes are performed in the same way as described above.
We continue the same process until the 8th nitrogen atom is added into the vacancy space.
All the obtained configurations, which are calculated by Abinit, are used as beginning configurations for VASP calculation. In the VASP calculation, we use the option IBRION = 2 with ISIF = 3 to optimize the ion position, the volume, and the cell-shape at the same time.
3. Results
The preferable position of impurity atoms in fcc Al lattice is defined by comparing the solution energy of each atom at octahedral and tetrahedral interstitial sites. The solution energy of impurities in the fcc Al can be calculated by the following formula:
ESX = E[XT/O + Al32] − E[Al32] − 1/2 × E[X2]
In Equation (1), ESX is the solution energy of the impurity atom X (X = H or N) in bulk Al. E[XT/O + Al32] is the total energy of the fcc Al super-cell consisting of 32 Al atoms at lattice sites and one atom X at the tetrahedral (T) or at the octahedral (O) interstitial site, respectively. E[Al32] is the total energy of fcc Al super-cell of 32 Al atoms at lattice sites. E[X2] is the total energy of H2 or N2 in vacuum.
We found that H and N atoms prefer to stay at the tetrahedral sites with the energy difference of about 0.1 eV. Hydrogen solution energies are 0.69 eV for Abinit and 0.70 eV for VASP. These values are close to the previous experimental and theoretical results [25,26,27,28] ranging from 0.65 to 0.71 eV.
3.1. The Formation of H–N Bond Near Al Vacancy
The stability of a configuration is firstly estimated by the binding energy calculated by Equations (1) and (2):
ECX–vacancy = E[XT + Al32] + E[vacancy + Al31] − E[Al32] − E[X–vacancy + Al31]
ECN–(H–vacancy) = E[NT + Al32] + E[H–vacancy + Al31] − E[Al32] − E[N–H–vacancy + Al31]
In Equations (2) and (3), ECX–vacancy is the binding of atom X (X = H, or N) with vacancy ; ECN–(H–vacancy) is the binding energy of nitrogen with the H–vacancy complex; E[Al32] is the total energy of pure Al super-cell of 32 Al atoms; E[XT + Al32] is the total energy of Al super-cell containing an impurity atom at tetrahedral interstitial site; E[vacancy + Al31] is the total energy of the Al super-cell with a lattice vacancy; E[X–vacancy + Al31] is the total energy of the Al super-cell with one lattice vacancy and one nearby impurity atom; E[N–H–vacancy + Al31] is the total energy of the Al super-cell with one lattice vacancy and two neighboring impurity atoms, which are close to the vacant position. The binding energies calculated by Abinit [7] and by VASP are presented in Table 1.
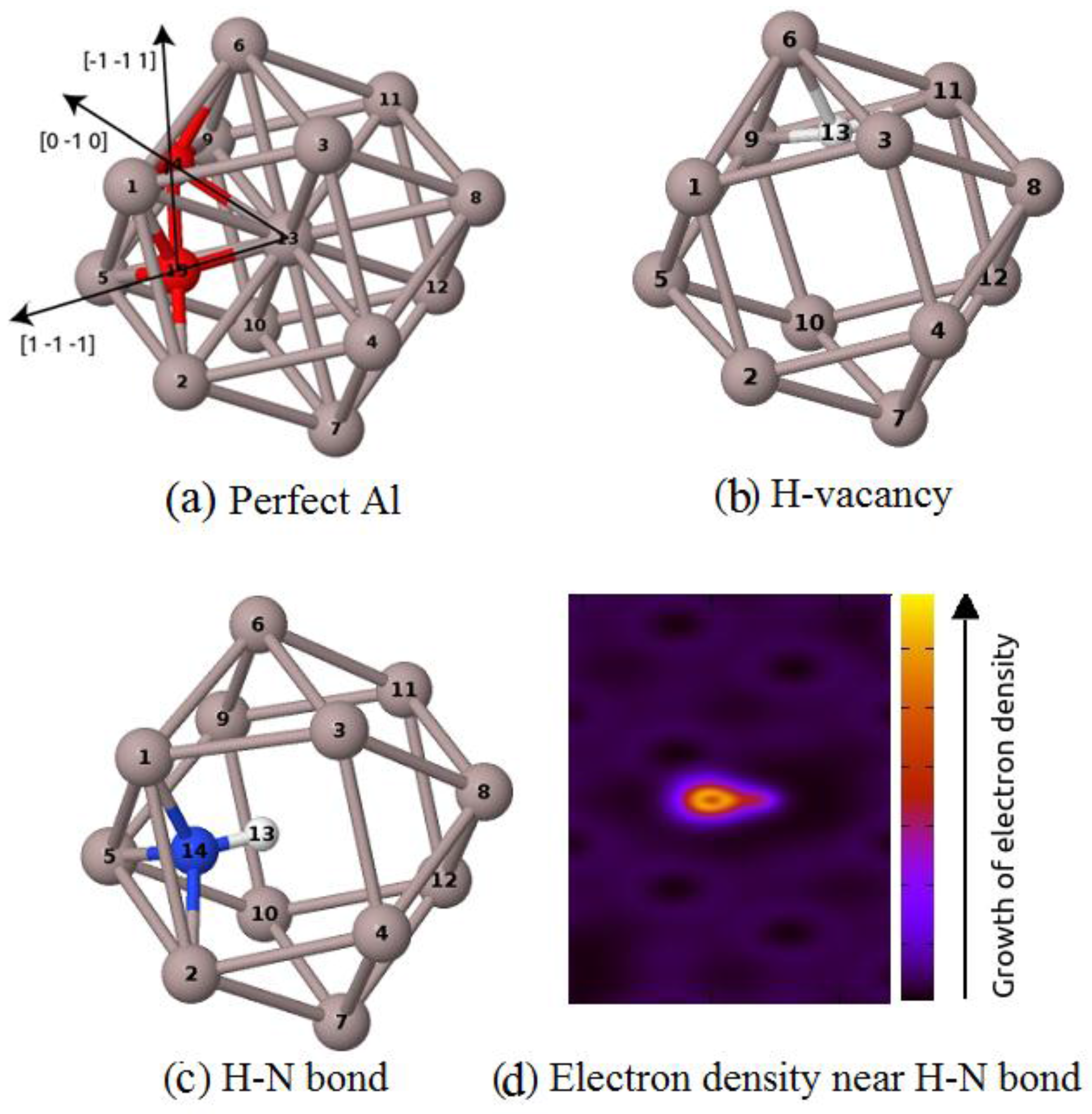
The occupation near the vacancy of H at the tetrahedral-like position (Figure 1a) was supported by its positive binding energies. In this case, VASP gives a higher H–vacancy binding energy than Abinit does. This value is higher than the theoretical results reported earlier (~0.33 eV and 0.39 eV) [29,13]. However, this value is closer to the experimental value, which is in the range of 0.43–0.53 eV [30,31,32,33]. The negative binding energy between the N impurity atom and the vacancy predicts a repulsive interaction between these point defects, which is in good agreement with theoretical prediction [6] and experimental measurement of Hogg et al. [12]. In the presence of H, the interaction between the N atom and the lattice vacancy becomes possible with the positive binding of 0.27 eV.
Figure 1 shows the starting position of impurities at tetrahedral and octahedral sites (Figure 1a). After relaxation and optimization processes for the case of Al containing both vacancies and impurities, we obtain an equilibrium configuration in which hydrogen locates at the tetrahedral-like site inside the vacancy (Figure 1b). In another equilibrium configuration, N locates at the center of a tetrahedral-like site formed by three Al atoms and one hydrogen atom (Figure 1c), the obtained N–H bond-length of 1.04 Å is close to the N–H bond in NH3, which is about 1.01 Å [34]. The increase in valence electron density along the line between the two impurities (Figure 1d) shows the covalent properties of the N–H bond.
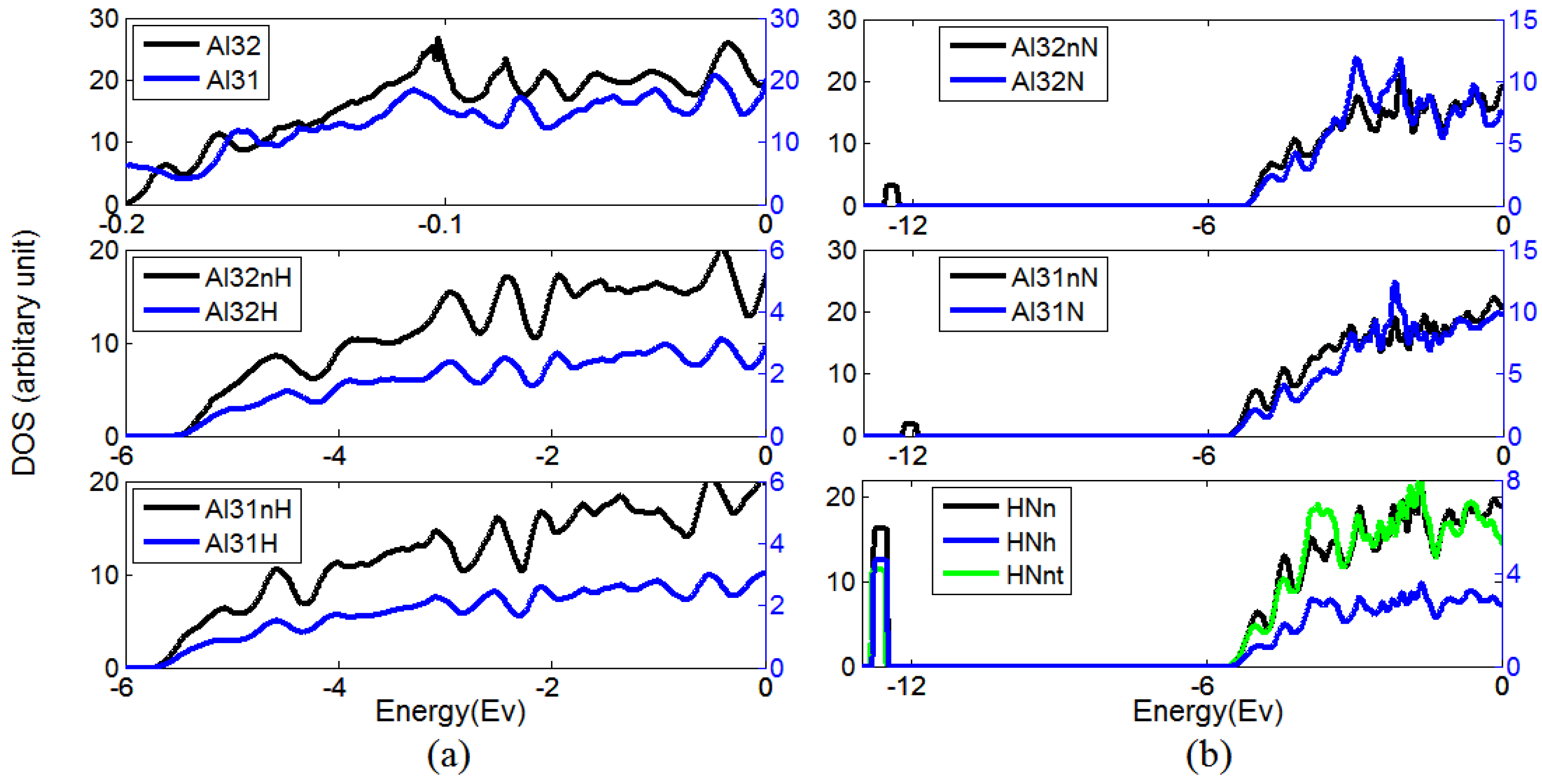
The changing in the density of state (DOS) of Al, H, and N in different configurations is shown in Figure 2, wherein the following symbols are used: Al31—DOS of Al near a vacancy; Al32—DOS of Al in pure Al; Al32H—DOS of interstitial H; Al32nH—DOS of Al near interstitial H; Al31H—DOS of H near a vacancy; Al31nH—DOS of Al near a vacancy and H in Figure 2a, Al32N—DOS of interstitial N; Al32nN—DOS of Al near interstitial N; Al31N—DOS of N near a vacancy; Al31nN—DOS of Al near N and vacancy; HNh—DOS of H near N and vacancy; HNn—DOS of Al near H, N, and vacancy; HNnt—DOS of N near H and vacancy in Figure 2b.
The DOS of H is much lower than the DOS of Al or N, so it is plotted by the right y-axis, which has a smaller scale than the left y-axis.
Figure 2a shows an insignificant difference between the DOS of Al in pure Al (Al32) and the DOS of Al near a vacancy (Al31). We notice that below Fermi level, the intensity of DOS of Al near hydrogen (Al32nH) is lower in comparison with Al32. The DOS of Al near both vacancy and hydrogen (Al31nH) is also lower than Al31. In both cases, there is a donation of electrons from Al to H, so the H–Al bonds in both pure Al and Al with vacancies are strong, and the vacancy–H complex is stable. Figure 2b shows that, despite the fact that N–vacancy complex is not stable given by our earlier calculated N–vacancy binding energy [13], the DOS of N near a vacancy (Al31N) is presented herein for comparison purposes. When N is located at the interstitial position or at the position near a vacancy, we observe in both cases that the appearance of a new state in DOS of Al near N arises from the doping of N. In the case of H bonding to N near a vacancy, a new occupation state occurs in the DOS representing the electron occupation of both N and H atoms. More specifically, it implies the hybridization state where N and H establish a polar covalent bond. In addition, this peak becomes higher for the DOS of Al near the point defects, indicating strong electronic interactions between H, N, and Al atoms.
3.2. The Ability of Vacancies to Trap Nitrogen Atoms
The hybridization state occurring in the DOS of N, H, and Al near the H–N–vacancy complex shows the high stability of this complex. Moreover, the binding energies listed in Table 1 show that H changes the N–vacancy interaction from repulsive (−0.51 eV) to attractive (0.59 eV). It is obvious that the H–vacancy–N complex is energetically favorable. Therefore, it is important to estimate the possibility of vacancy formation under the effect of H and N impurities, as well as the number of N atoms that H-induced vacancy can trap.
The formation energy of vacancy induced by H and N impurities is estimated by the following formula:
where E[vacancy + Al31] and E[Al32] are the total energies of the system containing vacancy and the perfect Al bulk, respectively. Based on Equation (4), we can calculate Evacancyf[H], i.e., the formation energy of vacancy, which is induced by the interstitial H impurities using the following formula:
or
Evacancyf = E[vacancy + Al31] − (31/32)E[Al32]
Evacancyf[H] = E[vacancy + H + Al31] + (1/32)E[Al32] − E[HT + Al32]
Evacancyf[H] = Evacancyf − ECH–vacancy
Since the hydrogen atom is proven to play an indispensable role in the interaction between N impurities and vacancies, we can finally calculate Evacancyf[N + H], the formation energy of vacancy, which is induced by both H and N impurities by the following formula:
Evacancyf[N + H] = Evacancyf[H] − ECN–(H–vacancy)
The formation energies calculated by formulas (5)–(7) are given in Table 2.
The vacancy formation energy calculated by Abinit is about 15% different from the experimental values (0.63–0.67 eV) [16,17], while the VASP gives a difference of about 1.5%. The presence of an H impurity leads to a decrease in vacancy formation from 0.64 to 0.15 eV. It is important to notice that, under the interaction of H and N, vacancies can be formed automatically with a negative formation energy of −0.12 eV, which results in a higher concentration of vacancies inside bulk Al.
The number of N atoms an H-induced vacancy is able to trap is estimated based on the formation energy of a vacancy–H–mN complex:
where m ranges from 1 to 8 because there are 8 equivalent tetrahedral-like positions inside the area of the vacancy. In principle, the vacancy containing (m−1)N atoms can trap one more N if the formation of vacancy–H–mN complex is energetically more favorable than the formation of both interstitial N and vacancy–H–(m−1)N complex. We name the difference in energy in this case as the trapping energy Etrap(m):
Efvacancy_H+mN = E[vacancy–H + mN] + mE[Al32] − mE[Al32 + NT] − E[vacancy–H]
Etrap(m) = Efvacancy−H+mN − Efvacancy−H+(m−1)N − Es[N]
The energetic and geometric characters of the vacancy–H complex containing nitrogen atoms are listed in Table 3.
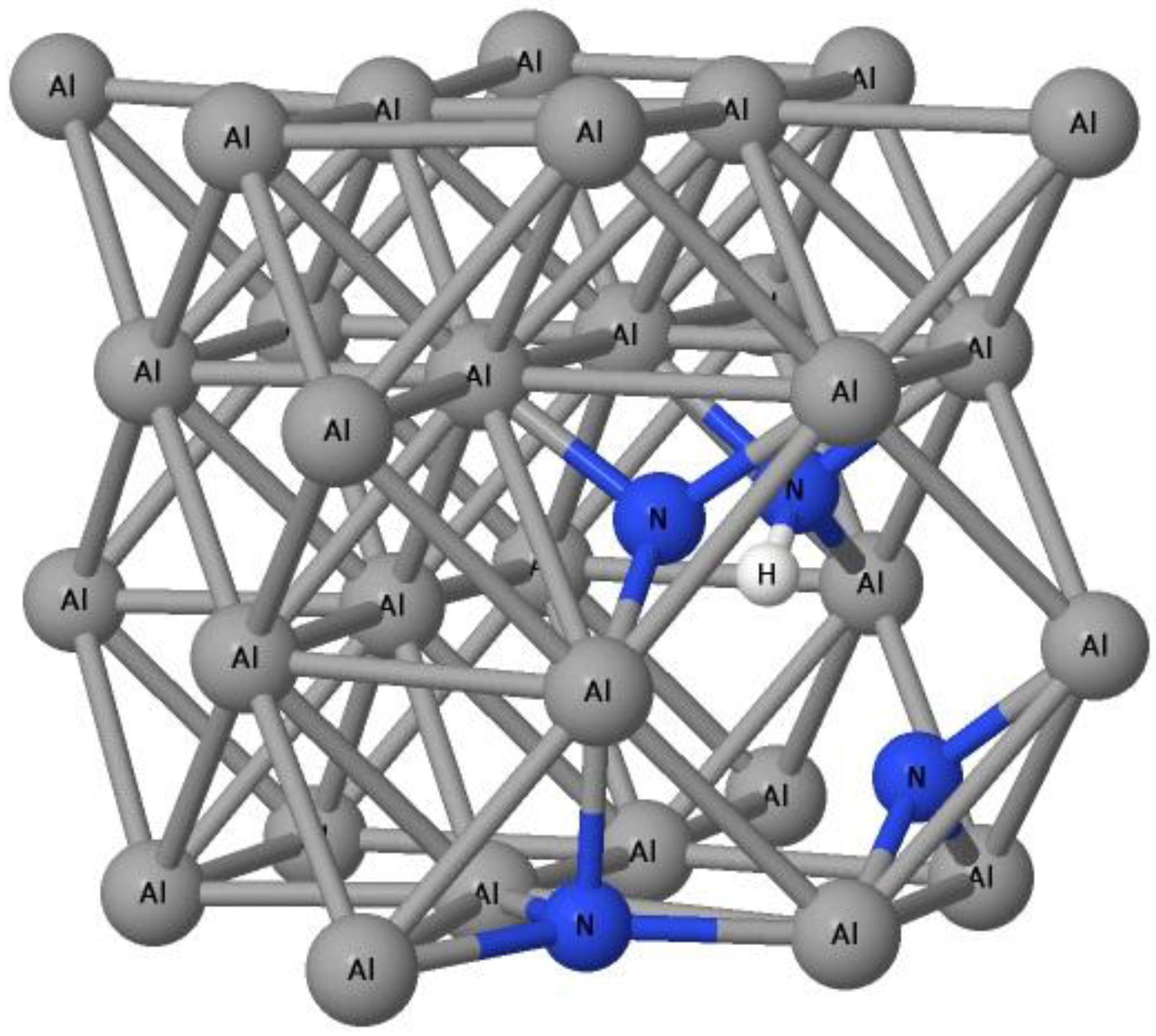
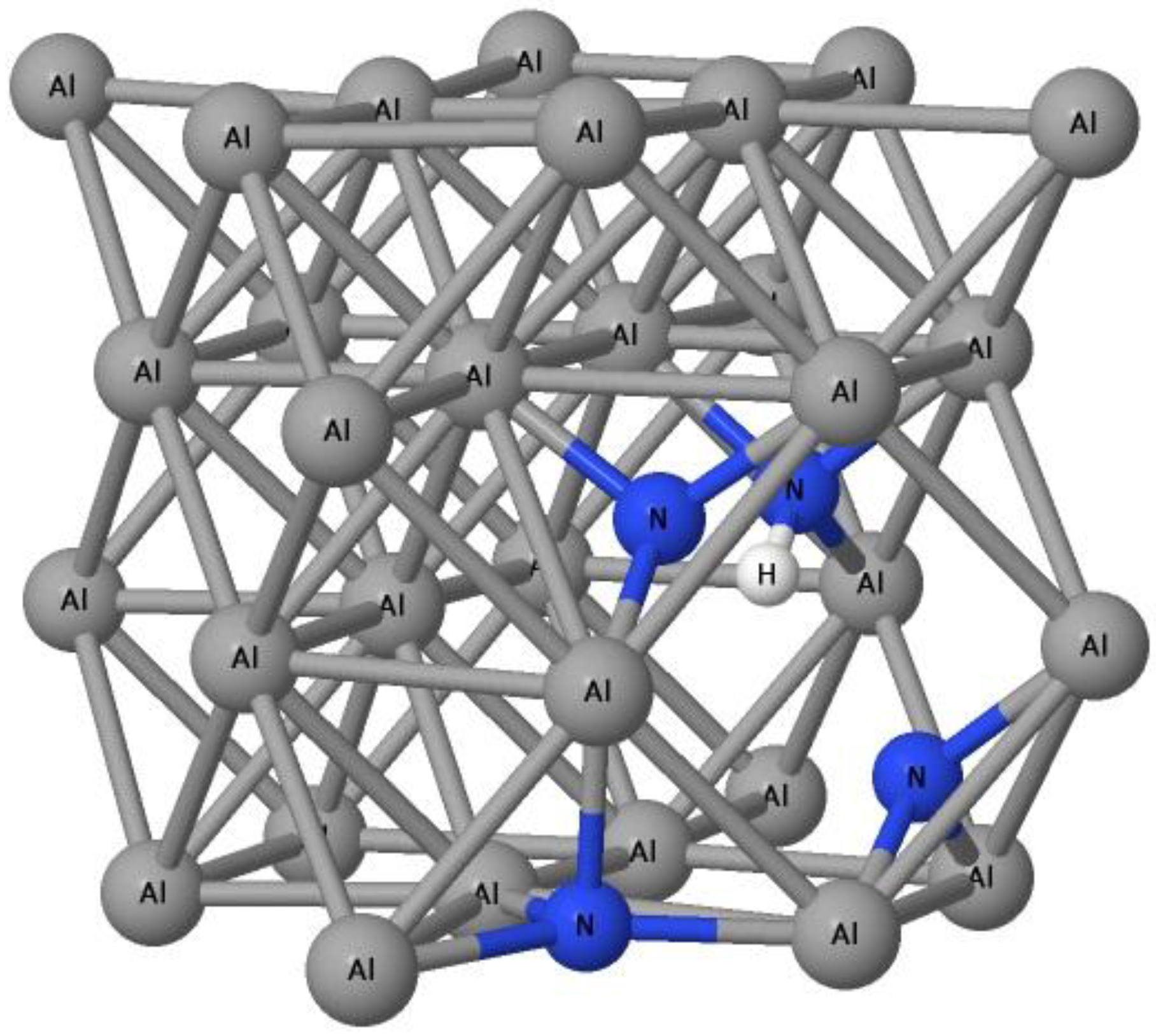
It is obvious that an H-induced vacancy can trap up to 4 N atoms with the Efvacancy−H+4N of −0.49 eV and the trapping energy Etrap(4) of −0.01 eV. Trapping a 5th N atom is not energetically favorable. It can be seen in Figure 3 that the 4th N atom is slightly out of the tetrahedral-like site of the vacancy area.
The bond length between neighboring Al atoms connecting to the 4th N atom is expanded by about 0.2%. Adding a 5th N atom may request more energy to reform the local structure of Al lattice. However, it is interesting that an H–vacancy complex containing 6 or 7 N atoms again has negative formation and trapping energies. We suggest that, if the N atoms receive energy enough to overcome the energy barrier, they will form, with H and a vacancy, a complex that can trap two more N atoms. The H-induced vacancy complex can trap an 8th N atom because the formation energy of H–vacancy–8N is negative, but the trapping energy with a positive energy of 1.80 eV is again the energy barrier for an N atom to dissolve into Al.
Trapping each nitrogen atom results in increasing the volume by 6–7 cubic Angstrom, and the cell-shape distortion is small (0.02–0.11 Angstrom). However, trapping 6 nitrogren atoms or more leads to high volume changing (4.92–16.37 cubic Angstrom) and serious distortion (0.30–0.80 Angstrom). The volume changes a little when H–vacancy complex traps the 5th nitrogen atom (1.43 cubic Angstrom), but the distortion is also high (0.38 Angstrom). From the obtained values in Table 3, we can say that at least 4 N atoms can be trapped by an H-induced vacancy.
4. Discussion and Conclusions
The DFT calculation of Abinit and VASP packages was performed to study the effect of H and N impurities on the properties of fcc Al. The VASP gives good agreement with experimental data for the case of vacancy formation energy and of the binding energy of the H–vacancy complex. Although there are differences in the values given by Abinit and VASP packages, both packages describe the same manner of H, N, and Al vacancy interaction:
- N–vacancy interaction is repulsive;
- hydrogen reduces the vacancy formation energy and makes the N–vacancy interaction attractive.
Going further than these results, we explain the stability of vacancy–H–N complex based on the DOS of atoms involving the interaction. This is the basis for studying the capability of H-induced vacancy that trap N atoms. We find that at least such vacancies can trap up to 4 N atoms. On the other hand, the presence of H and N impurities lowers the formation energy of the vacancy, which may lead to increases in vacancy concentration. This means that more and more N atoms can be trapped inside vacancy-based complexes that prevent the formation of Al nitride, which consequently affects the hardness of Al as well as the application of AlN coating in many areas of technology.
We also notice that the ternary interaction of point defects, or any interaction including more kinds of point defects, is very important for the properties of a material.
Acknowledgments
The authors thank the high-performance computing support from Belgorod State University. Aleksey G. Lipnitskii is grateful for the support from the Ministry of Education and Science of the Russian Federation under contract No. 3.1282.2014/K.
Author Contributions
Aleksey G. Lipnitskii and Nguyen Thoi Trung conceived and designed the experiments; Vo Duy Dat performed the experiments; Vo Duy Dat, Truong Khang Nguyen, and Nguyen Thoi Trung analyzed the data and wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zheng, X.; Ren, Z.; Li, X.; Wang, Y. Microstructural characterization and mechanical properties of nitrided layers on aluminum substrate prepared by nitrogen arc. Appl. Surf. Sci. 2012, 259, 508–514. [Google Scholar] [CrossRef]
- Figueroa, U.; Salas, O.; Oseguera, J. Deposition of AlN on Al substrates by reactive magnetron sputtering. Surf. Coat. Technol. 2005, 200, 1768–1776. [Google Scholar] [CrossRef]
- Sheppard, L.M. Aluminum nitride: A versatile but challenging material. Bull. Am. Ceram. Soc. 1990, 69, 1801–1812. [Google Scholar]
- Kuramoto, N.; Taniguchi, H. Transparent AIN ceramics. J. Mater. Sci. Lett. 1984, 3, 471–474. [Google Scholar] [CrossRef]
- Shu, L.; Peng, B.; Cui, Y.; Gong, D.; Yang, Z.; Liu, X.; Zhang, W. Effects of AlN Coating Layer on High Temperature Characteristics of Langasite SAW Sensors. Sensors 2016, 16, 1436. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; You, Z. Modeling and Experimental Analysis on the Temperature Response of AlN-Film Based SAWRs. Sensors 2016, 16, 1205. [Google Scholar] [CrossRef] [PubMed]
- Zamora, R.J.; Nair, A.K.; Hennig, R.G.; Warner, D.H. Ab initio prediction of environmental embrittlement at a crack tip in aluminum. Phys. Rev. B 2012, 86, 60101. [Google Scholar] [CrossRef]
- Aoki, Y.; Fujii, H.; Nogi, K. Effect of atomic oxygen exposure on bubble formation in aluminum alloy. J. Mater. Sci. 2004, 39, 1779–1783. [Google Scholar] [CrossRef]
- Turner, D.E.; Zhu, Z.Z.; Chan, C.T.; Ho, K.M. Energetics of vacancy and substitutional impurities in aluminum bulk and clusters. Phys. Rev. B 1997, 55, 13842–13852. [Google Scholar] [CrossRef]
- Chung, J.P.; Lee, J.S.; Kim, K.R.; Choi, B.H. The change of atomic distribution and hardness by nitrogen implantation into aluminum alloy. Rev. Sci. Instrum. 2008, 79, 02C509. [Google Scholar] [CrossRef] [PubMed]
- Ghoranneviss, M.; Sari, A.H.; Dorranian, D.; Khorshid, P.; Hajihosseini, G.H.S.; Shokouhy, A.; Hesabi, M. Nitrogen Ion Implantation in Pure Aluminium; Japan Society of Plasma Science and Nuclear Fusion Research: Nagoya, Japan, 2006. [Google Scholar]
- Hogg, B.G.; Paulin, R.; Troev, T.D. Detection of nitrogen impurities in aluminum by positron annihilation. Phys. Lett. 1979, 71, 240–242. [Google Scholar] [CrossRef]
- Andrey, I.K.; Dat Duy, V.; Lipnitskii, A.G. The interaction between light impurities and vacancies in titanium and aluminum metals: A DFT study. St. Petersburg Polytech. Univ. J. Phys. Math. 2016, 2, 96–102. [Google Scholar]
- Hehenkamp, T. Absolute vacancy concentrations in noble metals and some of their alloys. J. Phys. Chem. Solids 1994, 55, 907–915. [Google Scholar] [CrossRef]
- Lynn, K.G.; Schultz, P. Vacancy formation energy measurements in single crystal aluminum using a variable-energy positron beam. Appl. Phys. A 1985, 37, 31–36. [Google Scholar] [CrossRef]
- Fluss, M.J.; Smedskjaer, L.C.; Chason, M.K.; Legnini, D.G.; Siegel, R.W. Measurements of the vacancy formation enthalpy in aluminum using positron annihilation spectroscopy. Phys. Rev. B 1978, 17, 3444–3455. [Google Scholar] [CrossRef]
- Carling, K.M.; Wahnström, G.; Mattsson, T.R.; Sandberg, N.; Grimvall, G. Vacancy concentration in Al from combined first-principles and model potential calculations. Phys. Rev. B 2003, 67, 054101. [Google Scholar] [CrossRef]
- Poletaev, D.O.; Aksyonov, D.A.; Dat Duy, V.; Lipnitskii, A.G. Hydrogen solubility in hcp titanium with the account of vacancy complexes and hydrides: A DFT study. Comput. Mater. Sci. 2016, 114, 199–208. [Google Scholar] [CrossRef]
- Peter, E.B.; Jepsen, O.; Andersen, O.K. Improved tetrahedron method for Brillouin-zone integrations. Phys. Rev. B 1994, 49, 16223–16233. [Google Scholar]
- Perdew, J.P.; Kieron, B.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47. [Google Scholar] [CrossRef]
- Press, W.H.; Flannery, B.P.; Teukolsky, S.A.; Vetterling, W.T. Numerical Recipes; Cambridge University Press: New York, NY, USA, 1986. [Google Scholar]
- Jacobs, P.W.M.; Zhukovskii, Y.F.; Mastrikov, Y.; Shunin, Y.N. Bulk and surface properties of metallic aluminium: DFT simulations. Comput. Model. New Technol. 2002, 6, 7–28. [Google Scholar]
- Evans, D.J.; Holian, B.L. The Nose–Hoover thermostat. J. Chem. Phys. 1985, 83, 4069–4074. [Google Scholar] [CrossRef]
- Verlet, L. Computer “Experiments” on Classical Fluids. I. Thermodynamical Properties of Lennard-Jones Molecules. Phys. Rev. 1967, 159, 98–103. [Google Scholar] [CrossRef]
- Edwards, R.A.H.; Eichenauer, W. Reversible hydrogen trapping at grain boundaries in superpure aluminium. Scr. Metall. 1980, 14, 971–973. [Google Scholar] [CrossRef]
- Eichenauer, W.; Hattenbach, K.; Pebler, Z. The Solubility of Hydrogen in Solid and Liquid Aluminum. Z. Metallkunde 1961, 52, 682–684. [Google Scholar]
- Sugimoto, H.; Fukai, Y. Solubility of hydrogen in metals under high hydrogen pressures: Thermodynamical calculations. Acta Metall. Mater. 1992, 40, 2327–2336. [Google Scholar] [CrossRef]
- Ichimura, M.; Katsuta, H.; Sasajima, Y.; Imabayashi, M. Hydrogen and deuterium solubility in aluminum with voids. J. Phys. Chem. Solids 1988, 49, 1259–1267. [Google Scholar] [CrossRef]
- Wolverton, C.; Ozoliņš, V.; Asta, M. Hydrogen in aluminum: First-principles calculations of structure and thermodynamics. Phys. Rev. B 2004, 69, 144109. [Google Scholar] [CrossRef]
- Linderoth, S. Hydrogen diffusivity in aluminium. Philos. Mag. Lett. 1988, 57, 229–234. [Google Scholar] [CrossRef]
- Linderoth, S.; Rajainmäki, H.; Nieminen, R.M. Defect recovery in aluminum irradiated with protons at 20 K. Phys. Rev. B 1987, 35, 5524–5528. [Google Scholar] [CrossRef]
- Myers, S.M.; Besenbacher, F.; Norskov, J.K. Immobilization mechanisms for ion-implanted deuterium in aluminum. J. Appl. Phys. 1985, 58. [Google Scholar] [CrossRef]
- Demaison, J.; Margulès, L.; Boggs, J.E. The equilibrium N–H bond length. Chem. Phys. 2000, 260, 65–81. [Google Scholar] [CrossRef]
Figure 1.
The part of atomic configuration containing (a) impurities at tetrahedral and octahedral sites; (b) an H impurity at the tetrahedral site near a vacancy; (c) an N–H bond near a vacancy, and (d) the electron density surrounding the H–N bond.
Figure 1.
The part of atomic configuration containing (a) impurities at tetrahedral and octahedral sites; (b) an H impurity at the tetrahedral site near a vacancy; (c) an N–H bond near a vacancy, and (d) the electron density surrounding the H–N bond.
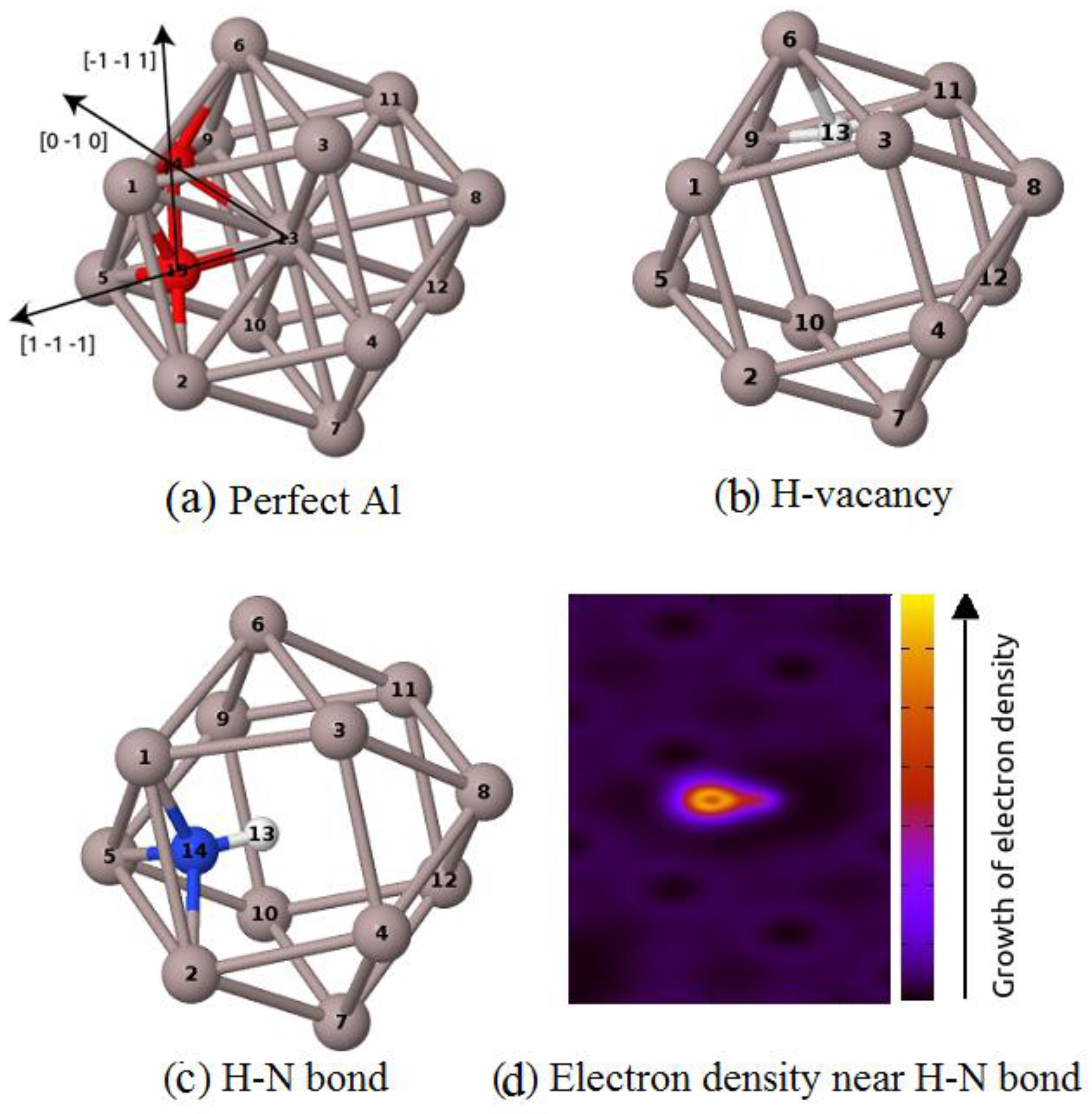
Figure 2.
(a) The density of state (DOS) of Al, and H when they locate in pure Al and near a vacancy, (b) The DOS of N, H and Al when they are in pure Al, near vacancy and near each other.
Figure 2.
(a) The density of state (DOS) of Al, and H when they locate in pure Al and near a vacancy, (b) The DOS of N, H and Al when they are in pure Al, near vacancy and near each other.
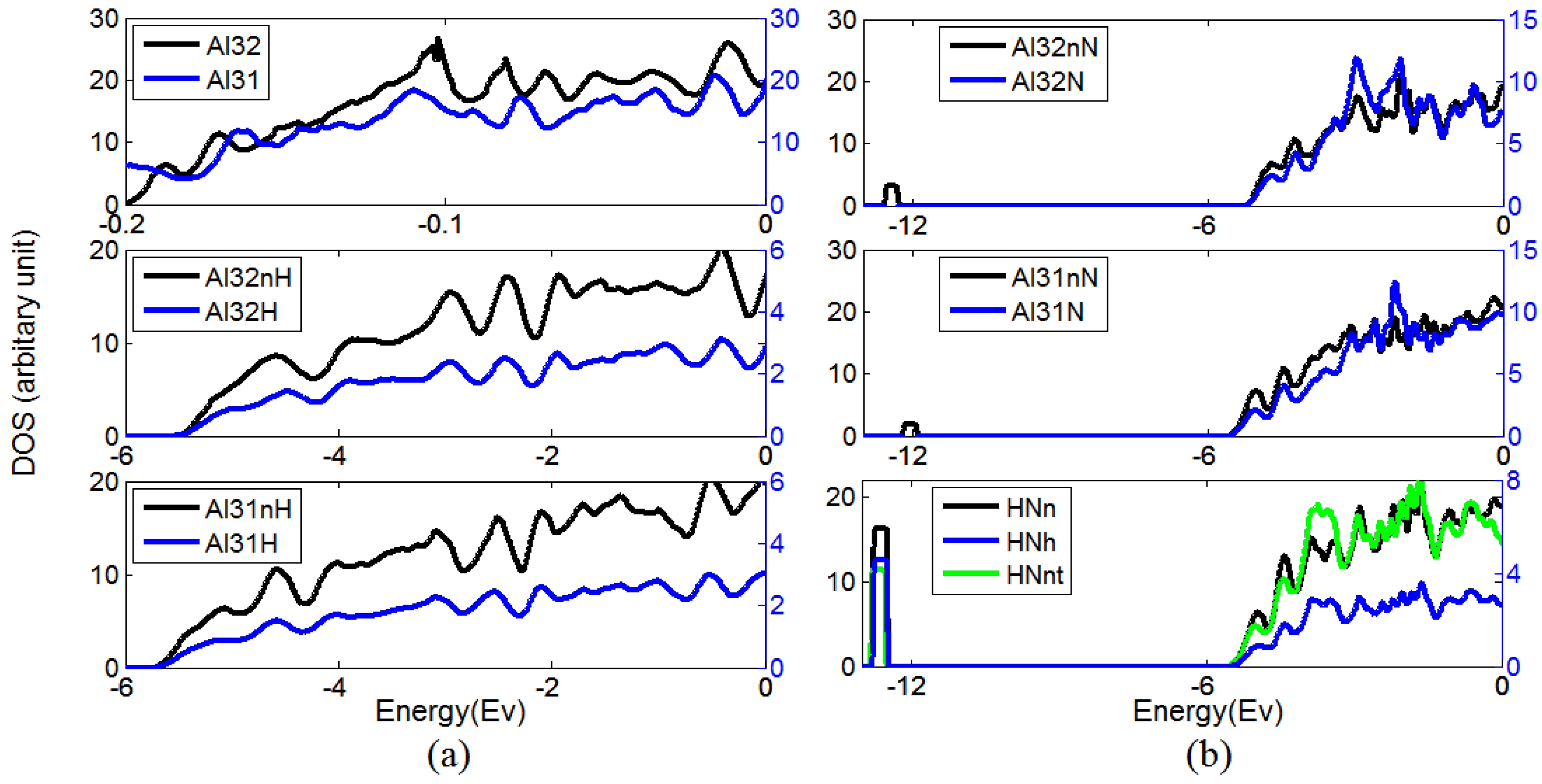
Figure 3.
The position of nitrogen atoms (blue balls) and hydrogen atoms (white balls) inside the Al (dark gray balls) lattice vacancy.
Figure 3.
The position of nitrogen atoms (blue balls) and hydrogen atoms (white balls) inside the Al (dark gray balls) lattice vacancy.

{kind=link}
{kind=link}
{kind=link}
Table 1.
Binding energy (eV) of impurity–vacancy complex (ECIMPURITY-vacancy) and impurity–hydrogeN–vacancy complex (ECIMPURITY–H–vacancy) in the Al lattice.
Table 1.
Binding energy (eV) of impurity–vacancy complex (ECIMPURITY-vacancy) and impurity–hydrogeN–vacancy complex (ECIMPURITY–H–vacancy) in the Al lattice.
| DFT package | ECH–vacancy | ECN–vacancy | ECN–(H–vacancy) |
|---|---|---|---|
| Abinit | 0.39 | −0.69 | 0.56 |
| VASP | 0.50 | −0.51 | 0.59 |
Table 2.
Calculated vacancy formation energy in case of pure Al (Evacancyf), Al with an H impurity (Evacancyf[H]), Al with H and N impurities (Evacancyf[H + N]). All values are given in eV.
Table 2.
Calculated vacancy formation energy in case of pure Al (Evacancyf), Al with an H impurity (Evacancyf[H]), Al with H and N impurities (Evacancyf[H + N]). All values are given in eV.
| DFT package | Evacancyf | Evacancyf[H] | Evacancyf[H + N] |
|---|---|---|---|
| Abinit | 0.55 | 0.16 | −0.38 |
| VASP | 0.65 | 0.15 | −0.44 |
Table 3.
The formation energy of vacancy–H–mN complex Efvacancy−H+mN, and the trapping energy Etrap(m) with m in the range from 1 to 8. All energies are in eV. The cell volume changing ΔV in cubic Angstrom, lattice changing Δa, and the average difference between lattice parameters of each configuration Δaver in Angstrom.
Table 3.
The formation energy of vacancy–H–mN complex Efvacancy−H+mN, and the trapping energy Etrap(m) with m in the range from 1 to 8. All energies are in eV. The cell volume changing ΔV in cubic Angstrom, lattice changing Δa, and the average difference between lattice parameters of each configuration Δaver in Angstrom.
| m | Efvacancy−H+mN | Etrap(m) | ΔV | Δa | Δaver |
|---|---|---|---|---|---|
| 1 | −0.59 | 0.00 | |||
| 2 | −0.54 | −0.16 | 5.69 | 0.04 | 0.02 |
| 3 | −0.69 | −0.35 | 6.12 | 0.07 | 0.17 |
| 4 | −0.49 | −0.01 | 7.47 | 0.05 | 0.11 |
| 5 | 1.57 | 1.90 | 1.43 | 0.15 | 0.38 |
| 6 | −0.08 | −1.90 | 4.92 | 0.10 | 0.47 |
| 7 | −2.62 | −2.74 | 10.91 | 0.24 | 0.80 |
| 8 | −0.62 | 1.80 | 16.37 | -0.30 | 0.30 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Vo, D.D.; Lipnitskii, A.G.; Nguyen, T.K.; Nguyen, T.T. Nitrogen Trapping Ability of Hydrogen-Induced Vacancy and the Effect on the Formation of AlN in Aluminum. Coatings 2017, 7, 79. https://doi.org/10.3390/coatings7060079
AMA Style
Vo DD, Lipnitskii AG, Nguyen TK, Nguyen TT. Nitrogen Trapping Ability of Hydrogen-Induced Vacancy and the Effect on the Formation of AlN in Aluminum. Coatings. 2017; 7(6):79. https://doi.org/10.3390/coatings7060079
Chicago/Turabian StyleVo, Duy Dat, Aleksey G. Lipnitskii, Truong Khang Nguyen, and Thoi Trung Nguyen. 2017. "Nitrogen Trapping Ability of Hydrogen-Induced Vacancy and the Effect on the Formation of AlN in Aluminum" Coatings 7, no. 6: 79. https://doi.org/10.3390/coatings7060079
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.