Swap-Driven Self-Adhesion and Healing of Vitrimers


1
Department of Applied Physics, Eindhoven University of Technology, Postbus 513, NL-5600 MB Eindhoven, The Netherlands
2
Institute for Complex Molecular Systems, Eindhoven University of Technology, Postbus 513, NL-5600 MB Eindhoven, The Netherlands
*
Author to whom correspondence should be addressed.
Coatings 2019, 9(2), 114; https://doi.org/10.3390/coatings9020114
Submission received: 16 January 2019
/
Revised: 6 February 2019
/
Accepted: 7 February 2019
/
Published: 12 February 2019
(This article belongs to the Special Issue Progress in Self-Healing Coatings)
Abstract
:Vitrimers are covalent network materials, comparable in structure to classical thermosets. Unlike normal thermosets, they possess a chemical bond swap mechanism that makes their structure dynamic and suitable for activated welding and even autonomous self-healing. The central question in designing such materials is the trade-off between autonomy and material stability: the swap mechanism facilitates the healing, but it also facilitates creep, which makes the perfectly stable self-healing solid a hard goal to reach. Here, we address this question for the case of self-healing vitrimers made from star polymers. Using coarse-grained molecular dynamics simulations, we studied the adhesion of two vitrimer samples and found that they bond together on timescales that are much shorter than the stress relaxation time. We showed that the swap mechanism allows the star polymers to diffuse through the material through coordinated swap events, but the healing process is much faster and does not depend on this mobility.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Equipping materials with a mechanism to repair themselves after damage is a cornerstone of the sustainable use of natural resources [1]. Over the years, strategies have been developed towards this goal for material classes as diverse as linear polymers [2], supramolecular networks [3,4], dendrimer-clay systems [5], metal ion–polymer systems [6,7], and multicomponent systems [8,9,10,11,12]. Notably, the worldwide corrosion authority NACE estimated the yearly cost of corrosion to be trillion [13], increasing the demand for coating materials capable of preventing and repairing damages of the the underlying structure.
Reversibly crosslinked polymeric materials have been at the heart of many developments [14] along those lines: polymers form a huge part of the materials industry, and physical crosslinks that can reform after being broken are a natural choice for self-healing. However, for strong and stable materials, polymer networks that are covalently crosslinked yet adaptable are preferred [15]. Vitrimers, in particular, have emerged as a new paradigm for combining malleability and recyclability with strength and solvent-resistance, based on the chemical swapping of bonds [16].
The ideal self-healing material would (1) have a fully controllable long-time solid-like mechanical behavior, (2) heal without any form of external intervention (3) to the point that the location of the damage can no longer be identified, and (4) even do so repeatedly if the same location gets damaged multiple times. We use the terms stability, autonomy, quality, and repeatability to denote these four desired properties. In practice, these demands are typically in conflict with each other, and choices need to be made regarding which demands to fulfill more strictly.
Vitrimers perform excellently regarding quality and repeatability, as they are rich in chemically active moieties that can drive the healing process, and the way the parts of the material are held together is chemically identical before and after healing. In this manuscript, we describe self-adhesion or welding of two fully equilibrated surfaces of vitrimers made of star polymers. The process relies on covalent bond-swapping across the cut. Because these networks will, on the longest of time scales, flow like extremely viscous liquids as long as the swap mechanism is active, the most obvious way to achieve stability is to sacrifice some autonomy and introduce a way to turn the swap mechanism on and off, e.g., using temperature [17]. Alternatively, one might want to keep the process fully autonomous in order to use vitrimers as coatings. In this case, the bond swapping has to be kept active, and the challenge is to make the ratio of stress relaxation time to self-healing time as large as possible, delaying the loss of stability that happens with the relaxation.
Specifically, we studied star-polymer vitrimers using a recently developed method to incorporate associative bond-swapping in molecular dynamics simulations [18,19]. We considered the situation in which two surfaces of this material are brought together after a long time, which for most healing strategies based on reversible crosslinking should be considered a worst-case scenario, and showed that two such surfaces develop a bulk-like mechanical connection on time scales that are much shorter than the diffusion time of the star polymers or the stress relaxation time of the material. Thus, we demonstrated the feasibility of autonomous self-healing in these materials for the case long-time stability is not needed, such as in the case of self-healing coatings, where the coated material provides the long-time rigidity. At the same time, we kept the possibility of attaining long-time stability at the cost of sacrificing autonomy.
The paper is organized as follows. First, we introduce the coarse-grained model for the polymer network and the basics of the simulation method. We then demonstrate the covalent vitrimer-based healing of two equilibrated surfaces. Longer simulations identify the mobility of the star polymers in the bond-swapping network, and we discuss how one might choose the building blocks such as to improve the autonomy–stability trade-off.
2. Model
The building blocks of our vitrimer networks are star polymers with eight arms which have the binding moieties at their ends. Star polymers are a versatile basis for adaptive networks [15] and are widely used for hydrogels [20]. In this work, we employed a coarse-grained representation which allows to distinguish between bonds in which the two binding moieties are identical, and bonds in which they are different. We focus on the latter type: Our binding groups represent, e.g., carboxyl and hydroxyl groups so that the bonds are ester bonds, and the swap mechanism is a transesterification reaction [16,21]. In Figure 1, we represent the two types of ends with black and red beads, respectively. Each arm of the star is a sequence of 5 beads connected by harmonic springs with rest length , which in our simulations serve as the unit of length. The beads interact with a repulsive Weeks–Chandler–Andersen (WCA) potential [22]:
with equal to the bond length . To model the bond forming attraction between black and red arm endings and their swap, we employed a combination of a generalized short-ranged attractive Lennard–Jones potential (binding energy ) with a three-body potential energy term [18]. The three-body potential serves the combined purpose of guaranteeing that only a single red and black bead bind to each other, and to facilitate the swapping of bonds. For a detailed description of the implementation of this method in the HOOMD-blue molecular dynamics package [23] and the stress relaxation behavior of this type of star polymer networks, we refer to [19]. The swapping method contains a parameter that can be used to tune the energy barrier to swapping , where is the binding energy of a red-black bond in case no other binding beads are nearby. Thus, represents the barrier-free energy landscape in which the swap rate is fully determined by the encounter rates between ester bonds and free hydroxyls, and values can be used to reduce the swap rate. We used a value of that is of order , so the “covalent” bonds will not break spontaneously and the only way for the network to rearrange is via the swap mechanism.
3. Simulation Protocol
All simulations start from equilibrated mixtures of stars with 7 “red”-functionalized ends and 1 “black”-functionalized end , and stars with the opposite composition . In cases where all minority ends formed a bond (a limit which we typically approach very closely since the binding energy is very high), we ended up with a network of 1500 star polymers with 4800 bonds and 2400 unbound ends of the majority (red) type. These unbound functional groups (hydroxyls in case of the transesterification swap) are the initiators of the swap reaction. The total number of monomer beads in each network is 49,500.
The size of the box provides a packing fraction of . This is below the glass transition, so the chains will be mobile. It is also high enough (2.2 times the overlap concentration) that the ends of the stars will encounter each other regularly, and a percolating network can be formed.
We generated networks with periodic boundaries in the -directions but with confining walls in the z-direction, which will allow to have equilibrated top and bottom surfaces. We equilibrated each sample for timesteps of size , where the time unit corresponds to the time it takes for the average number of swap events per bond to reach unity, in absence of barriers. We worked at fixed volume fraction and temperature , so that both surfaces were fully annealed and the swap rate was constant. To study the adhesion or healing process, we combined every pair of these initial networks by stacking them vertically, and continuing the simulation with fully periodic boundaries. Each adhesion simulation (of which we now have 300) therefore gives two healing cuts, one where the top of the first sample attaches to the bottom of the second sample, and one vice versa.
The combined system was then simulated without any swap barrier (), allowing swap events that create bonds between the two previously disconnected halves of the sample. We saved a series of configurations at different healing times . We then froze the topology of these configurations in order to have a visualization of the adhesion and to do a numerical mechanical test.
4. Results
The visualization was made using a vertical stretch and a small monomer–monomer attraction to enhance the view of the molecular bridges. The sample was then quenched to the nearest energy minumum. The result, which can be seen as a sequence of partially adhered pieces of stretched and dried elastomer, is shown in Figure 2.
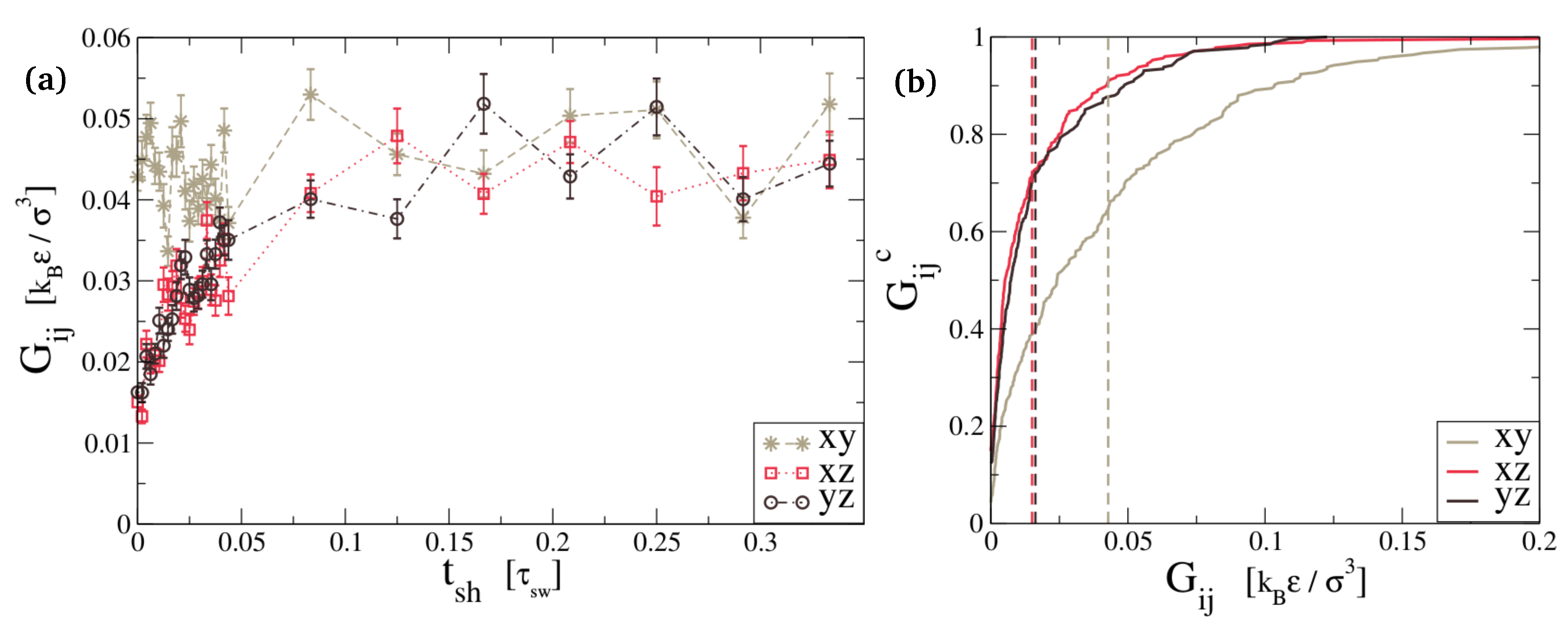
The mechanical test consists of a measurement of the stress relaxation modulus of the topologically frozen configuration, using the autocorrelation method [24]. This tells us how much mechanical resistance the sample has gained from the newly formed bonds. As expected for a solid-like material, the stress relaxation modulus shows a plateau for long times, corresponding to the static shear modulus. In Figure 3, we report the component of the stress relaxation modulus, which is not affected by the adhesion process because the surface across which it is happening is parallel to the -plane, in gray. The graph also shows the and components, which should start at a lower value of G because before the healing process, there are no bonds across the gap. The figure clearly shows that the component is independent of healing time, while the other two components develop a value of the order of the component within . This signals that the material has acquired a roughly isotropic elasticity, and mechanical testing can no longer reveal where the disconnected surface used to be.
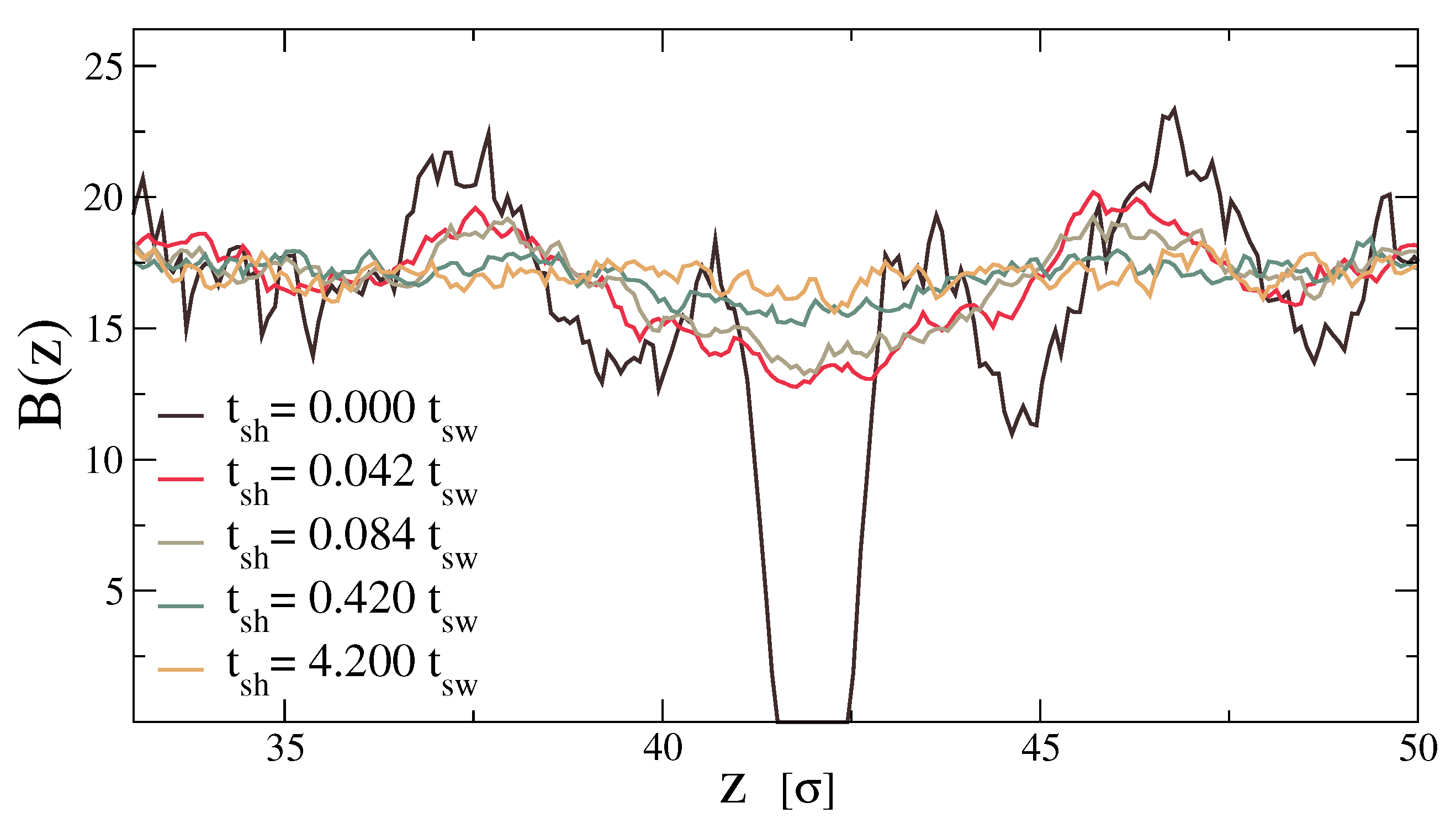
A complementary view of the healing process can be seen from the point of view of the bonds. In Figure 4, we show, as a function of coordinate z, the number of star–star bonds that cross a fictitious plane perpendicular to the z-axis at that position. Without any healing, there are no bonds that cross the region of the cut, around . There is also an evident layering effect at that produces zones of high density of bonds just around the cut, followed by a zone of low density and then an even higher concentration. This effect disappears after the third high density layer at the edge of Figure 4. The curves for higher values of demonstrate that the healing process eliminates the gap and reduces the layering, reinforcing the picture that the material is losing its memory of where the interface used to be.
5. Diffusive Long-Time Behavior
The different tests above demonstrate that our elastomer model is capable of swap-driven self-adhesion. For situations in which fully autonomous self-healing is not required, this may be sufficient.
To assess the suitability of these materials for autonomous self-healing, we needed to determine whether it is possible to have a large separation between the self-healing time and the stress relaxation time. After all, healing and stress relaxation both originate from the swap dynamics of the networks, so it is not possible to have one without the other, and a large separation of time scales would imply that we can have a material that can self-heal on timescales on which it is still a solid. Indeed, in our previous work, we demonstrated that stress relaxation in star polymer vitrimers takes many swap events, and can be controlled via the topology of the network [19].
Here, we studied the mobility of the star polymers that originate from each of the two halves of the self-adhering sample, and demonstrated that a simple diffusion model captures the essentials of the long-time behavior.
The central quantity in the analysis is the density of particles that were originally in the lowest half of the box. Translation symmetry in the -directions dictates that we can integrate over those and focus on the z-dependence, which we described using a diffusion equation:
here, D denotes the diffusion constant, which describes the rate at which bond swapping evens out spatial variations in .
For our adhesion simulations, the initial condition for this process is a square wave profile. This signifies that we ignore the layering effect which concerns fluctuations on short length scales, which will be washed out rapidly anyway. We note that any short-time behavior for which the layering is important will therefore not be captured by this model. In Appendix A, we describe two well known approaches to solving this equation: with a Fourier expansion and with its Green function. Here, we describe the outcome.
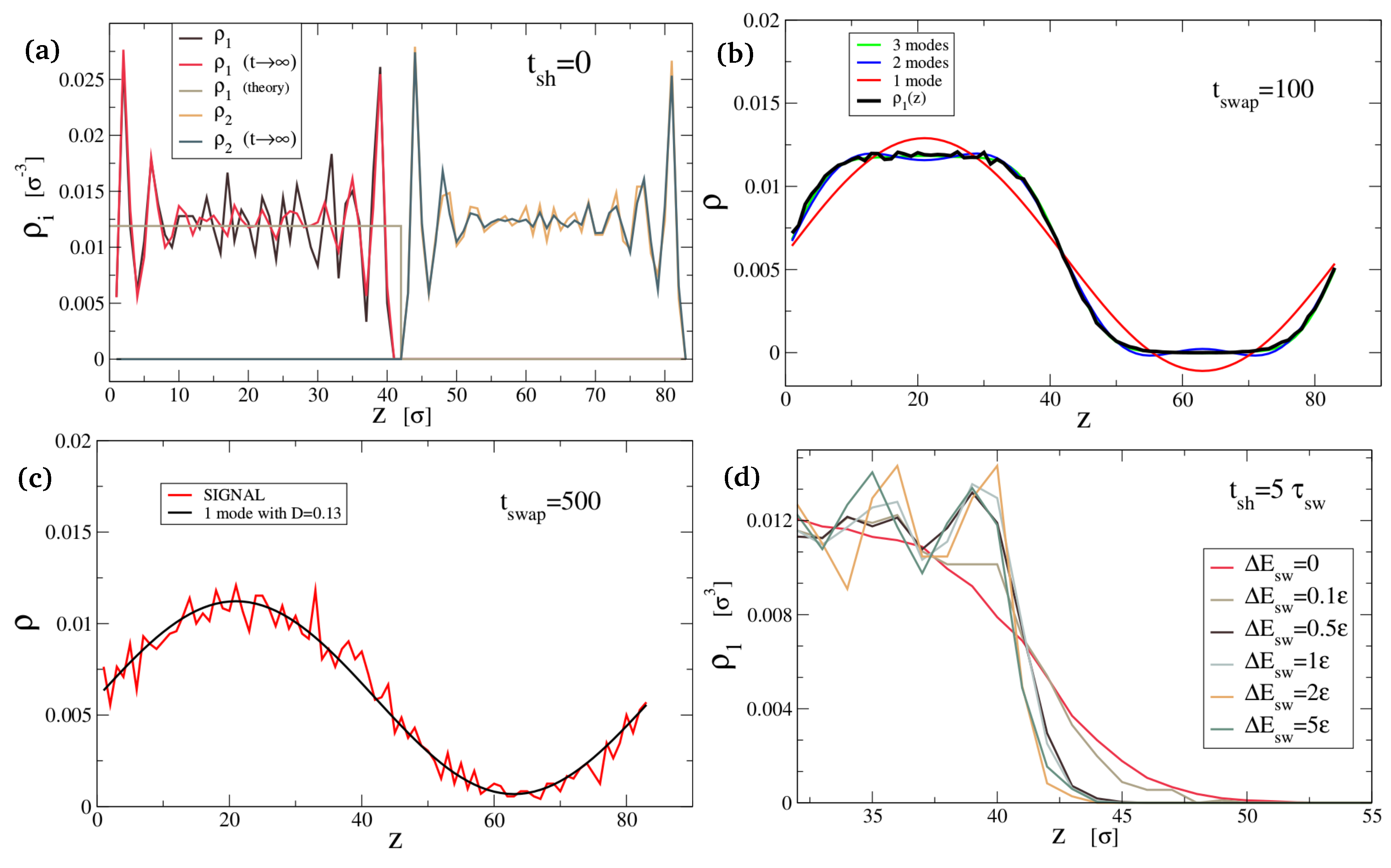
In Figure 5a–c, we show the density profiles that we measure in our simulations for 3 different values of the healing time (black curves). Figure 5a additionally shows that the density profile after a long equilibration with the swap mechanism turned off (red curve) is only marginally different from the profile before any equilibration, proving that bond swapping is necessary to have any mobility of the star polymers: when the swap mechanism is turned off. In panels b and c, we compared the density profiles from the simulation to the Fourier series solution of the diffusion Equation (1), providing a fit of the diffusion constant . These simulations were performed without any energy barrier to swapping. We could easily adjust the system to have an energy barrier by setting , in order to reduce the diffusion constant D. Figure 5d shows how this way of slowing down diffusion alters the density profile after a healing time of .
To assess the importance of the diffusion of star polymers through the network for the healing process, and to understand the time scales involved, we then extracted a measurable quantifier for the adhesion process from the diffusion model. Our approach is to estimate the number of bridges defined as the number of bonds between monomers coming from different halves of the system. We can directly measure this number in our simulation and we compare it to the following integral:
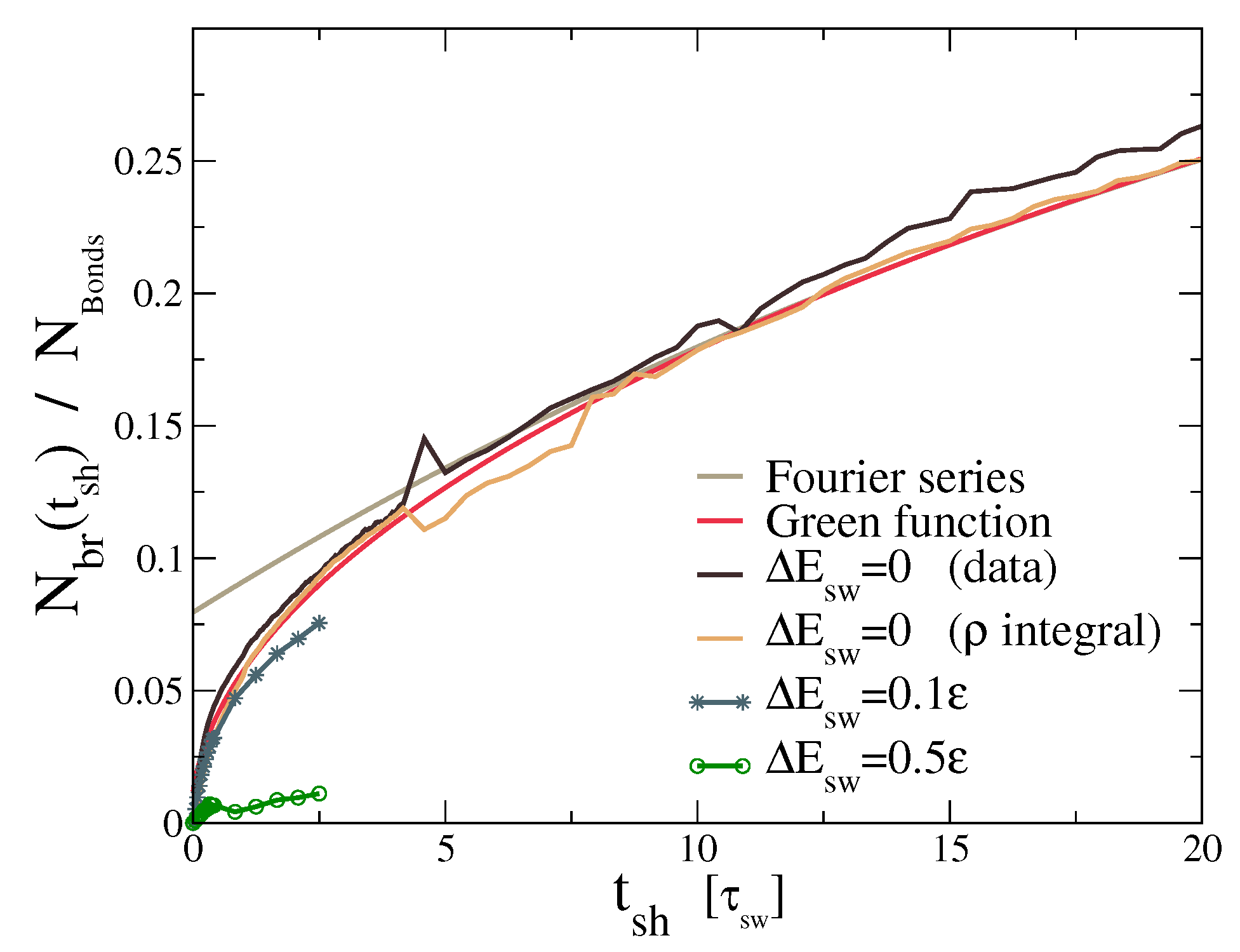
This is a functional of the density profiles at time t, and it assumes that the density of bridges is proportional to the product . The Factor 2 accounts for the two interfaces that we get from the periodic boundary conditions. Results are reported in Figure 6. The prediction derived from diffusion (red curve, Green function solution) perfectly reproduces its numerical equivalent (orange curve), while it slightly underestimates the actual number of bridges (black curve). Overall, the development of the number of bonds between stars that originate from the different halves of the sample is well described by the diffusion equation.
The crucial observation from the mobility study is that it takes about to achieve any appreciable interdiffusion of stars across the healing surface. The mechanical healing, on the other hand, took only , which is more than 30 times faster. More importantly, stress relaxation takes even longer than interdiffusion. Indeed, the same mixture showed a relaxation time of in [19], which is roughly three orders of magnitude slower than self-healing. Thus, without any further optimization of the polymer architecture, we achieved a material which autonomously heals three orders of magnitude faster than it flows.
6. Discussion
Vitrimers with an activated swap mechanism are continuously rearranging their structure. Bond swap events happen throughout the material. There is no enhanced concentration of active moieties at the surface and a picture of where swaps are happening shows no sign of the location of the healing surface (see Figure 7). The swap events drive both the healing and the stress relaxation, and the naive conclusion would therefore be that the only way to have a solid material with healing-like properties would be to resort to welding: Switch on the swap mechanism when it is needed to stick two surfaces together, and switch it off in order to have a well-behaved solid object.
Our work demonstrates a possible way out of this conundrum: The adhesion process only requires a very limited number of swap events, compared to full star polymer mobility (as we demonstrated by fitting to a diffusion model) and stress relaxation (as we showed in [19]). The reason is that healing does not require any mobility of the star polymers as a whole: A single swap event suffices to create a link between the two halves of the system, but mobility of the star requires each of its arms to swap several times. Thus, we should expect the number of arms per star polymer (i.e., their functionality) to be a major determinant for the size of the time scale gap between healing and mobility. Their architecture will also have a major role, because if the mixture is replaced by while keeping the total numbers of each binding moiety the same, the stress relaxation would grow by an order of magnitude, while the swap rate would be unaffected [19]. The rich design space of vitrimers will enable future work to optimize this time scale gap.
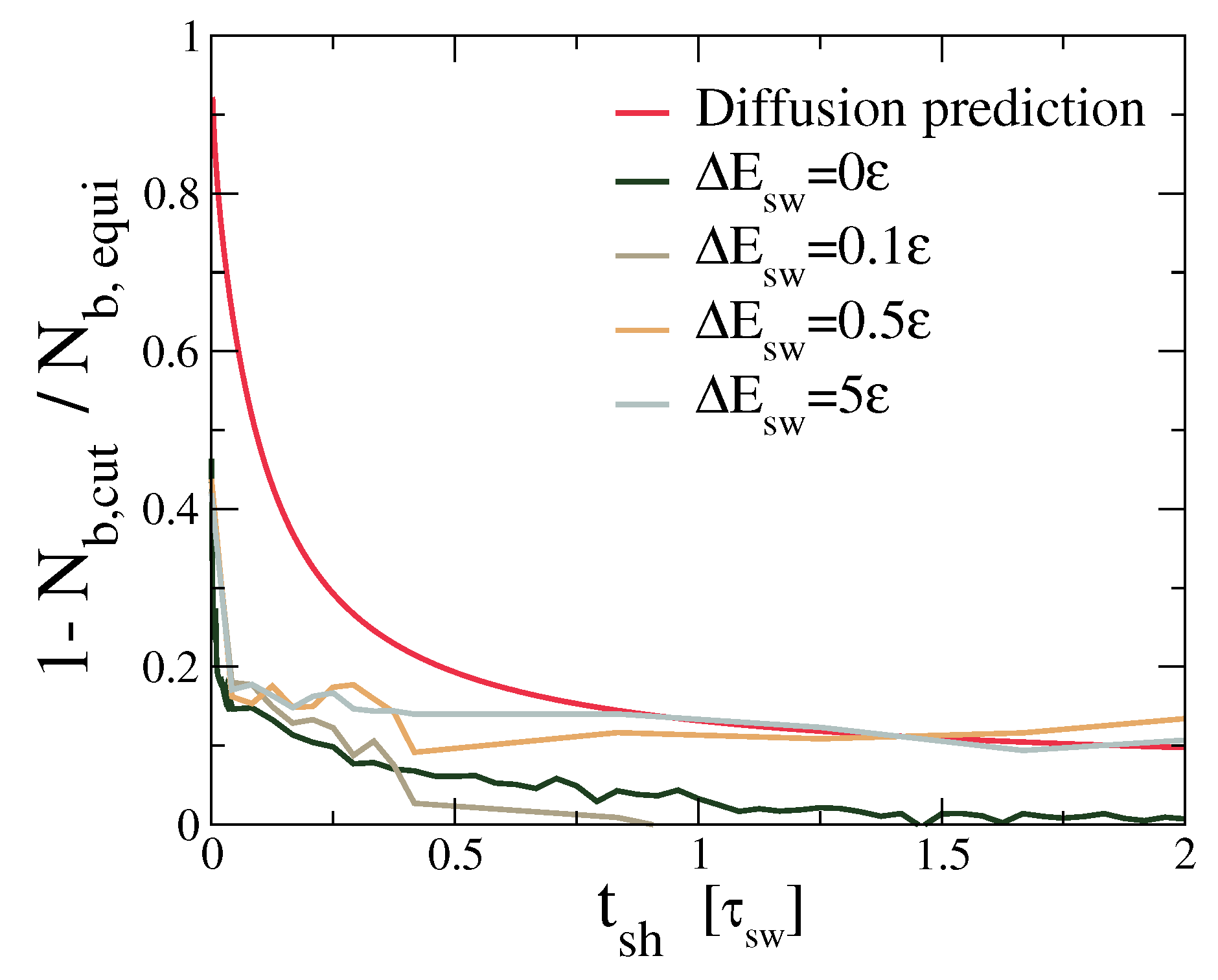
The fact that adhesion and healing do not require actual mobility of the star polymers can be further illustrated by considering the prediction for the self-healing time that would result from considering the diffusion model, with the diffusion constant D that we obtained by fitting the long-time density profiles. The diffusion model makes no predictions for the mechanical properties, but for this illustrative purpose, it suffices to consider the number of bonds in the region around the healing interface (). If diffusion were dominant for the healing, this model should work reasonably well. Instead, we note that the diffusion model greatly overestimates the healing time, as demonstrated in Figure 8. The actual numbers depend strongly on the chosen definition of healing time, but the difference between the actual number of bridges and the diffusion-based prediction is clear.
7. Conclusions
Some materials achieve self-healing trading off stability [3] while remaining fully autonomous. Others, instead, do not give up their mechanical properties but sacrifice autonomy to earn their healing ability [4], often involving secondary interactions [25,26,27,28]. Alternatively, it is possible to achieve autonomous self-healing embedding healing agents in the material [8,9,10], trading off repeatability and perhaps quality. Furthermore, self-healing in crosslinked networks is based on dangling chain motion, so it is favoured by the low density of crosslinks, which also corresponds to limited mechanical properties [29]. Vitrimers, naturally suited to be non-autonomous healers [16,17], revert this paradigm: They in fact heal through crosslink swaps, which are more probable when the density of crosslinks is higher.
Several models have been introduced to simulate vitrimers [30,31,32,33]. We employed a recently developed method [19] to reproduce vitrimers in Molecular Dynamics (MD) simulations and demonstrated their viability as self-healing materials. Network topology influences both mechanical properties [34] and healing speed [19]. More surprisingly, within the same type of material, both externally activated healing/welding and fully autonomous self-healing are possible, suggesting the feasibility of vitrimer coatings. Hence, depending on the demands on healing time and stability, the manufacturer can choose the most suited healing strategy and optimize the architecture: Favoring quick swaps to achieve extremely malleable vitrimers when activated, or hindering some of them through topological constraints that extend the relaxation time more than they do the self-healing time, thus retaining the autonomy required for coatings. The star polymer architecture we employed is particularly promising in this regard, as it will allow to directly tune the ratio between these time scales using the same structure: Healing is mostly driven by the fluctuations of single arms, while stress relaxation requires many swap events involving all arms on each star. For the same reason, we may also speculate that slowing down the swap rate would impact stress relaxation more, because it requires the swap of all of the arms of a star, expanding the separation between healing and relaxation timescales even more.
In summary, vitrimers are naturally suited to be activated healers, but they can be designed to be fully autonomous, separately tuning diffusion and healing properties. This provides a way to design tunable materials which are also genuinely self-healing.
Author Contributions
Conceptualization: S.C. and W.G.E.; Methodology: S.C. and W.G.E.; Software: S.C.; Validation: S.C.; Formal Analysis: S.C. and W.G.E.; Investigation: S.C. and W.G.E.; Resources: S.C. and W.G.E.; Data Curation: S.C.; Writing—Original Draft Preparation: S.C.; Writing—Review and Editing: S.C. and W.G.E.; Visualization: S.C. and W.G.E.; Supervision: W.G.E.; Project Administration: S.C. and W.G.E.; Funding Acquisition: W.G.E.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
Appendix A
Diffusion Equation
In this appendix we will briefly explain how we solved the diffusion equation that we use to model the self healing. We start assuming assume a flat periodic density profile, ignoring the layering effect, defined as the repetition of the block profile with period :
where is the average density. We rewrite the diffusion equation (Equation (1)) in Fourier space:
A Fourier expansion of the initial density then gives:
which is a superposition of waves of lengths that has to contain only even numbers for the symmetry. It shows that over time we will lose modes starting from the fastest (largest ). In the long time limit our solution will be dominated by the diffusion constant D and the longest mode available . This is the relation that gives us the estimation of . We can see from Figure 5 that it captures the dynamics very well.
The predicted number of bridges from Equation (2) (gray curve in Figure 6) is noticeably bad for the initial times, so we have to improve the theory. Rather than using the Fourier series of the density, we can exactly solve the diffusion equation using its Green function to build the solution from:
where,
The solution is a simple combination of gaussian error functions. From this we get the red curve in Figure 6 which is a reasonable approximation of our dynamics.
References
- Hager, M.D.; Greil, P.; Leyens, C.; van der Zwaag, S.; Schubert, U.S. Self-healing materials. Adv. Mater. 2010, 22, 5424–5430. [Google Scholar] [CrossRef] [PubMed]
- De Gennes, P.G. Reptation of a polymer chain in the presence of fixed obstacles. J. Chem. Phys. 1971, 55, 572–579. [Google Scholar] [CrossRef]
- Cordier, P.; Tournilhac, F.; Soulié-Ziakovic, C.; Leibler, L. Self-healing and thermoreversible rubber from supramolecular assembly. Nature 2008, 451, 977–980. [Google Scholar] [CrossRef] [PubMed]
- Burnworth, M.; Tang, L.; Kumpfer, J.R.; Duncan, A.J.; Beyer, F.L.; Fiore, G.L.; Rowan, S.J.; Weder, C. Optically healable supramolecular polymers. Nature 2011, 472, 334–337. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Mynar, J.L.; Yoshida, M.; Lee, E.; Lee, M.; Okuro, K.; Kinbara, K.; Aida, T. High-water-content mouldable hydrogels by mixing clay and a dendritic molecular binder. Nature 2010, 463, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.P.; Messersmith, P.B.; Israelachvili, J.N.; Waite, J.H. Mussel-inspired adhesives and coatings. Annu. Rev. Mater. Res. 2011, 41, 99–132. [Google Scholar] [CrossRef] [PubMed]
- Holten-Andersen, N.; Harrington, M.J.; Birkedal, H.; Lee, B.P.; Messersmith, P.B.; Lee, K.Y.C.; Waite, J.H. pH-induced metal-ligand cross-links inspired by mussel yield self-healing polymer networks with near-covalent elastic moduli. Proc. Natl. Acad. Sci. USA 2011, 108, 2651–2655. [Google Scholar] [CrossRef]
- Cho, S.H.; White, S.R.; Braun, P.V. Self-healing polymer coatings. Adv. Mater. 2009, 21, 645–649. [Google Scholar] [CrossRef]
- Toohey, K.S.; Sottos, N.R.; Lewis, J.A.; Moore, J.S.; White, S.R. Self-healing materials with microvascular networks. Nat. Mater. 2007, 6, 581–585. [Google Scholar] [CrossRef]
- Kolmakov, G.V.; Revanur, R.; Tangirala, R.; Emrick, T.; Russell, T.P.; Crosby, A.J.; Balazs, A.C. Using nanoparticle-filled microcapsules for site-specific healing of damaged substrates: Creating a “repair-and-go” system. ACS Nano 2010, 4, 1115–1123. [Google Scholar] [CrossRef]
- Chen, X.; Dam, M.A.; Ono, K.; Mal, A.; Shen, H.; Nutt, S.R.; Sheran, K.; Wudl, F. A thermally re-mendable cross-linked polymeric material. Science 2002, 295, 1698–1702. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, B.; Urban, M.W. Self-Repairing oxetane-substituted chitosan polyurethane networks. Science 2009, 323, 1458–1460. [Google Scholar] [CrossRef] [PubMed]
- International Measures of Prevention, Application, and Economics of Corrosion Technologies Study 2016; NACE International: Houston, TX, USA, 2016.
- Wojtecki, R.J.; Meador, M.A.; Rowan, S.J. Using the dynamic bond to access macroscopically responsive structurally dynamic polymers. Nat. Mater. 2011, 10, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Kloxin, C.J.; Bowman, C.N. Covalent adaptable networks: Smart, reconfigurable and responsive network systems. Chem. Soc. Rev. 2013, 42, 7161–7173. [Google Scholar] [CrossRef] [PubMed]
- Montarnal, D.; Capelot, M.; Tournilhac, F.; Leibler, L. Silica-like malleable materials from permanent organic networks. Science 2011, 334, 965–968. [Google Scholar] [CrossRef] [PubMed]
- Denissen, W.; Winne, J.M.; Du Prez, F.E. Vitrimers: Permanent organic networks with glass-like fluidity. Chem. Sci. 2016, 7, 30–38. [Google Scholar] [CrossRef]
- Sciortino, F. Three-body potential for simulating bond swaps in molecular dynamics. Eur. Phys. J. E 2017, 40, 3. [Google Scholar] [CrossRef]
- Ciarella, S.; Sciortino, F.; Ellenbroek, W.G. Dynamics of vitrimers: Defects as a highway to stress relaxation. Phys. Rev. Lett. 2018, 121, 058003. [Google Scholar] [CrossRef]
- Sakai, T.; Matsunaga, T.; Yamamoto, Y.; Ito, C.; Yoshida, R.; Suzuki, S.; Sasaki, N.; Shibayama, M.; Chung, U.I. Design and fabrication of a high-strength hydrogel with ideally homogeneous network structure from tetrahedron-like macromonomers. Macromolecules 2008, 41, 5379–5384. [Google Scholar] [CrossRef]
- Capelot, M.; Montarnal, D.; Tournilhac, F.F.; Leibler, L. Metal-catalyzed transesterification for healing and assembling of thermosets. J. Am. Chem. Soc. 2012, 134, 7664–7667. [Google Scholar] [CrossRef]
- Weeks, J.D.; Chandler, D.; Andersen, H.C. Role of Repulsive Forces in Determining the Equilibrium Structure of Simple Liquids. J. Chem. Phys. 1971, 54, 5237–5247. [Google Scholar] [CrossRef]
- Anderson, J.A.; Lorenz, C.D.; Travesset, A. General purpose molecular dynamics simulations fully implemented on graphics processing units. J. Comput. Phys. 2008, 227, 5342–5359. [Google Scholar] [CrossRef]
- Kriuchevskyi, I.; Wittmer, J.P.; Benzerara, O.; Meyer, H.; Baschnagel, J. Numerical determination of shear stress relaxation modulus of polymer glasses. Eur. Phys. J. E 2017, 40, 43. [Google Scholar] [CrossRef]
- Susa, A.; Mordvinkin, A.; Saalwächter, K.; van der Zwaag, S.; Garcia, S.J. Identifying the role of primary and secondary interactions on the mechanical properties and healing of densely branched polyimides. Macromolecules 2018, 51, 8333–8345. [Google Scholar] [CrossRef]
- Susa, A.; Bijleveld, J.; Hernandez Santana, M.; Garcia, S.J. Understanding the effect of the dianhydride structure on the properties of semiaromatic polyimides containing a biobased fatty diamine. ACS Sustain. Chem. Eng. 2018, 6, 668–678. [Google Scholar] [CrossRef]
- Susa, A.; Bose, R.K.; Grande, A.M.; Van Der Zwaag, S.; Garcia, S.J. Effect of the dianhydride/branched diamine ratio on the architecture and room temperature healing behavior of polyetherimides. ACS Appl. Mater. Interfaces 2016, 8, 34068–34079. [Google Scholar] [CrossRef]
- Phadke, A.; Zhang, C.; Arman, B.; Hsu, C.C.; Mashelkar, R.A.; Lele, A.K.; Tauber, M.J.; Arya, G.; Varghese, S. Rapid self-healing hydrogels. Proc. Natl. Acad. Sci. 2012, 109, 4383–4388. [Google Scholar] [CrossRef]
- Imbernon, L.; Norvez, S.; Leibler, L. Stress relaxation and self-adhesion of rubbers with exchangeable links. Macromolecules 2016, 49, 2172–2178. [Google Scholar] [CrossRef]
- Oyarzún, B.; Mognetti, B.M. Efficient sampling of reversible cross-linking polymers: Self-assembly of single-chain polymeric nanoparticles. J. Chem. Phys. 2018, 148, 114110. [Google Scholar] [CrossRef]
- Wittmer, J.P.; Kriuchevskyi, I.; Cavallo, A.; Xu, H.; Baschnagel, J. Shear-stress fluctuations in self-assembled transient elastic networks. Phys. Rev. E 2016, 93, 62611. [Google Scholar] [CrossRef] [PubMed]
- Stukalin, E.B.; Cai, L.H.; Kumar, N.A.; Leibler, L.; Rubinstein, M. Self-healing of unentangled polymer networks with reversible bonds. Macromolecules 2013, 46, 7525–7541. [Google Scholar] [CrossRef] [PubMed]
- Smallenburg, F.; Leibler, L.; Sciortino, F. Patchy particle model for vitrimers. Phys. Rev. Lett. 2013, 111, 188002. [Google Scholar] [CrossRef] [PubMed]
- Zhong, M.; Wang, R.; Kawamoto, K.; Olsen, B.D.; Johnson, J.A. Quantifying the impact of molecular defects on polymer network elasticity. Science 2016, 353, 1264–1268. [Google Scholar] [CrossRef] [PubMed]
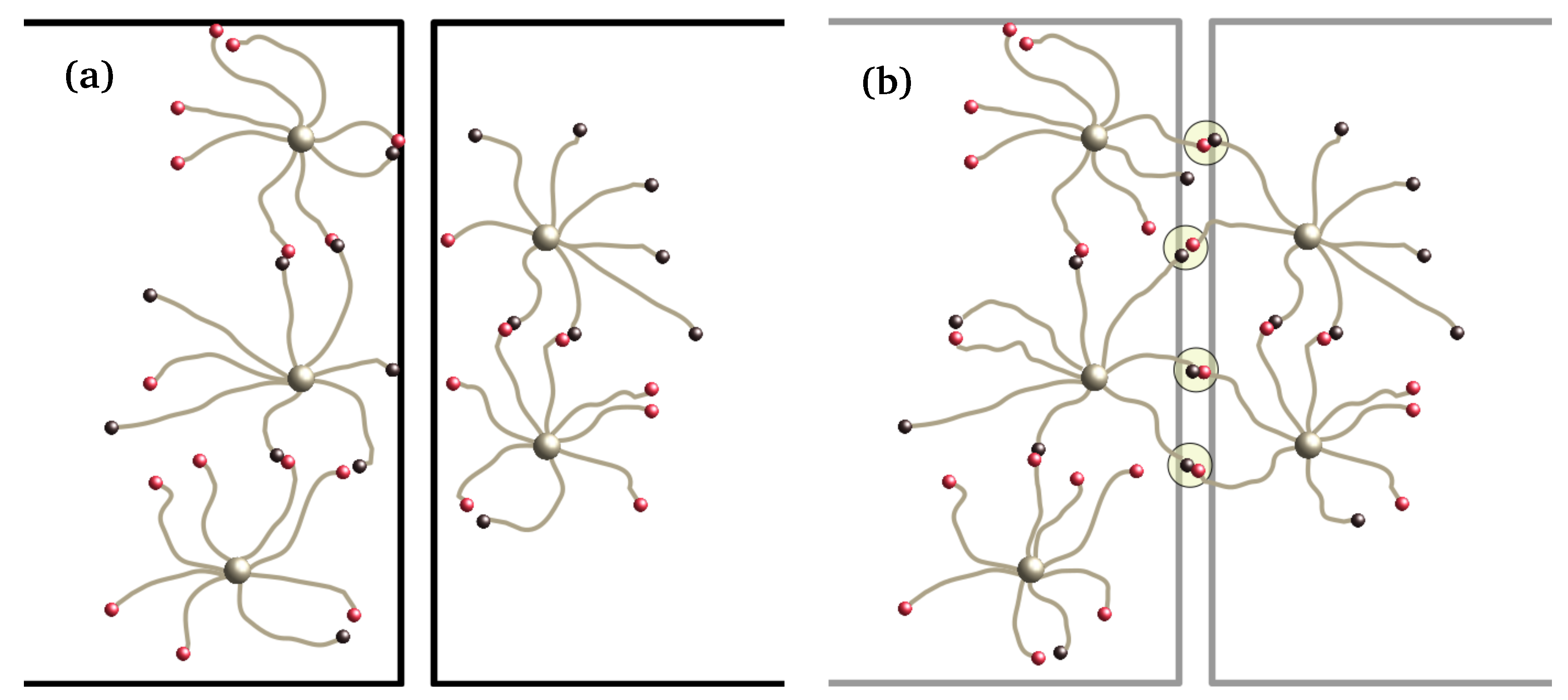
Figure 1.
Sketch of swap-driven self healing. (a) The two halves are put in contact, but there are no bonds to connect them so they cannot sustain stress. (b) Through swap moves, bridges start to form. After some time, the cut will be indistinguishable from bulk. Some of the stars loop back onto themselves, forming defects.
Figure 1.
Sketch of swap-driven self healing. (a) The two halves are put in contact, but there are no bonds to connect them so they cannot sustain stress. (b) Through swap moves, bridges start to form. After some time, the cut will be indistinguishable from bulk. Some of the stars loop back onto themselves, forming defects.

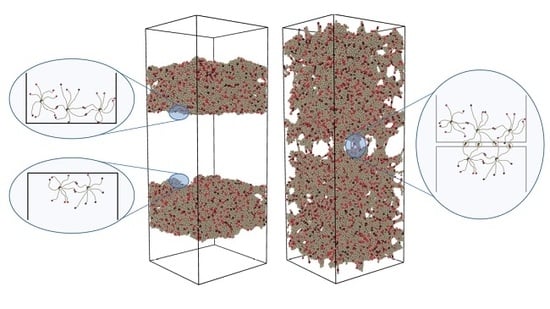
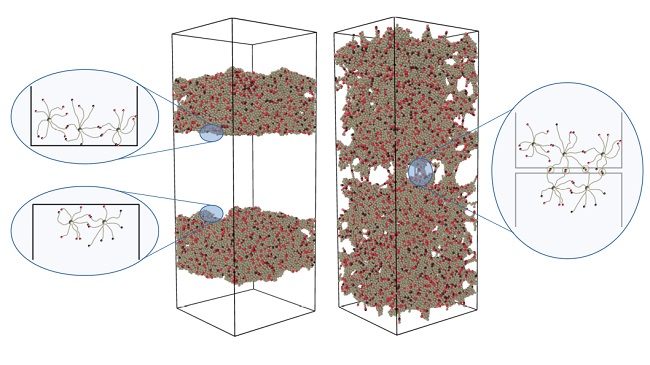
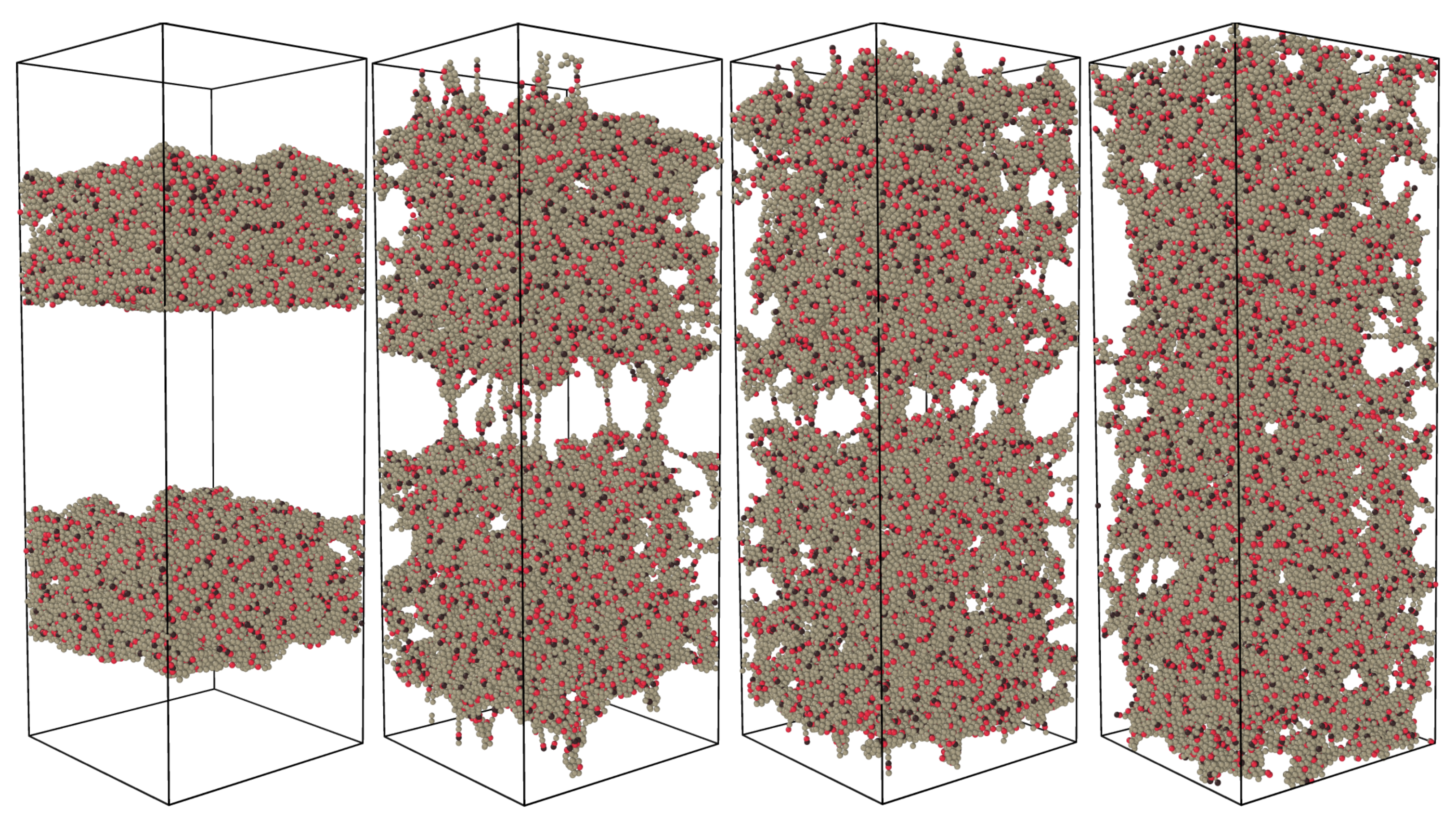
Figure 2.
Visualization of the formation of bridges between the two halves after healing times . The graphics were produced using energy minimization after a vertical strain, with a small monomer–monomer attraction to clear up the view. At , the material is completely separated into two pieces, but swaps are rapidly healing the fracture. Already at , traces of the fracture are only visible after straining. From , the healed cut is indistinguishable from the bulk.
Figure 2.
Visualization of the formation of bridges between the two halves after healing times . The graphics were produced using energy minimization after a vertical strain, with a small monomer–monomer attraction to clear up the view. At , the material is completely separated into two pieces, but swaps are rapidly healing the fracture. Already at , traces of the fracture are only visible after straining. From , the healed cut is indistinguishable from the bulk.

Figure 3.
Elastic plateau for different along three orthogonal directions, reported with their standard error. Because our simulations cover a finite amount of time, we see some remaining elasticity even without any healing (). The component is not affected by the cut or the healing process because it measures the resistance to shears in planes parallel to the cut. The and components denote the build-up of mechanical stiffness during healing/adhesion. In (b), we see the cumulative function that clearly shows the difference between directions perpendicular and orthogonal to the cut. Already around , the three components are comparable and thus, the network is capable of sustaining stress across the cut almost as the bulk. They become indistinguishable before reaching .
Figure 3.
Elastic plateau for different along three orthogonal directions, reported with their standard error. Because our simulations cover a finite amount of time, we see some remaining elasticity even without any healing (). The component is not affected by the cut or the healing process because it measures the resistance to shears in planes parallel to the cut. The and components denote the build-up of mechanical stiffness during healing/adhesion. In (b), we see the cumulative function that clearly shows the difference between directions perpendicular and orthogonal to the cut. Already around , the three components are comparable and thus, the network is capable of sustaining stress across the cut almost as the bulk. They become indistinguishable before reaching .

Figure 4.
Number of bonds that cut a z-plane for different healing times. At , a layering effect is visible, and we can distinguish three high density layers propagating from the cut. After a progressive healing process, the profile becomes flat.
Figure 4.
Number of bonds that cut a z-plane for different healing times. At , a layering effect is visible, and we can distinguish three high density layers propagating from the cut. After a progressive healing process, the profile becomes flat.

Figure 5.
Density profiles at . (a) At , layering is visible in the density profiles, so the square wave cannot capture the full details yet. The layers are stable if we equilibrate without healing (preventing swap). (b) At , the density profile is well-reproduced by the three slowest modes of the Fourier expansion. The system is diffusing with when . (c) At , only one mode is still present. Notice that the diffusion is far from reaching equilibrium. (d) The diffusion is slower when swaps are hindered by an energy barrier .
Figure 5.
Density profiles at . (a) At , layering is visible in the density profiles, so the square wave cannot capture the full details yet. The layers are stable if we equilibrate without healing (preventing swap). (b) At , the density profile is well-reproduced by the three slowest modes of the Fourier expansion. The system is diffusing with when . (c) At , only one mode is still present. Notice that the diffusion is far from reaching equilibrium. (d) The diffusion is slower when swaps are hindered by an energy barrier .

Figure 6.
Actual number of bridges (black) compared to the estimation of Equation (2) evaluated from the numerical simulations (orange), Fourier series (gray), and Green function (red). The Fourier and Green function curves use the previously fitted value , which represents the case of bond-swap without energy barriers. The plotted quantity should approach the limit of upon full mixing, which happens well beyond the limit of our simulations (see Figure 5). A barrier of is already enough to undermine the bridge formation. The gray curve is a truncated Fourier series that only takes into account the three slowest modes and is therefore not expected to match well at short times.
Figure 6.
Actual number of bridges (black) compared to the estimation of Equation (2) evaluated from the numerical simulations (orange), Fourier series (gray), and Green function (red). The Fourier and Green function curves use the previously fitted value , which represents the case of bond-swap without energy barriers. The plotted quantity should approach the limit of upon full mixing, which happens well beyond the limit of our simulations (see Figure 5). A barrier of is already enough to undermine the bridge formation. The gray curve is a truncated Fourier series that only takes into account the three slowest modes and is therefore not expected to match well at short times.

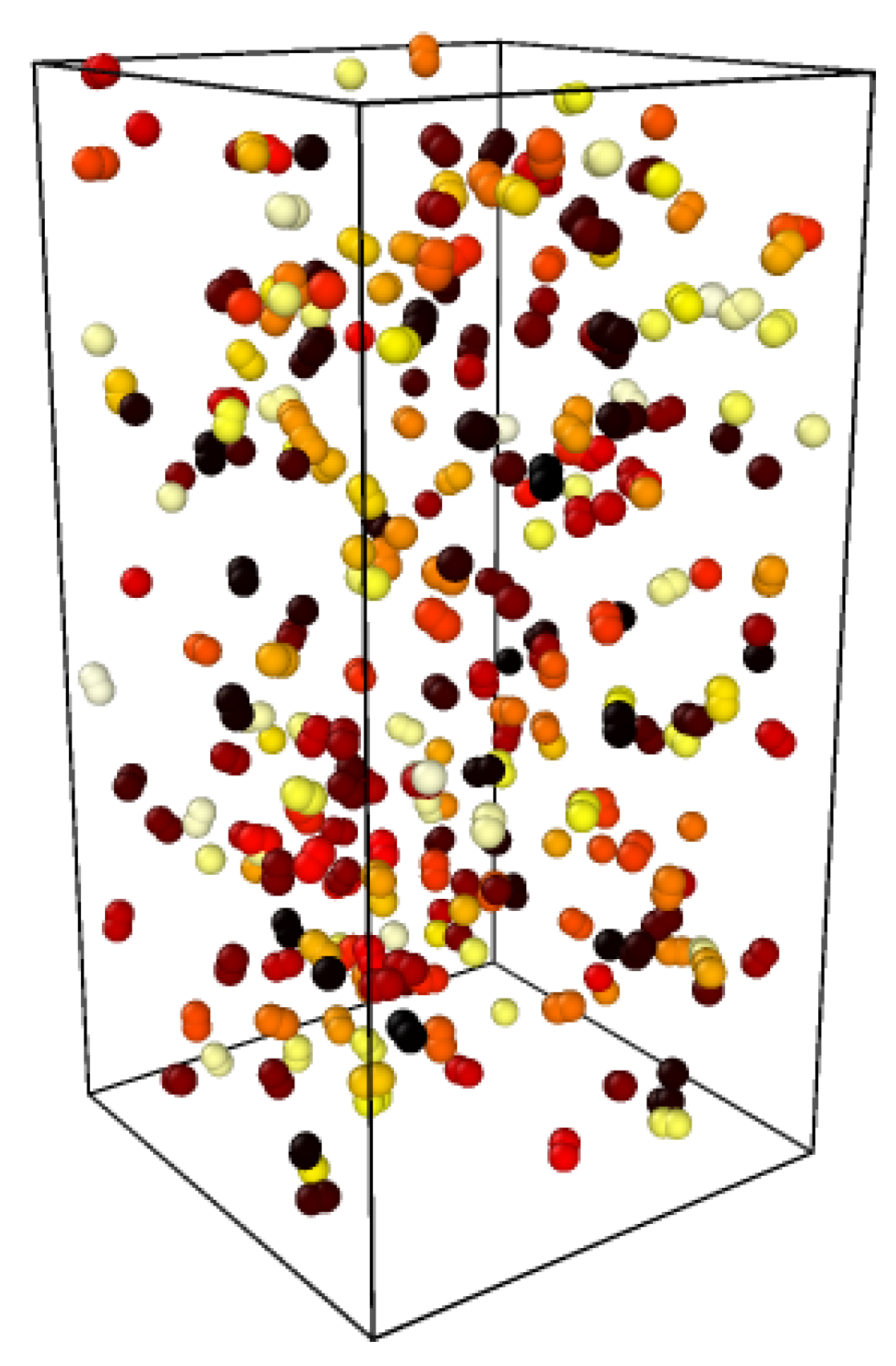
Figure 7.
Location of the first 100 swaps after the two pieces are put in contact. Near the cut, swap happens with the same probability as in the bulk. Healing is then determined by equilibrium swap rates.
Figure 7.
Location of the first 100 swaps after the two pieces are put in contact. Near the cut, swap happens with the same probability as in the bulk. Healing is then determined by equilibrium swap rates.

Figure 8.
The asymptotic approach of the number of bonds in the region around the cut () to its bulk equilibrium value. is the number of bonds in the region . It is normalized by the average number of bonds at equilibrium. The same quantity is underestimated by the diffusion with the maximum D corresponding to . This causes discrepancies between the actual healing and the one predicted by a purely diffusive argument. Even when swaps are hindered by energy barriers, some bridges emerge quickly across the cut, initiating a slower healing process.
Figure 8.
The asymptotic approach of the number of bonds in the region around the cut () to its bulk equilibrium value. is the number of bonds in the region . It is normalized by the average number of bonds at equilibrium. The same quantity is underestimated by the diffusion with the maximum D corresponding to . This causes discrepancies between the actual healing and the one predicted by a purely diffusive argument. Even when swaps are hindered by energy barriers, some bridges emerge quickly across the cut, initiating a slower healing process.

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ciarella, S.; Ellenbroek, W.G. Swap-Driven Self-Adhesion and Healing of Vitrimers. Coatings 2019, 9, 114. https://doi.org/10.3390/coatings9020114
AMA Style
Ciarella S, Ellenbroek WG. Swap-Driven Self-Adhesion and Healing of Vitrimers. Coatings. 2019; 9(2):114. https://doi.org/10.3390/coatings9020114
Chicago/Turabian StyleCiarella, Simone, and Wouter G. Ellenbroek. 2019. "Swap-Driven Self-Adhesion and Healing of Vitrimers" Coatings 9, no. 2: 114. https://doi.org/10.3390/coatings9020114
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.