The Role of DNA Methylation in Common Skeletal Disorders

Department of Internal Medicine, H.U. Marqués de Valdecilla-IFIMAV-University of Cantabria, Santander 39008, Spain
*
Author to whom correspondence should be addressed.
Biology 2012, 1(3), 698-713; https://doi.org/10.3390/biology1030698
Submission received: 25 September 2012
/
Revised: 31 October 2012
/
Accepted: 16 November 2012
/
Published: 22 November 2012
(This article belongs to the Special Issue Gene Expression and Regulation)
Abstract
:Bone is a complex connective tissue characterized by a calcified extracellular matrix. This mineralized matrix is constantly being formed and resorbed throughout life, allowing the bone to adapt to daily mechanical loads and maintain skeletal properties and composition. The imbalance between bone formation and bone resorption leads to changes in bone mass. This is the case of osteoporosis and osteoarthritis, two common skeletal disorders. While osteoporosis is characterized by a decreased bone mass and, consequently, higher susceptibly to fractures, bone mass tends to be higher in patients with osteoarthritis, especially in the subchondral bone region. It is known that these diseases are influenced by heritable factors. However, the DNA polymorphisms identified so far in GWAS explain less than 10% of the genetic risk, suggesting that other factors, and specifically epigenetic mechanisms, are involved in the pathogenesis of these disorders. This review summarizes current knowledge about the influence of epigenetic marks on bone homeostasis, paying special attention to the role of DNA methylation in the onset and progression of osteoporosis and osteoarthritis.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Bone Cells and Bone Remodeling in Health and Disease
Bone is a complex connective tissue composed of a calcified extracellular matrix in which different cell types are embedded. The complexity of bone is particularly evident in the cells present in this tissue and the multifaceted interactions between them [1]. Bone cells belong to two different families: the osteoblastic and the osteoclastic families (see Figure 1) [2,3].
Figure 1.
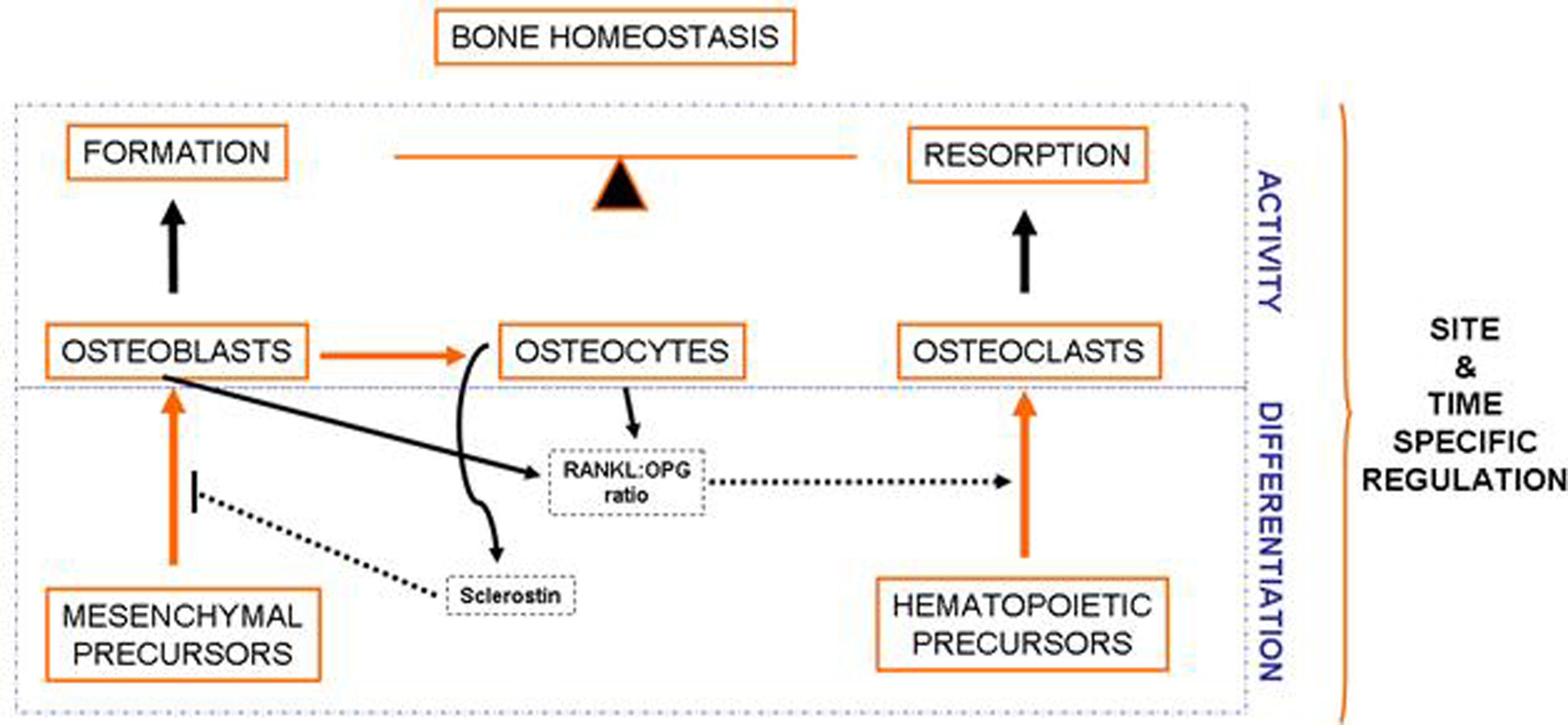
Bone is a complex and dynamic organ, which is constantly being remodeled by the balanced and coupled activity of the cells present in this tissue. Osteoblasts derive from mesenchymal precursors and eventually evolve into osteocytes and lining cells. Cells of the osteoblastic lineage are responsible for bone formation. Osteoclasts, derived from hematopoietic cells, are responsible for bone resorption. Interestingly, osteoclast differentiation is influenced by soluble factors secreted by osteoblasts and osteocytes, especially those related to the RANKL-RANK signaling pathway. On the other hand, osteocyte-derived sclerostin negatively modulates bone formation.
Figure 1.
Bone is a complex and dynamic organ, which is constantly being remodeled by the balanced and coupled activity of the cells present in this tissue. Osteoblasts derive from mesenchymal precursors and eventually evolve into osteocytes and lining cells. Cells of the osteoblastic lineage are responsible for bone formation. Osteoclasts, derived from hematopoietic cells, are responsible for bone resorption. Interestingly, osteoclast differentiation is influenced by soluble factors secreted by osteoblasts and osteocytes, especially those related to the RANKL-RANK signaling pathway. On the other hand, osteocyte-derived sclerostin negatively modulates bone formation.
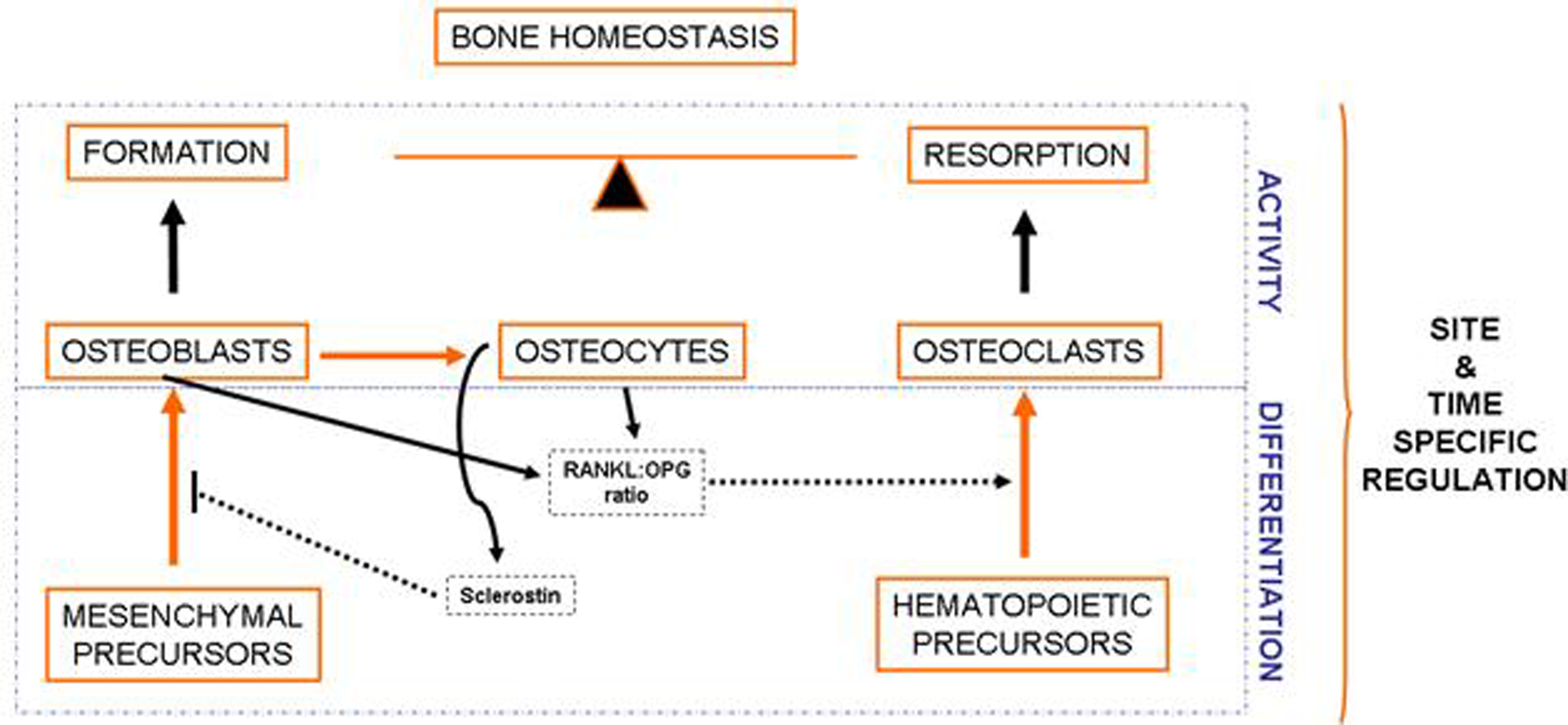
There are several cell types within the osteoblastic lineage, including osteoblasts, osteocytes and lining cells. All of them derive from mesenchymal precursors that differentiate into osteoblasts, which eventually evolve into osteocytes or lining cells [4]. Cells of the osteoblastic lineage modulate the proliferation and differentiation of cells belonging to the osteoclastic lineage, mainly by the RANKL-OPG-RANK signaling pathway [5,6,7]. Osteocytes derive from some osteoblasts that become embedded and surrounded by bone matrix [8]. These cells are emerging as the responders to mechanical stimuli that regulate bone formation and resorption, as well as key regulators of bone metabolism. Osteocytes modulate bone turnover through the modulation of the Wnt pathway and other pathways that influence the activity of both osteoblasts and osteoclasts [9,10,11].
Osteoclasts derive from hematopoietic precursors. These cells are formed by the fusion of cells of the monocyte-macrophage lineage and are responsible for bone resorption. As mentioned before, osteoclastogenesis is influenced by osteoblasts and osteocytes, which produce several factors critical for the differentiation of osteoclast precursors, such as the receptor activator of nuclear factor kappa-B ligand (RANKL) and the macrophage colony-stimulating factor (MCS-F) [7]. RANKL interacts with RANK (Receptor activator of nuclear factor kappa-B), present in the membrane of osteoclastic precursors, promoting the activation of the Nuclear factor Kappa B, which induces cell fusion and differentiation to originate multinucleated mature osteoclasts [12,13]. It is important to note that osteoblastic cells also express osteoprotegerin (OPG), which acts as a soluble decoy receptor for RANKL, thus impairing the RANKL-RANK interaction [14]. Mature osteoclasts locate on specific surfaces of bone, where they break down and resorb bone matrix, replacing old bone with new bone [12].
Bone is under constant turnover throughout life in order to maintain its properties. It is believed that this process, known as bone remodeling, occurs, in part, randomly. However, sometimes, osteocytes can mark the site where a remodeling cycle must be started in order to repair microcracks, which represent the so called “targeted remodeling” [15,16]. Bone remodeling is carried out by the cyclic and coupled activity of osteoclasts and osteoblasts. The process starts when osteoclast precursors are recruited to the bone surface, where they differentiate and remove a small volume of bone. Then osteoclasts undergo apoptosis and osteoblasts arrive to the region to fill up the defect with new bone [17]. Bone mass homeostasis depends on the balance between bone formation and bone resorption. Any disequilibrium between bone formation and bone resorption leads to changes in bone mass. This is the case of osteoporosis and osteoarthritis, two common skeletal diseases that tend to show changes of bone mass in opposite directions [18].
The maintenance of bone mass requires proper cell differentiation and cell activity. Osteoblast and osteoclast differentiation processes are highly organized and driven by deep changes in the gene expression patterns that in turn result in cells with different shapes and functions [3,19]. Since bone remodeling requires the sequential action of osteoclasts and osteoblasts at a given region, the differentiation of both cell types is controlled in a time and site-specific manner, and influenced not only by intrinsic factors, but also by some systemic and environmental factors. Emerging lines of evidence suggest that epigenetic mechanisms play an important role in establishing cellular identities and controlling gene expression. Herein, we summarize the current knowledge about the role of epigenetics, and specifically DNA methylation marks, in bone homeostasis and pathogenesis.
2. DNA Methylation Influences Gene Expression
Presently, epigenetics is defined as, “The study of stable genetic modifications that results in changes in gene expression without a corresponding alteration in DNA sequence [20]”. Importantly, epigenetic marks integrate intrinsic and environmental stimuli and confer both lineage commitment and phenotypic plasticity [21]. Thus, epigenetic marks could be considered as a link between genotype, environment, phenotype and disease. Epigenetic mechanisms comprise DNA methylation, post-translational histone modifications and non-coding RNAs [22,23]. Although the mechanisms of epigenetic inheritance during cell division are well established, heritable epigenetic patterns from parents to offspring are starting to be revealed [24,25].
In eukaryotes DNA methylation consists in the covalent addition of methyl groups to cytosines that precede guanines (CpG) [26]. In vertebrate genomes, CpG sites are predominantly methylated [27]. However, the globally methylated pattern is disrupted in regions known as CpG islands. These areas are present in approximately 70% of gene promoters. On average, CpG islands are 1 kb long, have an elevated C + G content and are frequently demethylated [28]. DNA methylation variations do not occur exclusively at CpG islands. Recently, it has been also shown that methylation occurs at a short distance from the CpG islands (at “CpG island shores”), rather than in the islands themselves [29]. The term CpG island shore refers to regions of lower CpG density that lie in the vicinity (~2 kb) of CpG islands. The addition of methyl groups to cytosines is catalyzed by DNA methyltransferases (DNMTs) [30]. DNMT1 was the first methyltransferase identified in mammals [31]. DNMT1 maintains DNA methylation during cell division by reading and copying the pattern in the hemimethylated strand [31]. Other members of the family are DNMT3A, DNMT3B, responsible for de novo methylation [32], and DNMT2, which has been recently shown to methylate tRNAs [33]. DNA methylation is considered an efficient repressor of transcriptional activity. Until recently, it was commonly thought that methyl groups directly prevent the binding of essential transcription factors to their targets. Although this is true for a specific set of transcription factors, it is not a general phenomenon. In fact, the binding sites for many transcription factors do not have CpGs. Emerging evidences support that the presence of methyl groups models the surrounding chromatin, inducing a DNA conformation less accessible to the transcription machinery. The mechanisms underlying this packing change are not fully understood yet, but many studies have concentrated on nucleosome structure, methyl-binding proteins, such as MECP2, MBD2 and MBD3, and interactions with chromatin remodeling enzymes [34,35,36]. Whatever the mechanism, on the basis of its potential to silence promoters, DNA methylation is supposed to play an important role in cell commitment and cell-specific gene expression.
How methylation is specifically targeted to a subset of promoters is generating intense debate. Apparently, methylation patterns are established early in the embryo [32,37,38]. Several studies support the idea that the ultimate methylation profile is determined by the underlying DNA sequence. In this sense, it has been shown that local DNA sequence is one of the main determinants for targeting DNA methylation to a specific locus. Thus, sequence variation between individuals might contribute to differential methylation patterns [38,39]. In fact, recent findings suggest that allele-specific methylation (ASM) is a common feature across the genome [40,41]. Notably, most of the ASM is strongly associated with SNPs genotypes [42,43]. Subsequent changes in the methylation pattern, generally of a tissue-specific nature, occur following implantation (i.e., repression of pluripotent genes) [44,45]. It has been proposed that tissue-specific changes occur through mechanisms apparently recruiting molecules needed for de novo methylation and demethylation (see Figure 2). Whereas demethylation may occur by active or repairing mechanisms [46,47], de novo methylation may be mediated by polycomb complexes [48].
3. Role of DNA Methylation in Establishing a Bone Cell Phenotype
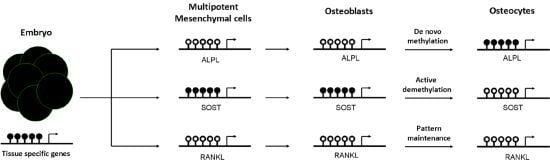
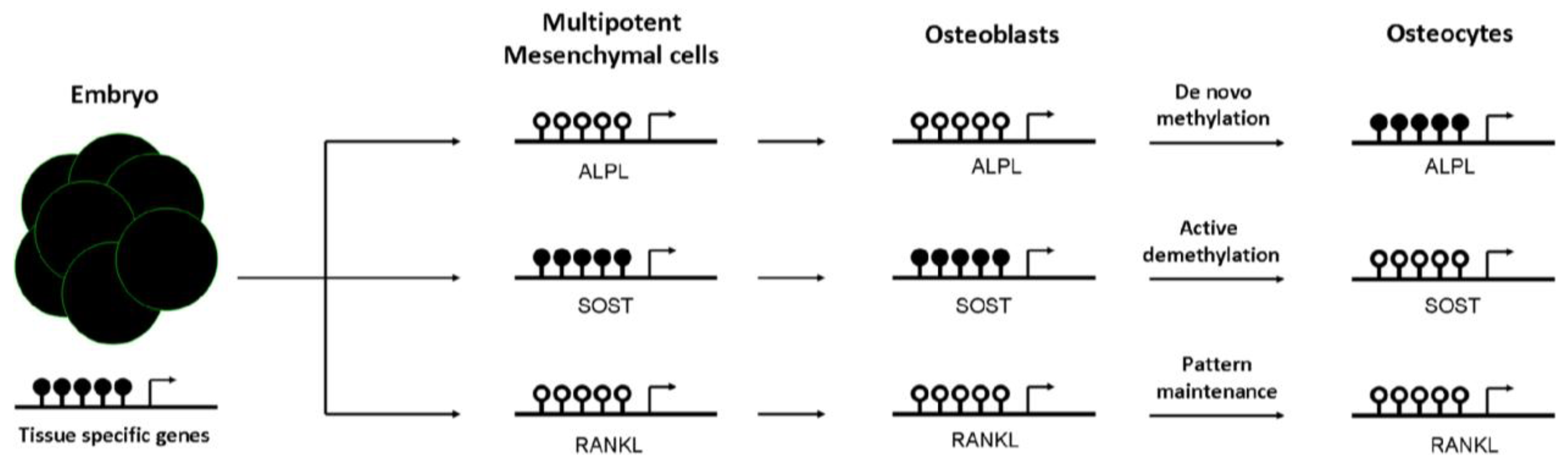
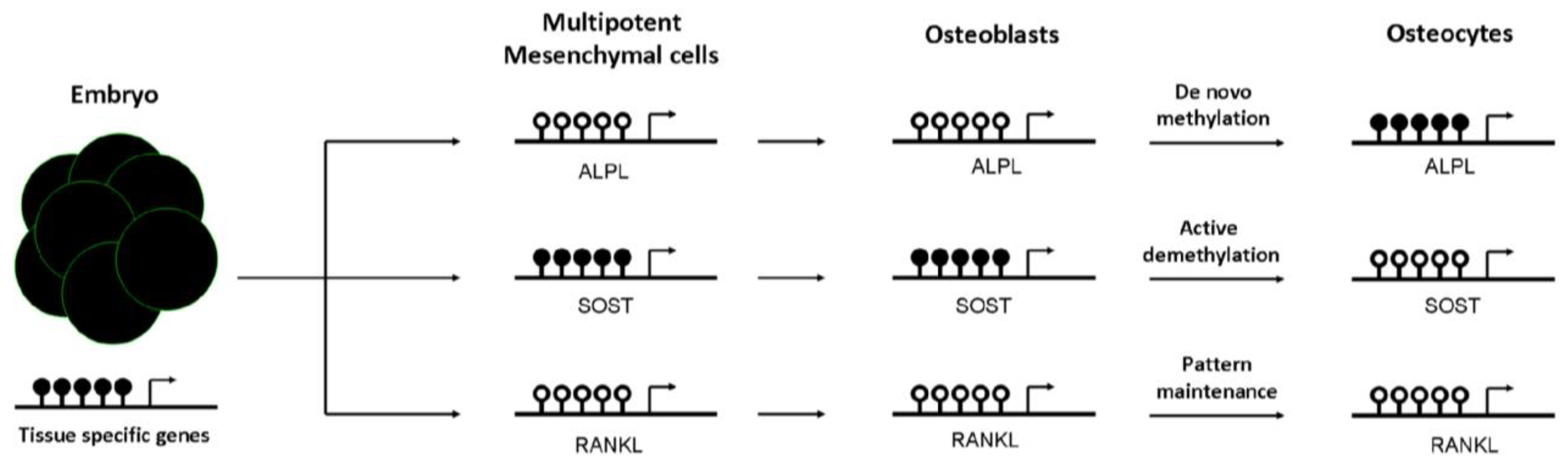
An impaired mesenchymal differentiation negatively affects bone mass. Several studies suggest that the osteogenic capacity of mesenchymal cells decrease with aging [49]. However, the contribution of this event to the decline of bone mass associated with aging is not clear yet. Osteogenic differentiation of mesenchymal cells towards osteoprogenitor and osteoblastic cells is regulated by several mechanisms, including DNA methylation. Kang et al. demonstrated that promoter methylation changes during mesenchymal cell differentiation [50]. Likewise, it has been proposed that active demethylation of gene promoters (i.e., osteocalcin, osterix, or Runx2), via GADD45-dependent mechanisms, is involved in the ostegenic differentiation of mesenchymal cells [51]. In fact, as reported by Locklin et al., osteogenic differentiation may be modulated by demethylating agents [52]. DNA methylation marks are not only important in osteoblastogenesis, but also afterwards in the osteoblast to osteocyte transition. Our group demonstrated that CpG methylation at regulatory regions controls the expression of the alkaline phosphatase and sclerostin genes. We observed that the hypermethylation of ALPL and SOST promoters was inversely correlated with gene expression in osteoblastic cells. Furthermore, we showed that the presence of methyl groups at the proximal promoter of SOST markedly decreased the transcriptional activity of this sequence, presumably by impairing the binding of essential transcription factors to the core promoter. In addition, we demonstrated that the methylation of those promoters changes during osteoblast differentiation towards osteocytes and controls gene expression in a cell-specific manner [53,54]. DNA methylation at ALPL promoter increased progressively during osteoblast differentiation, silencing ALPL expression in osteocytes. DNA methylation represses SOST expression in osteoblasts, whereas the physiological demethylation of its promoter favored the expression of this gene in osteocytes (see Figure 2). Consistent with this observation, SOST promoter remains methylated in other cell types that do not express sclerostin [53]. The results of other investigators also support that the expression of a number of genes important for osteogenic differentiation and osteoblast/osteocyte activity (including podoplanin, osteopontin, Brachury transcription factor, estrogen receptor, aromatase, collagen cross-linking enzyme lysyl oxidase or the homeobox protein Dlx-5) is regulated by DNA methylation [55,56,57,58,59,60,61,62].
Figure 2.
Dynamics of tissue-specific DNA methylation of bone cells. DNA methylation is established early in the embryo. Generally, at this stage, pluripotent genes and house-keeping genes are unmethylated, whereas tissue-specific genes are largely methylated. Then, the differentiation to mesenchymal cells promotes not only demethylation of many of these specific cell lineage genes, but also de novo methylation events to silence multipotent-specific genes. During mesenchymal commitment to the osteoblastic lineage, some unmethylated genes undergo de novo methylation to be silenced in osteocytes, such as alkaline phosphatase gene (ALPL). However, other methylated genes are actively demethylated to promote its expression in the late osteocyte [sclerostin (SOST)]. Lastly, other genes already demethylated in mesenchymal cells remain demethylated in differentiated cells, such as the Receptor activator of nuclear factor kappa-B ligand (RANKL). Black and white circles represent DNA methylation and hypomethylation, respectively.
Figure 2.
Dynamics of tissue-specific DNA methylation of bone cells. DNA methylation is established early in the embryo. Generally, at this stage, pluripotent genes and house-keeping genes are unmethylated, whereas tissue-specific genes are largely methylated. Then, the differentiation to mesenchymal cells promotes not only demethylation of many of these specific cell lineage genes, but also de novo methylation events to silence multipotent-specific genes. During mesenchymal commitment to the osteoblastic lineage, some unmethylated genes undergo de novo methylation to be silenced in osteocytes, such as alkaline phosphatase gene (ALPL). However, other methylated genes are actively demethylated to promote its expression in the late osteocyte [sclerostin (SOST)]. Lastly, other genes already demethylated in mesenchymal cells remain demethylated in differentiated cells, such as the Receptor activator of nuclear factor kappa-B ligand (RANKL). Black and white circles represent DNA methylation and hypomethylation, respectively.
On the other hand, DNA methylation contributes to regulate self-renewal and differentiation capacity of hematopoietic cells, the early precursors of osteoclasts [63]. The subsequent differentiation of osteoclast precursors towards mature osteoclasts is tightly regulated by soluble factors secreted by osteoblasts and osteocytes, such as RANKL, OPG and MCS-F [7,64]. As first reported by Kitazawa et al., and recently confirmed by our group in human samples, RANKL and OPG expression is regulated by DNA methylation in osteoblastic cells [65,66]. CpG methylation at the regulatory regions of RANKL and OPG genes is associated with low transcript levels. In turn, the demethylation of their promoters mediated by 5-azadeoxycitidine, a demethylating agent, induces the expression of both genes. Based upon these evidences, epigenetic mechanisms appear to be important for osteoclast differentiation (reviewed by Yasui et al. [67]). However, it is worth emphasizing that besides DNA methylation, other epigenetic marks, such as microRNAs or chromatin modifiers, are also involved in determining the differentiation and activity of bone cells, recently reviewed by Delgado-Calle et al., Kato S et al. and Earl CS et al. [68,69,70].
4. Methylation Marks and Common Skeletal Diseases
4.1. Osteoporosis
Osteoporosis is characterized by reduced bone mass and/or abnormal bone microarchitecture, which decrease bone strength and augment the susceptibility to fracture. Common osteoporotic fractures include those of the vertebral bodies, hip, pelvis, proximal arm and wrist. Any imbalance in the activity of osteoclasts and osteoblasts in a way such that bone formation is smaller than bone resorption results in osteoporosis. In some cases, osteoporosis is secondary to an underlying disease, but quite often it is just an exaggeration of the universal age-associated decrease in bone mass. In part, it is related to the diminished availability of sex steroids taking place in women after menopause and in elderly men [71], but other incompletely known factors associated with aging are likely involved [49,72]. Whatever those mechanisms might be, osteoporosis is the final result of the complex interplay between genetic and acquired factors in which epigenetic marks may also participate.
As discussed above, experimental evidence shows that the methylation of several genes play a major role in the differentiation of bone cells, which is required to sustain a normal bone remodeling. Therefore, it might be hypothesized that DNA methylation is involved in the pathogenesis of osteoporosis, though there is little evidence directly supporting this hypothesis so far. However, it has been recently proposed that reduced Dnmt1 activity decreases bone mineral density and body weight [73]. A number of studies suggest that environmental influences may contribute to shaping the methylation pattern of the individual and that the pattern may change in association with aging [74,75,76,77,78]. Several animal studies have related the environmental factors during early phases of development with DNA methylation and skeletal status. In fact, maternal dietary intake has been shown to influence bone mass of the offspring, both in experimental animals and in humans [79]. In some cases, DNA methylation may be involved. For instance, dietary restriction of pregnant rats induces changes in the methylation of genes that are important for bone cell differentiation and activity, such as the glucocorticoid receptor and the peroxisomal-activated receptor genes [80,81,82,83].
4.2. Osteoarthritis
Whereas osteoporosis is primarily a bone disorder, osteoarthritis is the most common form of joint disease. In fact, damage of the articular cartilage is the hallmark of osteoarthritis. However, osteoarthritis involves an abnormal remodeling of other tissues in affected joints, such as the synovium and the subchondral bone [84,85]. Thus, other typical changes in the joints of patients with osteoarthritis (besides the narrowing of joint space secondary to cartilage thinning), include the formation of osteophytes (bone excrescences at the periphery of the joints) and sclerosis of the subchondral bone. The extracellular matrix of the articular cartilage contains several types of collagen (II, IX and XI) and proteoglycanes, such as aggrecan. Several investigators have shown an altered homeostasis in the diseased cartilage, with increased expression of catabolic genes, accompanied by a diminished synthesis of components of the cartilaginous matrix. Matrix metalloproteases (MMPs) and aggrecanases (ADAMTS-4 and 5) are regarded as major enzymes mediating the destructive process of cartilage. The sclerosis of subchondral bone used to be considered a secondary reactive change, but in recent years, the concept is emerging that subchondral bone may play more than a passive role in the pathogenesis of the disease. In line with this concept, it has been postulated that cytokines released by bone cells influence the activity of chondrocytes, and vice-versa [86]. Furthermore, subchondral bone influences the overlying cartilage, not only through its biomechanical properties, but also through the synthesis of various humoral factors (see recent reviews [86,87,88,89,90]).
After the seminal work by Roach et al. [91,92], it has been demonstrated that, in some cases, changes in chondrocyte gene expression are associated with inverse changes in DNA methylation. For instance, Zimmerman et al. reported that the induction of type X collagen expression during chondrogenic differentiation of mesenchymal stem cells is associated with the demethylation of specific CpGs in its promoter [93]. However, in adult chondrocytes, the promoter was methylated, which correlated with the absence of gene expression. Inverse correlations between methylation and gene expression have been reported for other genes, including osteogenic protein 1 (OP-1), interleukin 1 beta, MMP3, MMP9, MMP13, leptin and ADMTS4 [58,94,95]. The exact molecular mechanisms have not been completely elucidated, but in some cases they may include methylation-dependent differences in the ability to recruit transcription factors, such as CREB, to gene regulatory regions [96]. However, gene expression and DNA methylation are not always inversely correlated. Thus, the reduced expression of genes, such as type II collagen or aggrecan reported in diseased cartilage, does not seem to be associated with increased methylation of these genes [94,97]. On the other hand, the functional consequences of gene methylation have also been revealed by in vitro experiments, showing that inducing DNA demethylation by 5-azadeoxycitidine modulates the differentiation of articular chondrocytes [98].
Some data suggest that the shape of the bones may also influence osteoarthritis. For instance, epidemiological studies showed that the certain morphological characteristics of the femoral epiphysis and the pelvis determine the risk of hip osteoarthritis [99,100,101]. This has given support to the hypothesis of a developmental origin of osteoarthritis [102,103,104]. The importance of developmental factors is also emphasized by recent results from genome-wide association studies [105,106]. Somewhat unexpectedly, these studies have not revealed significant associations between osteoarthritis and genes typically involved in cartilage homeostasis, such as those encoding proteases. Instead, they have shown association between some polymorphisms of genes involved in joint development, such as GDF5, and osteoarthritis of the large joints, particularly the knee and the hip [107,108]. It is currently thought that the epigenome is the consequence of environmental factors, genetic features and stochastic variations. In this regard, it is interesting to note that some GDF5 single nucleotide polymorphisms (SNPs) showed differential allelic expression. Interestingly, the functional effect of these “CpGs SNPs” on gene expression is modulated by DNA methylation [108].
Our group has recently published a genome-wide methylation study in bone samples obtained from patients with severe hip osteoarthritis and compared the results with those obtained in patients with osteoporotic hip fractures [109]. Our results revealed several genes showing differential methylation between osteoporotic and osteoarthritic patients. Somewhat unexpectedly, genes showing differential methylation where not typical bone candidates. However, functional network analysis revealed that epigenetic changes in “upstream” regulatory genes may have downstream influences on well-known bone-related genes. Interestingly, and in tune with the developmental hypothesis, we found that genes showing differential methylation were overrepresented in pathways related to skeletal development, and particularly those of the homeobox family. Globally, DNA methylation correlated negatively with gene expression in both groups of patients. However, as observed in other studies, we found a subset of genes in which there was no inverse correlation between DNA methylation and the abundance of gene transcripts, thus pointing out that factors other than methylation play an important role in the fine tuning of gene expression in adult tissues.
4.3. Tumors and Bone
Some tumors originate in the skeleton, and many others have a propensity to metastasize in bone. In fact, bone metastases are very common in a variety of advanced cancers and contribute importantly to cancer morbidity. The mechanisms influencing the metastatic potential of tumor cells and their tropism for certain tissues are being actively investigated. They are likely complex and include factors related to the tumor itself and others which depend on the tissue hosting the metastases. In some cases, they may include certain methylation patterns that result in specific gene expression signatures that facilitate the initiation or the growth of the metastases. Prostate cancer cells are among those with a stronger tropism for bone. Saha et al. reported that the hypomethylation of the E-cadherin gene, and the subsequent reduction of gene expression, was associated with metastatic prostate cancer cells in bone [110]. Also, reduced DNA methylation is associated with increased expression of the parathyroid hormone-related protein (PTHrP) by breast cancer cells, which in turn may facilitate osteolysis and the growth of the metastatic niche [111]. On the other hand, the fact that certain tumors have specific gene methylation patterns, different from those of normal tissues, has raised the possibility of using the analysis of DNA remnants present in serum as a biomarker to help in diagnosing specific types of cancer.
5. Concluding Remarks
Common skeletal disorders, such as osteoporosis and osteoarthritis, are the result of a complex interplay of genetic and acquired factors. However, despite tremendous efforts, including several GWAS, the genetic factors so far identified explain less than 10% of the genetic risk. This suggests that mechanisms not related to DNA sequence may be involved in the development of these diseases. This could be the case of DNA methylation and its mediators. DNA methylation marks are heritable, at least through cell divisions, control gene response to environment, change with aging and underlie cell commitment and the spatiotemporal control of gene expression. DNA methylation plays an important role in the differentiation of cells of the osteoblastic and osteoclastic lineages. Therefore, it is tempting to speculate that the aberrant phenotypes observed in bone diseases might be the consequence of a combination of intrinsic and environmental factors, including gene sequence variations and epigenetic signatures (see Figure 3). However, it is important to note that DNA methylation is only one of the mechanisms underlying gene expression. Thus, the integration of knowledge from both epigenenomics and genomics, together with other “omics” (i.e., transcriptomics, proteomics) will be essential for the full understanding of the underlying mechanisms that govern the initiation and progression of bone diseases. Although still a long way to go, further studies in bone epigenetics may open a new door for drug development combining genetic and epigenetic strategies.
Figure 3.
Factors involved in epigenetic variability. Epigenetic marks can change throughout life and, thereby, determine the adult phenotypes of the cells present in bone. Several factors (i.e., starving) may influence epigenetic marks at different stages of intrauterus life. For instance, genetic inheritance might influence the de novo methylation that occurs before implantation. Likewise, stochastic variations and environmental cues may also influence the epigenetic pattern. During the post-natal life, epigenetic variability may depend on environmental (i.e., toxic habits, food...) and intrinsic factors (genetic predisposition), as well as on the occurrence of DNA somatic mutations. Epigenetic signatures are directly linked to the control of cell differentiation and gene expression, thus accumulated epigenetic variability can lead to aberrant phenotypes and, consequently, induce skeletal disease.
Figure 3.
Factors involved in epigenetic variability. Epigenetic marks can change throughout life and, thereby, determine the adult phenotypes of the cells present in bone. Several factors (i.e., starving) may influence epigenetic marks at different stages of intrauterus life. For instance, genetic inheritance might influence the de novo methylation that occurs before implantation. Likewise, stochastic variations and environmental cues may also influence the epigenetic pattern. During the post-natal life, epigenetic variability may depend on environmental (i.e., toxic habits, food...) and intrinsic factors (genetic predisposition), as well as on the occurrence of DNA somatic mutations. Epigenetic signatures are directly linked to the control of cell differentiation and gene expression, thus accumulated epigenetic variability can lead to aberrant phenotypes and, consequently, induce skeletal disease.

Acknowledgments
Work on epigenetics in our lab is supported by a grant from Instituto de Salud Carlos III-Fondo de Investigaciones Sanitarias (09/539). JDC is recipient of a contract from IFIMAV.
References
- Matsuo, K.; Irie, N. Osteoclast-osteoblast communication. Arch. Biochem. Biophys. 2008, 473, 201–209. [Google Scholar] [CrossRef]
- Jackson, L.; Jones, D.R.; Scotting, P.; Sottile, V. Adult mesenchymal stem cells: Differentiation potential and therapeutic applications. J. Postgrad. Med. 2007, 53, 121–127. [Google Scholar]
- Vaananen, H.K.; Zhao, H.; Mulari, M.; Halleen, J.M. The cell biology of osteoclast function. J. Cell. Sci. 2000, 113, 377–381. [Google Scholar]
- Dallas, S.L.; Bonewald, L.F. Dynamics of the transition from osteoblast to osteocyte. Ann. NY Acad. Sci. 2010, 1192, 437–443. [Google Scholar]
- Boyce, B.F.; Xing, L. Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch. Biochem. Biophys. 2008, 473, 139–146. [Google Scholar] [CrossRef]
- Nakashima, T.; Hayashi, M.; Fukunaga, T.; Kurata, K.; Oh-Hora, M.; Feng, J.Q.; Bonewald, L.F.; Kodama, T.; Wutz, A.; et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat. Med. 2011, 17, 1231–1234. [Google Scholar] [CrossRef]
- Xiong, J.; Onal, M.; Jilka, R.L.; Weinstein, R.S.; Manolagas, S.C.; O’Brien, C.A. Matrix-embedded cells control osteoclast formation. Nat. Med. 2011, 17, 1235–1241. [Google Scholar] [CrossRef]
- Bonewald, L.F. The amazing osteocyte. J. Bone Miner. Res. 2011, 26, 229–238. [Google Scholar] [CrossRef]
- Zarrinkalam, M.R.; Mulaibrahimovic, A.; Atkins, G.J.; Moore, R.J. Changes in osteocyte density correspond with changes in osteoblast and osteoclast activity in an osteoporotic sheep model. Osteoporos. Int. 2012, 23, 1329–1336. [Google Scholar]
- O’Brien, C.A.; Plotkin, L.I.; Galli, C.; Goellner, J.J.; Gortazar, A.R.; Allen, M.R.; Robling, A.G.; Bouxsein, M.; Schipani, E.; Turner, C.H.; et al. Control of bone mass and remodeling by PTH receptor signaling in osteocytes. PLoS One 2008, 3, e2942. [Google Scholar]
- Winkler, D.G.; Sutherland, M.K.; Geoghegan, J.C.; Yu, C.; Hayes, T.; Skonier, J.E.; Shpektor, D.; Jonas, M.; Kovacevich, B.R.; Staehling-Hampton, K.; et al. Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J. 2003, 22, 6267–6276. [Google Scholar]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nature 2003, 423, 337–342. [Google Scholar] [CrossRef]
- Boyce, B.F.; Xing, L. The RANKL/RANK/OPG pathway. Curr. Osteoporos. Rep. 2007, 5, 98–104. [Google Scholar] [CrossRef]
- Kong, Y.Y.; Yoshida, H.; Sarosi, I.; Tan, H.L.; Timms, E.; Capparelli, C.; Morony, S.; Oliveira-dos-Santos, A.J.; van, G.; Itie, A.; et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature 1999, 397, 315–323. [Google Scholar]
- Hadjidakis, D.J.; Androulakis, I.I. Bone remodeling. Ann. NY Acad. Sci. 2006, 1092, 385–396. [Google Scholar] [CrossRef]
- Sims, N.A.; Gooi, J.H. Bone remodeling: Multiple cellular interactions required for coupling of bone formation and resorption. Semin. Cell Dev. Biol. 2008, 19, 444–451. [Google Scholar]
- Raggatt, L.J.; Partridge, N.C. Cellular and molecular mechanisms of bone remodeling. J. Biol. Chem. 2010, 285, 25103–25108. [Google Scholar] [CrossRef]
- Dequeker, J.; Aerssens, J.; Luyten, F.P. Osteoarthritis and osteoporosis: Clinical and research evidence of inverse relationship. Aging Clin. Exp. Res. 2003, 15, 426–439. [Google Scholar]
- Franz-Odendaal, T.A.; Hall, B.K.; Witten, P.E. Buried alive: how osteoblasts become osteocytes. Dev. Dyn. 2006, 235, 176–190. [Google Scholar] [CrossRef]
- Probst, A.V.; Dunleavy, E.; Almouzni, G. Epigenetic inheritance during the cell cycle. Nat. Rev. Mol. Cell. Biol. 2009, 10, 192–206. [Google Scholar]
- Feinberg, A.P. Phenotypic plasticity and the epigenetics of human disease. Nature 2007, 447, 433–440. [Google Scholar] [CrossRef]
- Esteller, M. Epigenetics in cancer. N. Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Tycko, B. The history of cancer epigenetics. Nat. Rev. Cancer 2004, 4, 143–153. [Google Scholar] [CrossRef]
- Buiting, K.; Barnicoat, A.; Lich, C.; Pembrey, M.; Malcolm, S.; Horsthemke, B. Disruption of the bipartite imprinting center in a family with Angelman syndrome. Am. J. Hum. Genet. 2001, 68, 1290–1294. [Google Scholar]
- Buiting, K.; Gross, S.; Lich, C.; Gillessen-Kaesbach, G.; El Maarri, O.; Horsthemke, B. Epimutations in Prader-Willi and Angelman syndromes: A molecular study of 136 patients with an imprinting defect. Am. J. Hum. Genet. 2003, 72, 571–577. [Google Scholar] [CrossRef]
- Miranda, T.B.; Jones, P.A. DNA methylation: The nuts and bolts of repression. J. Cell. Physiol. 2007, 213, 384–390. [Google Scholar] [CrossRef]
- Bird, A.P. DNA methylation and the frequency of CpG in animal DNA. Nucleic Acids Res. 1980, 8, 1499–1504. [Google Scholar]
- Illingworth, R.S.; Gruenewald-Schneider, U.; Webb, S.; Kerr, A.R.; James, K.D.; Turner, D.J.; Smith, C.; Harrison, D.J.; Andrews, R.; Bird, A.P. Orphan CpG islands identify numerous conserved promoters in the mammalian genome. PLoS Genet. 2010, 6, e1001134. [Google Scholar]
- Irizarry, R.A.; Ladd-Acosta, C.; Wen, B.; Wu, Z.; Montano, C.; Onyango, P.; Cui, H.; Gabo, K.; Rongione, M.; Webster, M.; et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet. 2009, 41, 178–186. [Google Scholar] [CrossRef]
- Hermann, A.; Gowher, H.; Jeltsch, A. Biochemistry and biology of mammalian DNA methyltransferases. Cell. Mol. Life. Sci. 2004, 61, 2571–2587. [Google Scholar]
- Leonhardt, H.; Page, A.W.; Weier, H.U.; Bestor, T.H. A targeting sequence directs DNA methyltransferase to sites of DNA replication in mammalian nuclei. Cell 1992, 71, 865–873. [Google Scholar] [CrossRef] [Green Version]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef]
- Goll, M.G.; Kirpekar, F.; Maggert, K.A.; Yoder, J.A.; Hsieh, C.L.; Zhang, X.; Golic, K.G.; Jacobsen, S.E.; Bestor, T.H. Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science 2006, 311, 395–398. [Google Scholar]
- Hashimshony, T.; Zhang, J.; Keshet, I.; Bustin, M.; Cedar, H. The role of DNA methylation in setting up chromatin structure during development. Nat. Genet. 2003, 34, 187–192. [Google Scholar]
- Razin, A.; Cedar, H. Distribution of 5-methylcytosine in chromatin. Proc. Natl. Acad. Sci. USA 1977, 74, 2725–2728. [Google Scholar] [CrossRef]
- Klose, R.J.; Bird, A.P. Genomic DNA methylation: The mark and its mediators. Trends Biochem. Sci. 2006, 31, 89–97. [Google Scholar] [CrossRef]
- Frank, D.; Keshet, I.; Shani, M.; Levine, A.; Razin, A.; Cedar, H. Demethylation of CpG islands in embryonic cells. Nature 1991, 351, 239–241. [Google Scholar]
- Straussman, R.; Nejman, D.; Roberts, D.; Steinfeld, I.; Blum, B.; Benvenisty, N.; Simon, I.; Yakhini, Z.; Cedar, H. Developmental programming of CpG island methylation profiles in the human genome. Nat. Struct. Mol. Biol. 2009, 16, 564–571. [Google Scholar] [CrossRef]
- Lienert, F.; Wirbelauer, C.; Som, I.; Dean, A.; Mohn, F.; Schubeler, D. Identification of genetic elements that autonomously determine DNA methylation states. Nat. Genet. 2011, 43, 1091–1097. [Google Scholar]
- Epsztejn-Litman, S.; Feldman, N.; Abu-Remaileh, M.; Shufaro, Y.; Gerson, A.; Ueda, J.; Deplus, R.; Fuks, F.; Shinkai, Y.; Cedar, H.; et al. De novo DNA methylation promoted by G9a prevents reprogramming of embryonically silenced genes. Nat. Struct. Mol. Biol. 2008, 15, 1176–1183. [Google Scholar]
- Feldman, N.; Gerson, A.; Fang, J.; Li, E.; Zhang, Y.; Shinkai, Y.; Cedar, H.; Bergman, Y. G9a-mediated irreversible epigenetic inactivation of Oct-3/4 during early embryogenesis. Nat. Cell. Biol. 2006, 8, 188–194. [Google Scholar] [CrossRef]
- Niehrs, C.; Schafer, A. Active DNA demethylation by Gadd45 and DNA repair. Trends Cell Biol. 2012, 22, 220–227. [Google Scholar] [CrossRef]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef]
- Kerkel, K.; Spadola, A.; Yuan, E.; Kosek, J.; Jiang, L.; Hod, E.; Li, K.; Murty, V.V.; Schupf, N.; Vilain, E.; et al. Genomic surveys by methylation-sensitive SNP analysis identify sequence-dependent allele-specific DNA methylation. Nat. Genet. 2008, 40, 904–908. [Google Scholar] [CrossRef]
- Tycko, B. Allele-specific DNA methylation: Beyond imprinting. Hum. Mol. Genet. 2010, 19, R210–R220. [Google Scholar] [CrossRef]
- Bell, J.T.; Pai, A.A.; Pickrell, J.K.; Gaffney, D.J.; Pique-Regi, R.; Degner, J.F.; Gilad, Y.; Pritchard, J.K. DNA methylation patterns associate with genetic and gene expression variation in HapMap cell lines. Genome Biol. 2011, 12, R10. [Google Scholar]
- Hellman, A.; Chess, A. Extensive sequence-influenced DNA methylation polymorphism in the human genome. Epigenetics Chromatin 2010, 3, 11. [Google Scholar] [CrossRef]
- Vire, E.; Brenner, C.; Deplus, R.; Blanchon, L.; Fraga, M.; Didelot, C.; Morey, L.; van Eynde, A.; Bernard, D.; Vanderwinden, J.M.; et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 2006, 439, 871–874. [Google Scholar] [CrossRef]
- Jiang, Y.; Mishima, H.; Sakai, S.; Liu, Y.K.; Ohyabu, Y.; Uemura, T. Gene expression analysis of major lineage-defining factors in human bone marrow cells: Effect of aging, gender, and age-related disorder. J. Orthop. Res. 2008, 26, 910–917. [Google Scholar] [CrossRef]
- Kang, M.I.; Kim, H.S.; Jung, Y.C.; Kim, Y.H.; Hong, S.J.; Kim, M.K.; Baek, K.H.; Kim, C.C.; Rhyu, M.G. Transitional CpG methylation between promoters and retroelements of tissue-specific genes during human mesenchymal cell differentiation. J. Cell. Biochem. 2007, 102, 224–239. [Google Scholar] [CrossRef]
- Zhang, R.P.; Shao, J.Z.; Xiang, L.X. GADD45A protein plays an essential role in active DNA demethylation during terminal osteogenic differentiation of adipose-derived mesenchymal stem cells. J. Biol. Chem. 2011, 286, 41083–41094. [Google Scholar]
- Locklin, R.M.; Oreffo, R.O.; Triffitt, J.T. Modulation of osteogenic differentiation in human skeletal cells in vitro by 5-azacytidine. Cell Biol. Int. 1998, 22, 207–215. [Google Scholar] [CrossRef]
- Delgado-Calle, J.; Sanudo, C.; Bolado, A.; Fernandez, A.F.; Arozamena, J.; Pascual-Carra, M.A.; Rodriguez-Rey, J.C.; Fraga, M.F.; Bonewald, L.F.; Riancho, J.A. DNA methylation contributes to the regulation of sclerostin expression in human osteocytes. J. Bone Miner. Res. 2012, 27, 926–937. [Google Scholar] [CrossRef]
- Delgado-Calle, J.; Sanudo, C.; Sanchez-Verde, L.; Garcia-Renedo, R.J.; Arozamena, J.; Riancho, J.A. Epigenetic regulation of alkaline phosphatase in human cells of the osteoblastic lineage. Bone 2011, 49, 830–838. [Google Scholar] [CrossRef]
- Arnsdorf, E.J.; Tummala, P.; Castillo, A.B.; Zhang, F.; Jacobs, C.R. The epigenetic mechanism of mechanically induced osteogenic differentiation. J. Biomech. 2010, 43, 2881–2886. [Google Scholar] [CrossRef]
- Villagra, A.; Gutierrez, J.; Paredes, R.; Sierra, J.; Puchi, M.; Imschenetzky, M.; Wijnen, A.A.; Lian, J.; Stein, G.; Stein, J.; et al. Reduced CpG methylation is associated with transcriptional activation of the bone-specific rat osteocalcin gene in osteoblasts. J. Cell Biochem. 2002, 85, 112–122. [Google Scholar] [CrossRef]
- Dansranjavin, T.; Krehl, S.; Mueller, T.; Mueller, L.P.; Schmoll, H.J.; Dammann, R.H. The role of promoter CpG methylation in the epigenetic control of stem cell related genes during differentiation. Cell Cycle 2009, 8, 916–924. [Google Scholar] [CrossRef]
- Loeser, R.F.; Im, H.J.; Richardson, B.; Lu, Q.; Chubinskaya, S. Methylation of the OP-1 promoter: Potential role in the age-related decline in OP-1 expression in cartilage. Osteoarthr. Cartil. 2009, 17, 513–517. [Google Scholar]
- Lee, J.Y.; Lee, Y.M.; Kim, M.J.; Choi, J.Y.; Park, E.K.; Kim, S.Y.; Lee, S.P.; Yang, J.S.; Kim, D.S. Methylation of the mouse DIx5 and Osx gene promoters regulates cell type-specific gene expression. Mol. Cells 2006, 22, 182–188. [Google Scholar]
- Penolazzi, L.; Lambertini, E.; Giordano, S.; Sollazzo, V.; Traina, G.; del Senno, L.; Piva, R. Methylation analysis of the promoter F of estrogen receptor alpha gene: Effects on the level of transcription on human osteoblastic cells. J. Steroid Biochem.Mol. Biol. 2004, 91, 1–9. [Google Scholar] [CrossRef]
- Demura, M.; Bulun, S.E. CpG dinucleotide methylation of the CYP19 I.3/II promoter modulates cAMP-stimulated aromatase activity. Mol. Cell Endocrinol. 2008, 283, 127–132. [Google Scholar] [CrossRef]
- Thaler, R.; Agsten, M.; Spitzer, S.; Paschalis, E.P.; Karlic, H.; Klaushofer, K.; Varga, F. Homocysteine suppresses the expression of the collagen cross-linker lysyl oxidase involving IL-6, Fli1, and epigenetic DNA methylation. J. Biol. Chem. 2011, 286, 5578–5588. [Google Scholar]
- Teitell, M.A.; Mikkola, H.K. Transcriptional activators, repressors, and epigenetic modifiers controlling hematopoietic stem cell development. Pediatr. Res. 2006, 59, 33–39. [Google Scholar] [CrossRef]
- Riancho, J.A.; Delgado-Calle, J. Osteoblast-osteoclast interaction mechanisms. Reumatol. Clin. 2011, 7, S1–S4. [Google Scholar]
- Delgado-Calle, J.; Sanudo, C.; Fernandez, A.F.; Garcia-Renedo, R.; Fraga, M.F.; Riancho, J.A. Role of DNA methylation in the regulation of the RANKL-OPG system in human bone. Epigenetics 2012, 7, 83–91. [Google Scholar] [CrossRef]
- Kitazawa, R.; Kitazawa, S. Methylation status of a single CpG locus 3 bases upstream of TATA-box of receptor activator of nuclear factor-kappaB ligand RANKL. Gene promoter modulates cell- and tissue-specific RANKL expression and osteoclastogenesis. Mol. Endocrinol. 2007, 21, 148–158. [Google Scholar]
- Yasui, T.; Hirose, J.; Aburatani, H.; Tanaka, S. Epigenetic regulation of osteoclast differentiation. Ann. NY Acad. Sci. 2011, 1240, 7–13. [Google Scholar]
- Delgado-Calle, J.; Garmilla, P.; Riancho, J.A. Do epigenetic marks govern bone homeostasis? Curr. Genomics 2012, 13, 252–263. [Google Scholar]
- Kato, S.; Inoue, K.; Youn, M.I. Emergence of the osteo-epigenome in bone biology. IBMS BoneKEy 2010, 7, 314–324. [Google Scholar] [CrossRef]
- Earl, S.C.; Harvey, N.; Cooper, C. The epigenetic regulation of bone mass. IBMS BoneKEy 2010, 7, 54–62. [Google Scholar] [CrossRef]
- Riggs, B.L.; Khosla, S.; Melton, L.J. Sex steroids and the construction and conservation of the adult skeleton. Endocr. Rev. 2002, 23, 279–302. [Google Scholar] [CrossRef]
- Manolagas, S.C. From estrogen-centric to aging and oxidative stress: A revised perspective of the pathogenesis of osteoporosis. Endocr. Rev. 2010, 31, 266–300. [Google Scholar] [CrossRef]
- Liu, L.; van Groen, T.; Kadish, I.; Li, Y.; Wang, D.; James, S.R.; Karpf, A.R.; Tollefsbol, T.O. Insufficient DNA methylation affects healthy aging and promotes age-related health problems. Clin. Epigenetics 2011, 2, 349–360. [Google Scholar] [CrossRef]
- Rodriguez-Rodero, S.; Fernandez-Morera, J.L.; Fernandez, A.F.; Menendez-Torre, E.; Fraga, M.F. Epigenetic regulation of aging. Discov. Med. 2010, 10, 225–233. [Google Scholar]
- Fraga, M.F.; Esteller, M. Epigenetics and aging: The targets and the marks. Trends Genet. 2007, 23, 413–418. [Google Scholar] [CrossRef]
- Fraga, M.F. Genetic and epigenetic regulation of aging. Curr. Opin. Immunol. 2009, 21, 446–453. [Google Scholar] [CrossRef]
- Calvanese, V.; Lara, E.; Kahn, A.; Fraga, M.F. The role of epigenetics in aging and age-related diseases. Ageing Res. Rev. 2009, 8, 268–276. [Google Scholar]
- Huidobro, C.; Fernandez, A.F.; Fraga, M.F. Aging epigenetics: Causes and consequences. Mol. Asp. Med. 2012, in press. [Google Scholar]
- Mahon, P.; Harvey, N.; Crozier, S.; Inskip, H.; Robinson, S.; Arden, N.; Swaminathan, R.; Cooper, C.; Godfrey, K. Low maternal vitamin D status and fetal bone development: Cohort study. J. Bone Miner. Res. 2010, 25, 14–19. [Google Scholar] [CrossRef]
- Oreffo, R.O.; Lashbrooke, B.; Roach, H.I.; Clarke, N.M.; Cooper, C. Maternal protein deficiency affects mesenchymal stem cell activity in the developing offspring. Bone 2003, 33, 100–107. [Google Scholar] [CrossRef]
- Lillycrop, K.A.; Phillips, E.S.; Torrens, C.; Hanson, M.A.; Jackson, A.A.; Burdge, G.C. Feeding pregnant rats a protein-restricted diet persistently alters the methylation of specific cytosines in the hepatic PPAR alpha promoter of the offspring. Br. J. Nutr. 2008, 100, 278–282. [Google Scholar]
- Lillycrop, K.A.; Slater-Jefferies, J.L.; Hanson, M.A.; Godfrey, K.M.; Jackson, A.A.; Burdge, G.C. Induction of altered epigenetic regulation of the hepatic glucocorticoid receptor in the offspring of rats fed a protein-restricted diet during pregnancy suggests that reduced DNA methyltransferase-1 expression is involved in impaired DNA methylation and changes in histone modifications. Br. J. Nutr. 2007, 97, 1064–1073. [Google Scholar] [CrossRef]
- Lillycrop, K.A.; Phillips, E.S.; Jackson, A.A.; Hanson, M.A.; Burdge, G.C. Dietary protein restriction of pregnant rats induces and folic acid supplementation prevents epigenetic modification of hepatic gene expression in the offspring. J. Nutr. 2005, 135, 1382–1386. [Google Scholar]
- Brandt, K.D.; Dieppe, P.; Radin, E.L. Etiopathogenesis of osteoarthritis. Rheum. Dis. Clin. North Am. 2008, 34, 531–559. [Google Scholar]
- Loeser, R.F.; Goldring, S.R.; Scanzello, C.R.; Goldring, M.B. Osteoarthritis: A disease of the joint as an organ. Arthritis Rheum. 2012, 64, 1697–1707. [Google Scholar]
- Goldring, S.R. The role of bone in osteoarthritis pathogenesis. Rheum.Dis. Clin. North Am. 2008, 34, 561–571. [Google Scholar] [CrossRef]
- Bellido, M.; Lugo, L.; Roman-Blas, J.A.; Castaneda, S.; Calvo, E.; Largo, R.; Herrero-Beaumont, G. Improving subchondral bone integrity reduces progression of cartilage damage in experimental osteoarthritis preceded by osteoporosis. Osteoarthr. Cartil. 2011, 19, 1228–1236. [Google Scholar] [CrossRef]
- Goldring, M.B.; Goldring, S.R. Articular cartilage and subchondral bone in the pathogenesis of osteoarthritis. Ann. NY Acad. Sci. 2010, 1192, 230–237. [Google Scholar] [CrossRef]
- Herrero-Beaumont, G.; Roman-Blas, J.A.; Largo, R.; Berenbaum, F.; Castaneda, S. Bone mineral density and joint cartilage: Four clinical settings of a complex relationship in osteoarthritis. Ann.Rheum. Dis. 2011, 70, 1523–1525. [Google Scholar] [CrossRef]
- Suri, S.; Walsh, D.A. Osteochondral alterations in osteoarthritis. Bone 2012, 51, 204–211. [Google Scholar] [CrossRef]
- Roach, H.I.; Yamada, N.; Cheung, K.S.; Tilley, S.; Clarke, N.M.; Oreffo, R.O.; Kokubun, S.; Bronner, F. Association between the abnormal expression of matrix-degrading enzymes by human osteoarthritic chondrocytes and demethylation of specific CpG sites in the promoter regions. Arthritis Rheum. 2005, 52, 3110–3124. [Google Scholar] [CrossRef]
- Roach, H.I.; Aigner, T. DNA methylation in osteoarthritic chondrocytes: A new molecular target. Osteoarthr. Cartil. 2007, 15, 128–137. [Google Scholar] [CrossRef]
- Zimmermann, P.; Boeuf, S.; Dickhut, A.; Boehmer, S.; Olek, S.; Richter, W. Correlation of COL10A1 induction during chondrogenesis of mesenchymal stem cells with demethylation of two CpG sites in the COL10A1 promoter. Arthritis Rheum. 2008, 589, 2743–2753. [Google Scholar]
- Barter, M.J.; Bui, C.; Young, D.A. Epigenetic mechanisms in cartilage and osteoarthritis: DNA methylation, histone modifications and microRNAs. Osteoarthr. Cartil. 2012, 20, 339–349. [Google Scholar]
- Goldring, M.B.; Marcu, K.B. Epigenomic and microRNA-mediated regulation in cartilage development, homeostasis, and osteoarthritis. Trends Mol. Med. 2012, 18, 109–118. [Google Scholar] [CrossRef]
- Bui, C.; Barter, M.J.; Scott, J.L.; Xu, Y.; Galler, M.; Reynard, L.N.; Rowan, A.D.; Young, D.A. cAMP response element-binding CREB recruitment following a specific CpG demethylation leads to the elevated expression of the matrix metalloproteinase 13 in human articular chondrocytes and osteoarthritis. FASEB J. 2012, 26, 3000–3011. [Google Scholar]
- Poschl, E.; Fidler, A.; Schmidt, B.; Kallipolitou, A.; Schmid, E.; Aigner, T. DNA methylation is not likely to be responsible for aggrecan down regulation in aged or osteoarthritic cartilage. Ann. Rheum. Dis. 2005, 64, 477–480. [Google Scholar]
- Zuscik, M.J.; Baden, J.F.; Wu, Q.; Sheu, T.J.; Schwarz, E.M.; Drissi, H.; O’Keefe, R.J.; Puzas, J.E.; Rosier, R.N. 5-azacytidine alters TGF-beta and BMP signaling and induces maturation in articular chondrocytes. J. Cell Biochem. 2004, 922, 316–331. [Google Scholar]
- Javaid, M.K.; Lane, N.E.; Mackey, D.C.; Lui, L.Y.; Arden, N.K.; Beck, T.J.; Hochberg, M.C.; Nevitt, M.C. Changes in proximal femoral mineral geometry precede the onset of radiographic hip osteoarthritis: The study of osteoporotic fractures. Arthritis Rheum. 2009, 60, 2028–2036. [Google Scholar] [CrossRef]
- Baker-Lepain, J.C.; Lynch, J.A.; Parimi, N.; McCulloch, C.E.; Nevitt, M.C.; Corr, M.; Lane, N.E. Variant alleles of the WNT antagonist FRZB are determinants of hip shape and modify the relationship between hip shape and osteoarthritis. Arthritis Rheum. 2012, 64, 1457–1465. [Google Scholar] [CrossRef]
- Schiffern, A.N.; Stevenson, D.A.; Carroll, K.L.; Pimentel, R.; Mineau, G.; Viskochil, D.H.; Roach, J.W. Total hip arthroplasty; hip osteoarthritis; total knee arthroplasty; and knee osteoarthritis in patients with developmental dysplasia of the hip and their family members: A kinship analysis report. J. Pediatr. Orthop. 2012, 32, 609–612. [Google Scholar]
- Sandell, L.J. Etiology of osteoarthritis: genetics and synovial joint development. Nat. Rev. Rheumatol. 2012, 8, 77–89. [Google Scholar]
- Bos, S.D.; Slagboom, P.E.; Meulenbelt, I. New insights into osteoarthritis: Early developmental features of an ageing-related disease. Curr. Opin. Rheumatol. 2008, 20, 553–559. [Google Scholar] [CrossRef]
- Aspden, R.M. Osteoarthritis: A problem of growth not decay? Rheumatology 2008, 47, 1452–1460. [Google Scholar] [CrossRef]
- Panoutsopoulou, K.; Southam, L.; Elliott, K.S.; Wrayner, N.; Zhai, G.; Beazley, C.; Thorleifsson, G.; Arden, N.K.; Carr, A.; Chapman, K.; et al. Insights into the genetic architecture of osteoarthritis from stage 1 of the arcOGEN study. Ann. Rheum. Dis. 2011, 70, 864–867. [Google Scholar] [CrossRef] [Green Version]
- Arcogen Consortium. Identification of new susceptibility loci for osteoarthritis arcOGEN: A genome-wide association study. Lancet 2012, 380, 815–823. [CrossRef] [Green Version]
- Valdes, A.M.; Spector, T.D. Genetic epidemiology of hip and knee osteoarthritis. Nat. Rev. Rheumatol. 2011, 7, 23–32. [Google Scholar] [CrossRef]
- Reynard, L.N.; Bui, C.; Canty-Laird, E.G.; Young, D.A.; Loughlin, J. Expression of the osteoarthritis-associated gene GDF5 is modulated epigenetically by DNA methylation. Hum. Mol. Genet. 2011, 20, 3450–3460. [Google Scholar] [CrossRef]
- Delgado-Calle, J.; Fernandez, A.F.; Sainz, J.; Zarrabeitia, M.T.; Garcia-Renedo, R.J.; Perez-Nunez, M.I.; Garcia-Ibarbia, C.; Fraga, M.F.; Riancho, J.A. Genome-wide profiling of bone reveals differentially methylated regions in osteoporosis and osteoarthritis. Arthritis Rheum. 2012. [Google Scholar] [CrossRef]
- Saha, B.; Kaur, P.; Tsao-Wei, D.; Naritoku, W.Y.; Groshen, S.; Datar, R.H.; Jones, L.W.; Imam, S.A. Unmethylated E-cadherin gene expression is significantly associated with metastatic human prostate cancer cells in bone. Prostate 2008, 68, 1681–1688. [Google Scholar]
- Tost, J.; Hamzaoui, H.; Busato, F.; Neyret, A.; Mourah, S.; Dupont, JM.; Bouizar, Z. Methylation of specific CpG sites in the P2 promoter of parathyroid hormone-related protein determines the invasive potential of breast cancer cell lines. Epigenetics 2011, 6, 1035–1046. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Delgado-Calle, J.; Riancho, J.A. The Role of DNA Methylation in Common Skeletal Disorders. Biology 2012, 1, 698-713. https://doi.org/10.3390/biology1030698
AMA Style
Delgado-Calle J, Riancho JA. The Role of DNA Methylation in Common Skeletal Disorders. Biology. 2012; 1(3):698-713. https://doi.org/10.3390/biology1030698
Chicago/Turabian StyleDelgado-Calle, Jesús, and José A. Riancho. 2012. "The Role of DNA Methylation in Common Skeletal Disorders" Biology 1, no. 3: 698-713. https://doi.org/10.3390/biology1030698