Causes and Consequences of Age-Related Changes in DNA Methylation: A Role for ROS?

{kind=link}
{kind=link}
Abstract
:1. Introduction
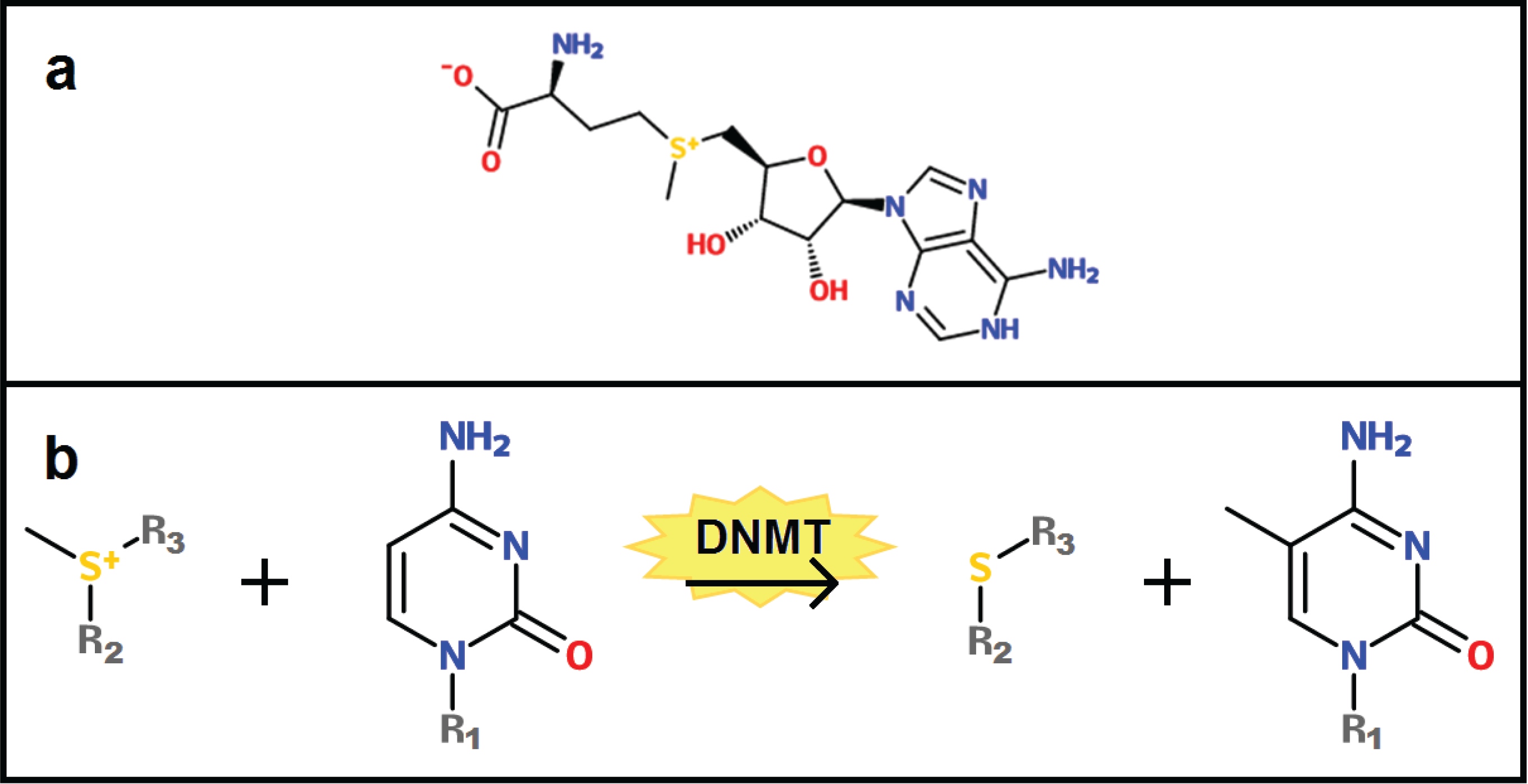
2. Basics of DNA Methylation
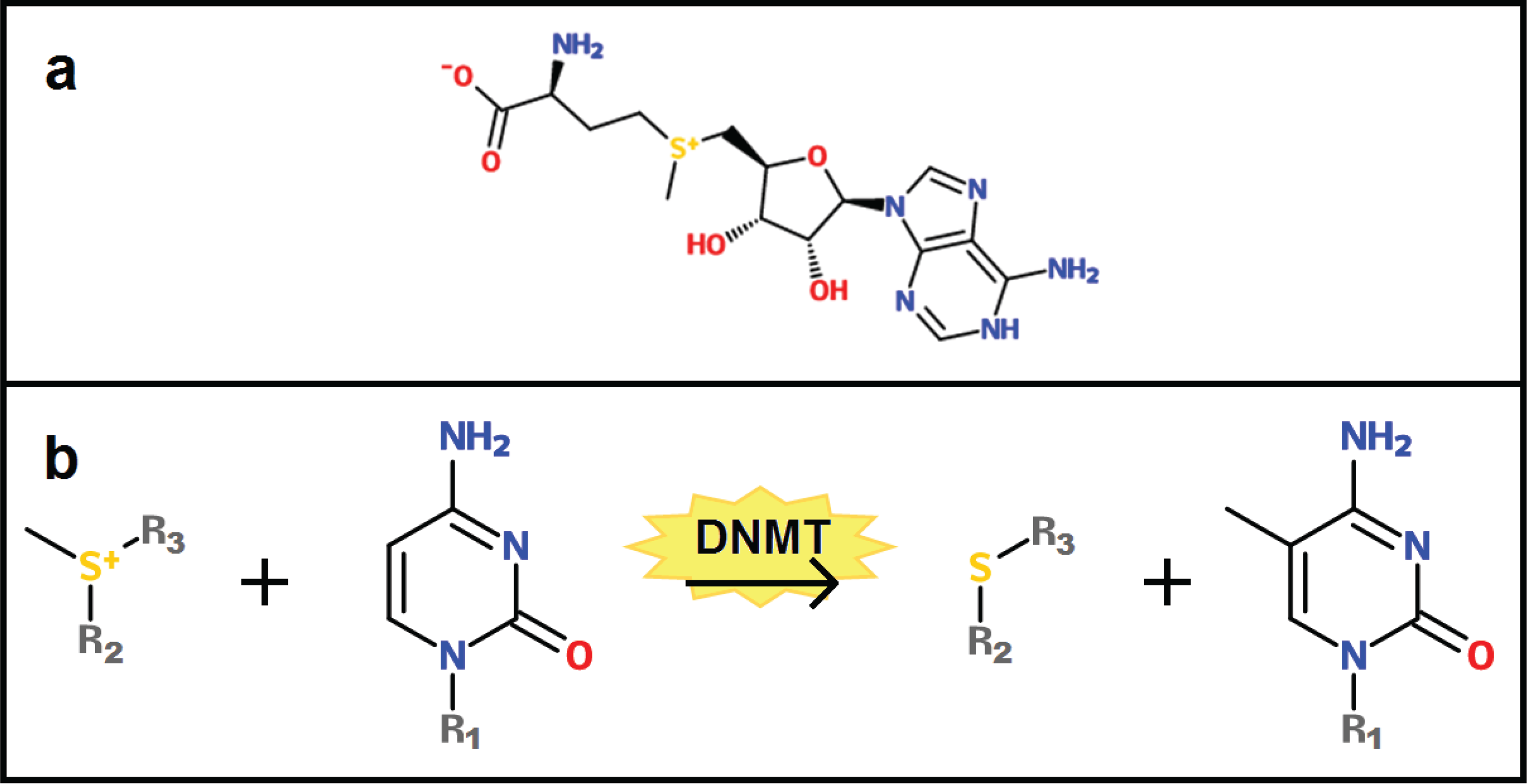
3. Changes in the DNA Methylome with Age
4. Influence of ROS on DNA Methylation
4.1. Reactive Oxygen Species in Aging
4.2. Formation, Regulation and Function of ROS
4.3. ROS-Induced Changes in DNA Methylation
4.4. Mechanisms of ROS-Induced Changes in DNA Methylation
4.4.1. ROS-Induced DNA Damage Influences DNMT Activity
4.4.2. ROS as Catalysts of DNA Methylation
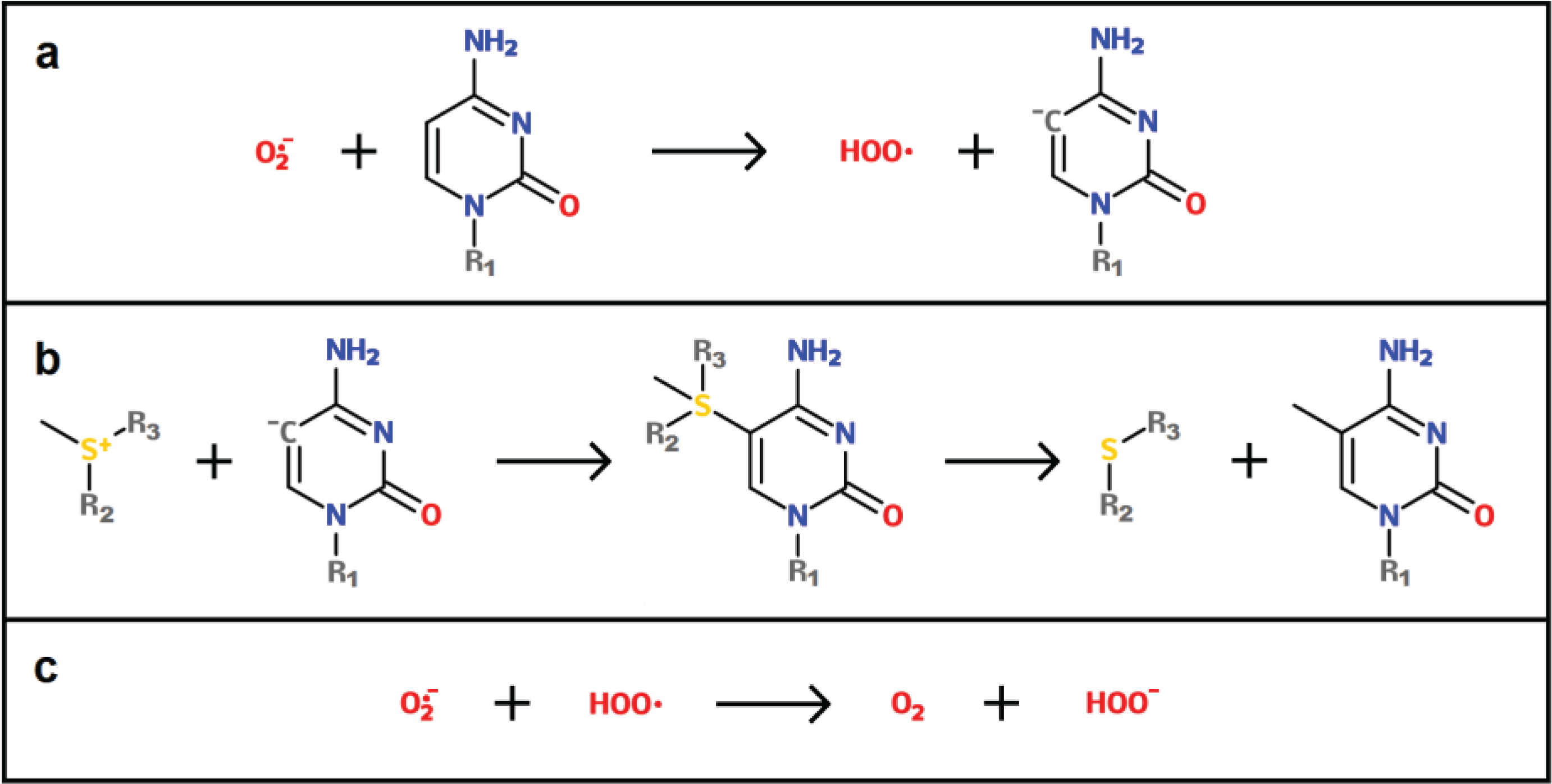
4.4.3. ROS-Induced Differential Binding of a DNMT Containing Complex
4.4.4. ROS-Regulated Expression of DNMTs
4.4.5. Other Mechanisms
5. Link to Disease
6. Conclusion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Szilard, L. On the Nature of the Aging Process. Proc. Natl. Acad. Sci. USA 1959, 45, 30–45. [Google Scholar] [CrossRef]
- Orgel, L.E. The Maintenance of the Accuracy of Protein Synthesis and Its Relevance to Aging. Proc. Natl. Acad. USA. 1963, 49, 517–521. [Google Scholar] [CrossRef]
- Kruk, P.A.; Rampino, N.J.; Bohr, V.A. DNA Damage and Repair in Telomeres: Relation to Aging. Proc. Natl. Acad. Sci. USA. 1995, 92, 258–262. [Google Scholar] [CrossRef]
- Christensen, B.C.; Houseman, E.A.; Marsit, C.J.; Zheng, S.; Wrensch, M.R.; Wiemels, J.L.; Nelson, H.H.; Karagas, M.R.; Padbury, J.F.; Bueno, R.; et al. Aging and Environmental Exposures Alter Tissue-Specific DNA Methylation Dependent upon CpG Island Context. PLoS Genet. 2009, 5, e1000602. [Google Scholar] [CrossRef]
- Rakyan, V.K.; Down, T.A.; Maslau, S.; Andrew, T.; Yang, T.-P.; Beyan, H.; Whittaker, P.; McCann, O.T.; Finer, S.; Valdes, A.M.; et al. Human Aging-Associated DNA Hypermethylation Occurs Preferentially at Bivalent Chromatin Domains. Genome Res. 2010, 20, 434–439. [Google Scholar] [CrossRef]
- Johansson, A.; Enroth, S.; Gyllensten, U. Continuous Aging of the Human DNA Methylome Throughout the Human Lifespan. PLoS One 2013, 8, e67378. [Google Scholar] [CrossRef]
- Xu, Z.; Taylor, J.A. Genome-Wide Age-Related DNA Methylation Changes in Blood and Other Tissues Relate to Histone Modification, Expression and Cancer. Carcinogen 2014, 35, 356–364. [Google Scholar] [CrossRef]
- Teschendorff, A.E.; Menon, U.; Gentry-Maharaj, A.; Ramus, S.J.; Weisenberger, D.J.; Shen, H.; Campan, M.; Noushmehr, H.; Bell, C.G.; Maxwell, A.P.; et al. Age-Dependent DNA Methylation of Genes That Are Suppressed in Stem Cells Is a Hallmark of Cancer. Genome Res. 2010, 20, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Johnson, K.C.; Koestler, D.C.; Cheng, C.; Christensen, B.C. Age-Related DNA Methylation in Normal Breast Tissue and Its Relationship with Invasive Breast Tumor Methylation. Epigenetics 2013, 9, 268–275. [Google Scholar]
- Boks, M.P.; Derks, E.M.; Weisenberger, D.J.; Strengman, E.; Janson, E.; Sommer, I.E.; Kahn, R.S.; Ophoff, R.A. The Relationship of DNA Methylation with Age, Gender and Genotype in Twins and Healthy Controls. PLoS One 2009, 4, e6767. [Google Scholar] [CrossRef]
- Bell, J.T.; Tsai, P.-C.; Yang, T.-P.; Pidsley, R.; Nisbet, J.; Glass, D.; Mangino, M.; Zhai, G.; Zhang, F.; Valdes, A.; et al. Epigenome-Wide Scans Identify Differentially Methylated Regions for Age and Age-Related Phenotypes in a Healthy Ageing Population. PLoS Genet. 2012, 8, e1002629. [Google Scholar] [Green Version]
- Harman, D. Aging: A Theory Based on Free Radical and Radiation Chemistry. J Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef]
- Sohal, R.S.; Weindruch, R. Oxidative Stress, Caloric Restriction, and Aging. Science 1996, 273, 59–63. [Google Scholar]
- Campos, A.C.E.; Molognoni, F.; Melo, F.H.M.; Galdieri, L.C.; Carneiro, C.R.W.; D’Almeida, V.; Correa, M.; Jasiulionis, M.G. Oxidative Stress Modulates DNA Methylation during Melanocyte Anchorage Blockade Associated with Malignant Transformation. Neoplasia 2007, 9, 1111–1121. [Google Scholar] [CrossRef]
- Lim, S.-O.; Gu, J.-M.; Kim, M.S.; Kim, H.-.; Park, Y.N.; Park, C.K.; Cho, J.W.; Park, Y.M.; Jung, G. Epigenetic Changes Induced by Reactive Oxygen Species in Hepatocellular Carcinoma: Methylation of the E-Cadherin Promoter. Gastroenterology 2008, 135, 2128–2140. [Google Scholar] [CrossRef]
- Quan, X.; Lim, S.-O.; Jung, G. Reactive Oxygen Species Downregulate Catalase Expression via Methylation of a CpG Island in the Oct-1 Promoter. FEBS Lett. 2011, 585, 3436–3441. [Google Scholar] [CrossRef]
- Bird, A.P.; Taggart, M.; Frommer, M.; Miller, O.J.; Macleod, D. A Fraction of the Mouse Genome That Is Derived from Islands of Nonmethylated, CpG-Rich DNA. Cell 1985, 40, 91–99. [Google Scholar] [CrossRef]
- Bird, A.P.; Wolffe, A.P. Methylation-Induced Repression-Belts, Braces, and Chromatin. Cell 1999, 99, 451–454. [Google Scholar] [CrossRef]
- Hellman, A.; Chess, A. Gene Body-Specific Methylation on the Active X Chromosome. Science 2007, 315, 1141–1143. [Google Scholar] [CrossRef]
- Suzuki, M.M.; Bird, A. DNA Methylation Landscapes: Provocative Insights from Epigenomics. Nat. Rev. Genet. 2008, 9, 465–476. [Google Scholar] [CrossRef]
- Rountree, M.R.; Selker, E.U. DNA Methylation Inhibits Elongation but Not Initiation of Transcription in Neurospora Crassa. Genes Dev. 1997, 11, 2383–2395. [Google Scholar] [CrossRef]
- Klose, R.; Bird, A. Genomic DNA Methylation: The Mark and Its Mediators. Trends Biochem. Sci. 2006, 31, 89–97. [Google Scholar] [CrossRef]
- Yoder, J.A.; Walsh, C.P.; Bestor, T.H. Cytosine Methylation and the Ecology of Intragenomic Parasites. Trends Genet. 1997, 13, 335–340. [Google Scholar] [CrossRef]
- Laurent, L.; Wong, E.; Li, G.; Huynh, T.; Tsirigos, A.; Ong, C.T.; Low, H.M.; Sung, K.W.K.; Rigoutsos, I.; Loring, J.; Wei, C.L. Dynamic Changes in the Human Methylome during Differentiation. Genome Res. 2010, 20, 320–331. [Google Scholar] [CrossRef]
- Schmidl, C.; Klug, M.; Boeld, T.J.; Andreesen, R.; Hoffmann, P.; Edinger, M.; Rehli, M. Lineage-Specific DNA Methylation in T Cells Correlates with Histone Methylation and Enhancer Activity. Genome Res. 2009, 19, 1165–1174. [Google Scholar] [CrossRef]
- Wiench, M.; John, S.; Baek, S.; Johnson, T.A.; Sung, M.-H.; Escobar, T.; Simmons, C.A.; Pearce, K.H.; Biddie, S.C.; Sabo, P.J.; Thurman, R.E.; Stamatoyannopoulos, J.A.; Hager, G.L. DNA Methylation Status Predicts Cell Type-Specific Enhancer Activity. EMBO J. 2011, 30, 3028–3039. [Google Scholar] [CrossRef]
- Xu, G.L.; Bestor, T.H.; Bourc’his, D.; Hsieh, C.L.; Tommerup, N.; Bugge, M.; Hulten, M.; Qu, X.; Russo, J.J.; Viegas-Péquignot, E. Chromosome Instability and Immunodeficiency Syndrome Caused by Mutations in a DNA Methyltransferase Gene. Nature 1999, 402, 187–191. [Google Scholar] [CrossRef]
- Gopalakrishnan, S.; Sullivan, B.A.; Trazzi, S.; della Valle, G.; Robertson, K.D. DNMT3B Interacts with Constitutive Centromere Protein CENP-C to Modulate DNA Methylation and the Histone Code at Centromeric Regions. Hum. Mol. Genet. 2009, 18, 3178–3193. [Google Scholar] [CrossRef]
- Bestor, T.; Laudano, A.; Mattaliano, R.; Ingram, V. Cloning and Sequencing of a cDNA Encoding DNA Methyltransferase of Mouse Cells. J. Mol. Biol. 1988, 203, 971–983. [Google Scholar] [CrossRef]
- Okano, M.; Xie, S.; Li, E. Cloning and Characterization of a Family of Novel Mammalian DNA (cytosine-5) Methyltransferases. Nat. Genet. 1998, 19, 219–220. [Google Scholar] [CrossRef]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA Methyltransferases Dnmt3a and Dnmt3b Are Essential for De Novo Methylation and Mammalian Development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef]
- Chen, T.; Ueda, Y.; Dodge, J.E.; Wang, Z.; Li, E. Establishment and Maintenance of Genomic Methylation Patterns in Mouse Embryonic Stem Cells by Dnmt3a and Dnmt3b. Mol. Cell. Biol. 2003, 23, 5594–5605. [Google Scholar] [CrossRef]
- Bestor, T.H. Activation of Mammalian DNA Methyltransferase by Cleavage of a Zn Binding Regulatory Domain. EMBO J. 1992, 11, 2611–2617. [Google Scholar]
- Leonhardt, H.; Page, A.W.; Weier, H.-U.; Bestor, T.H. A Targeting Sequence Directs DNA Methyltransferase to Sites of DNA Replication in Mammalian Nuclei. Cell 1992, 71, 865–873. [Google Scholar] [CrossRef]
- Gowher, H.; Liebert, K.; Hermann, A.; Xu, G.; Jeltsch, A. Mechanism of Stimulation of Catalytic Activity of Dnmt3A and Dnmt3B DNA-(cytosine-C5)-Methyltransferases by Dnmt3L. J. Biol. Chem. 2005, 280, 13341–13348. [Google Scholar] [CrossRef]
- Takusagawa, F.; Kamitori, S.; Markham, G.D. Structure and Function of S-Adenosylmethionine Synthetase: Crystal Structures of S-Adenosylmethionine Synthetase with ADP, BrADP, and PPi at 28 Angstroms Resolution. Biochem. 1996, 35, 2586–2596. [Google Scholar] [CrossRef]
- Challen, G.A.; Sun, D.; Jeong, M.; Luo, M.; Jelinek, J.; Berg, J.S.; Bock, C.; Vasanthakumar, A.; Gu, H.; Xi, Y.; et al. Dnmt3a Is Essential for Hematopoietic Stem Cell Differentiation. Nat. Genet. 2012, 44, 23–31. [Google Scholar]
- Riggs, A.D. X Inactivation, Differentiation, and DNA Methylation. Cytogenet. Genome Res. 1975, 14, 9–25. [Google Scholar] [CrossRef]
- Razin, A.; Riggs, A. DNA Methylation and Gene Function. Science 1980, 210, 604–610. [Google Scholar]
- Feinberg, A.P.; Vogelstein, B. Hypomethylation Distinguishes Genes of Some Human Cancers from Their Normal Counterparts. Nature 1983, 301, 89–92. [Google Scholar] [CrossRef]
- Issa, J.P.; Ottaviano, Y.L.; Celano, P.; Hamilton, S.R.; Davidson, N.E.; Baylin, S.B. Methylation of the Oestrogen Receptor CpG Island Links Ageing and Neoplasia in Human Colon. Nat. Genet. 1994, 7, 536–540. [Google Scholar] [CrossRef]
- Ahuja, N.; Li, Q.; Mohan, A.L.; Baylin, S.B.; Issa, J.-P.J. Aging and DNA Methylation in Colorectal Mucosa and Cancer. Cancer Res. 1998, 58, 5489–5494. [Google Scholar]
- Wang, S.-C.; Oelze, B.; Schumacher, A. Age-Specific Epigenetic Drift in Late-Onset Alzheimer’s Disease. PLoS One 2008, 3, e2698. [Google Scholar]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.-B.; Gao, Y.; et al. Genome-Wide Methylation Profiles Reveal Quantitative Views of Human Aging Rates. Mol. Cell 2013, 49, 359–367. [Google Scholar] [CrossRef]
- Hartwig, A.; Schlepegrell, R. Induction of Oxidative DNA Damage by Ferric Iron in Mammalian Cells. Carcinogenesis 1995, 16, 3009–3013. [Google Scholar] [CrossRef]
- Karthikeyan, G.; Lewis, K.; Resnick, M.A. The Mitochondrial Protein Frataxin Prevents Nuclear Damage. Hum. Mol. Genet. 2002, 11, 1351–1362. [Google Scholar] [CrossRef]
- Kim, D.-H.; Nelson, H.H.; Wiencke, J.K.; Zheng, S.; Christiani, D.C.; Wain, J.C.; Mark, E.J.; Kelsey, K.T. p16INK4a and Histology-Specific Methylation of CpG Islands by Exposure to Tobacco Smoke in Non-Small Cell Lung Cancer. Cancer Res. 2001, 61, 3419–3424. [Google Scholar]
- Zöchbauer-Müller, S.; Lam, S.; Toyooka, S.; Virmani, A.K.; Toyooka, K.O.; Seidl, S.; Minna, J.D.; Gazdar, A.F. Aberrant Methylation of Multiple Genes in the Upper Aerodigestive Tract Epithelium of Heavy Smokers. Int. J. Cancer 2003, 107, 612–616. [Google Scholar] [CrossRef]
- Dammann, R.; Strunnikova, M.; Schagdarsurengin, U.; Rastetter, M.; Papritz, M.; Hattenhorst, U.E.; Hofmann, H.-S.; Silber, R.-E.; Burdach, S.; Hansen, G. CpG Island Methylation and Expression of Tumour-Associated Genes in Lung Carcinoma. Eur. J. Cancer 2005, 41, 1223–1236. [Google Scholar] [CrossRef]
- Enokida, H.; Shiina, H.; Urakami, S.; Terashima, M.; Ogishima, T.; Li, L.-C.; Kawahara, M.; Nakagawa, M.; Kane, C.J.; Carroll, P.R.; Igawa, M.; Dahiya, R. Smoking Influences Aberrant CpG Hypermethylation of Multiple Genes in Human Prostate Carcinoma. Cancer 2006, 106, 79–86. [Google Scholar] [CrossRef]
- Vaissière, T.; Hung, R.J.; Zaridze, D.; Moukeria, A.; Cuenin, C.; Fasolo, V.; Ferro, G.; Paliwal, A.; Hainaut, P.; Brennan, P.; et al. Quantitative Analysis of DNA Methylation Profiles in Lung Cancer Identifies Aberrant DNA Methylation of Specific Genes and Its Association with Gender and Cancer Risk Factors. Cancer Res. 2009, 69, 243–252. [Google Scholar] [CrossRef]
- Hsiung, D.T.; Marsit, C.J.; Houseman, E.A.; Eddy, K.; Furniss, C.S.; McClean, M.D.; Kelsey, K.T. Global DNA Methylation Level in Whole Blood as a Biomarker in Head and Neck Squamous Cell Carcinoma. Cancer Epidemiol. Biomarkers Prev. 2007, 16, 108–114. [Google Scholar] [CrossRef]
- Hillemacher, T.; Frieling, H.; Moskau, S.; Muschler, M.A.N.; Semmler, A.; Kornhuber, J.; Klockgether, T.; Bleich, S.; Linnebank, M. Global DNA Methylation Is Influenced by Smoking Behaviour. Eur. Neuropsychopharmacol. 2008, 18, 295–298. [Google Scholar] [CrossRef] [Green Version]
- Oka, D.; Yamashita, S.; Tomioka, T.; Nakanishi, Y.; Kato, H.; Kaminishi, M.; Ushijima, T. The Presence of Aberrant DNA Methylation in Noncancerous Esophageal Mucosae in Association with Smoking History: A Target for Risk Diagnosis and Prevention of Esophageal Cancers. Cancer 2009, 115, 3412–3426. [Google Scholar] [CrossRef]
- Christensen, B.C.; Houseman, E.A.; Godleski, J.J.; Marsit, C.J.; Longacker, J.L.; Roelofs, C.R.; Karagas, M.R.; Wrensch, M.R.; Yeh, R.-F.; Nelson, H.H.; et al. Epigenetic Profiles Distinguish Pleural Mesothelioma from Normal Pleura and Predict Lung Asbestos Burden and Clinical Outcome. Cancer Res. 2009, 69, 227–234. [Google Scholar]
- Christensen, B.C.; Godleski, J.J.; Marsit, C.J.; Houseman, E.A.; Lopez-Fagundo, C.Y.; Longacker, J.L.; Bueno, R.; Sugarbaker, D.J.; Nelson, H.H.; et al. Asbestos Exposure Predicts Cell Cycle Control Gene Promoter Methylation in Pleural Mesothelioma. Carcinogen. 2008, 29, 1555–1559. [Google Scholar] [CrossRef]
- Rahman, I.; Morrison, D.; Donaldson, K.; MacNee, W. Systemic Oxidative Stress in Asthma, COPD, and Smokers. Am. J. Respir. Crit. Care Med. 1996, 154, 1055–1060. [Google Scholar] [CrossRef]
- Panduri, V.; Weitzman, S.A.; Chandel, N.S.; Kamp, D.W. Mitochondrial-Derived Free Radicals Mediate Asbestos-Induced Alveolar Epithelial Cell Apoptosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 286, 1220–1227. [Google Scholar] [CrossRef]
- Harman, D. The Biologic Clock: The Mitochondria? J. Am. Geriatr. Soc. 1972, 20, 145–147. [Google Scholar]
- Lapointe, J.; Hekimi, S. When a Theory of Aging Ages Badly. Cell. Mol. Life Sci. 2010, 67, 1–8. [Google Scholar] [CrossRef]
- Huang, T.T.; Carlson, E.J.; Gillespie, A.M.; Shi, Y.; Epstein, C.J. Ubiquitous Overexpression of CuZn Superoxide Dismutase Does Not Extendlife Span in Mice. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2000, 55, B5–B9. [Google Scholar] [CrossRef]
- Pérez, V.I.; Van Remmen, H.; Bokov, A.; Epstein, C.J.; Vijg, J.; Richardson, A. The Overexpression of Major Antioxidant Enzymes Does Not Extend the Lifespan of Mice. Aging Cell 2009, 8, 73–75. [Google Scholar] [CrossRef]
- Sohal, R.S.; Sohal, B.H. Hydrogen Peroxide Release by Mitochondria Increases during Aging. Mech. Ageing Dev. 1991, 57, 187–202. [Google Scholar] [CrossRef]
- Hamilton, M.L.; Van Remmen, H.; Drake, J.A.; Yang, H.; Guo, Z.M.; Kewitt, K.; Walter, C.A.; Richardson, A. Does Oxidative Damage to DNA Increase with Age? Proc. Natl. Acad. Sci. USA 2001, 98, 10469–10474. [Google Scholar] [CrossRef]
- Mansouri, A.; Muller, F.L.; Liu, Y.; Ng, R.; Faulkner, J.; Hamilton, M.; Richardson, A.; Huang, T.-T.; Epstein, C.J.; Van Remmen, H. Alterations in Mitochondrial Function, Hydrogen Peroxide Release and Oxidative Damage in Mouse Hind-Limb Skeletal Muscle during Aging. Mech. Ageing Dev. 2006, 127, 298–306. [Google Scholar] [CrossRef]
- Hekimi, S.; Lapointe, J.; Wen, Y. Taking a “Good” Look at Free Radicals in the Aging Process. Trends Cell Biol. 2011, 21, 569–576. [Google Scholar] [CrossRef]
- Sundaresan, M.; Yu, Z.-X.; Ferrans, V.J.; Irani, K.; Finkel, T. Requirement for Generation of H2O2 for Platelet-Derived Growth Factor Signal Transduction. Science 1995, 270, 296–299. [Google Scholar]
- Bae, Y.S.; Kang, S.W.; Seo, M.S.; Baines, I.C.; Tekle, E.; Chock, P.B.; Rhee, S.G. Epidermal Growth Factor (EGF)-Induced Generation of Hydrogen Peroxide. Role in EGF Receptor-Mediated Tyrosine Phosphorylation. J. Biol. Chem. 1997, 272, 217–221. [Google Scholar]
- Sena, L.A.; Li, S.; Jairaman, A.; Prakriya, M.; Ezponda, T.; Hildeman, D.A.; Wang, C.-R.; Schumacker, P.T.; Licht, J.D.; Perlman, H.; et al. Mitochondria Are Required for Antigen-Specific T Cell Activation through Reactive Oxygen Species Signaling. Immunity 2013, 38, 225–236. [Google Scholar] [CrossRef]
- Dickinson, B.C.; Peltier, J.; Stone, D.; Schaffer, D.V.; Chang, C.J. Nox2 Redox Signaling Maintains Essential Cell Populations in the Brain. Nat. Chem. Biol. 2011, 7, 106–112. [Google Scholar] [CrossRef]
- Kirkland, R.; Adibhatla, R.; Hatcher, J.; Franklin, J. Loss of Cardiolipin and Mitochondria during Programmed Neuronal Death: Evidence of a Role for Lipid Peroxidation and Autophagy. Neurosci. 2002, 115, 587–602. [Google Scholar] [CrossRef]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive Oxygen Species Are Essential for Autophagy and Specifically Regulate the Activity of Atg4. EMBO J. 2007, 26, 1749–1760. [Google Scholar] [CrossRef]
- Salmon, A.B.; Richardson, A.; Pérez, V.I. Update on the Oxidative Stress Theory of Aging: Does Oxidative Stress Play a Role in Aging or Healthy Aging? Free Radic. Biol. Med. 2010, 48, 642–655. [Google Scholar] [CrossRef]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free Radicals, Metals and Antioxidants in Oxidative Stress-Induced Cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef]
- Kuthan, H.; Ullrich, V. Oxidase and Oxygenase Function of the Microsomal Cytochrome P450 Monooxygenase System. Eur. J. Biochem. 1982, 126, 583–588. [Google Scholar] [CrossRef]
- De Duve, C.; Baudhuin, P. Peroxisomes (microbodies and Related Particles). Physiol. Rev. 1966, 46, 323–357. [Google Scholar]
- Boveris, A.; Oshino, N.; Chance, B. The Cellular Production of Hydrogen Peroxide. 1972, 128, 617–630. [Google Scholar]
- Mackaness, G.B. The Monocyte in Cellular Immunity. Semin. Hematol. 1970, 7, 172–184. [Google Scholar]
- Adams, D.O.; Johnson, W.J.; Marino, P.A. Mechanisms of Target Recognition and Destruction in Macrophage-Mediated Tumor Cytotoxicity. Fed. Proc. 1982, 41, 2212–2221. [Google Scholar]
- Halliwell, B.; Gutteridge, J.M. Oxygen Radicals in Biological Systems Part B: Oxygen Radicals and Antioxidants. Meth. Enzymol. 1990, 186, 1–85. [Google Scholar] [CrossRef]
- Davies, K. Oxidative Stress: The Paradox of Aerobic Life. Biochem. Soc. Synmp. 1995, 61, 1–31. [Google Scholar]
- Borish, E.T.; Cosgrove, J.P.; Church, D.F.; Deutsch, W.A.; Pryor, W.A. Cigarette Tar Causes Single-Strand Breaks in DNA. Biochem. Biophys. Res. Commun. 1985, 133, 780–786. [Google Scholar] [CrossRef]
- Ariza, R.R.; Serrano, A.; Pueyo, C. Study on the Mutagenicity of Brandy with the Ara Test. Mutagenesis 1992, 7, 77–81. [Google Scholar] [CrossRef]
- Lauffer, R.B. Iron and Human Disease, 1st ed.; CRC Press: Boca Raton, FL, USA, 1992; p. 560. [Google Scholar]
- Zhang, X.; Rosenstein, B.S.; Wang, Y.; Lebwohl, M.; Wei, H. Identification of Possible Reactive Oxygen Species Involved in Ultraviolet Radiation-Induced Oxidative DNA Damage. Free Radic. Biol. Med. 1997, 23, 980–985. [Google Scholar] [CrossRef]
- Leach, J.K.; Van Tuyle, G.; Lin, P.-S.; Schmidt-Ullrich, R.; Mikkelsen, R.B. Ionizing Radiation-Induced, Mitochondria-Dependent Generation of Reactive Oxygen/Nitrogen. Cancer Res. 2001, 61, 3894–3901. [Google Scholar]
- Monari, M.; Foschi, J.; Calabrese, C.; Liguori, G.; Di Febo, G.; Rizzello, F.; Gionchetti, P.; Trinchero, A.; Serrazanetti, G.P. Implications of Antioxidant Enzymes in Human Gastric Neoplasms. Int. J. Mol. Med. 2009, 24, 693–700. [Google Scholar]
- Martindale, J.L.; Holbrook, N.J. Cellular Response to Oxidative Stress: Signaling for Suicide and Survival. J. Cell. Physiol. 2002, 192, 1–15. [Google Scholar] [CrossRef]
- Rhee, S.G.; Bae, Y.S.; Lee, S.R.; Kwon, J. Hydrogen Peroxide: A Key Messenger That Modulates Protein Phosphorylation through Cysteine Oxidation. Sci. STKE 2000, 53, pe1. [Google Scholar]
- Ziech, D.; Franco, R.; Georgakilas, A.G.; Georgakila, S.; Malamou-Mitsi, V.; Schoneveld, O.; Pappa, A.; Panayiotidis, M.I. The Role of Reactive Oxygen Species and Oxidative Stress in Environmental Carcinogenesis and Biomarker Development. Chem. Biol. Interact. 2010, 188, 334–339. [Google Scholar] [CrossRef]
- Garcia-Garcia, A.; Rodriguez-Rocha, H.; Madayiputhiya, N.; Pappa, A.; Panayiotidis, M.I.; Franco, R. Biomarkers of Protein Oxidation in Human Disease. Curr. Mol. Med. 2012, 12, 681–697. [Google Scholar] [CrossRef]
- Cocuzza, M.; Athayde, K.S.; Agarwal, A.; Sharma, R.; Pagani, R.; Lucon, A.M.; Srougi, M.; Hallak, J. Age-Related Increase of Reactive Oxygen Species in Neat Semen in Healthy Fertile Men. Urology 2008, 71, 490–494. [Google Scholar] [CrossRef]
- Driver, A.S.; Kodavanti, P.R.; Mundy, W.R. Age-Related Changes in Reactive Oxygen Species Production in Rat Brain Homogenates. Neurotoxicol. Teratol. 2000, 22, 175–181. [Google Scholar] [CrossRef]
- Mecocci, P.; Fanó, G.; Fulle, S.; MacGarvey, U.; Shinobu, L.; Polidori, M.C.; Cherubini, A.; Vecchiet, J.; Senin, U.; Beal, M.F. Age-Dependent Increases in Oxidative Damage to DNA, Lipids, and Proteins in Human Skeletal Muscle. Free Radic. Biol. Med. 1999, 26, 303–308. [Google Scholar] [CrossRef]
- Bejma, J.; Ramires, P.; Ji, L.L. Free Radical Generation and Oxidative Stress with Ageing and Exercise: Differential Effects in the Myocardium and Liver. Acta Physiol. Scand. 2000, 169, 343–351. [Google Scholar] [CrossRef]
- Franco, R.; Schoneveld, O.; Georgakilas, A.G.; Panayiotidis, M.I. Oxidative Stress, DNA Methylation and Carcinogenesis. Cancer Lett. 2008, 266, 6–11. [Google Scholar] [CrossRef]
- Ziech, D.; Franco, R.; Pappa, A.; Panayiotidis, M.I. Reactive Oxygen Species (ROS)--Induced Genetic and Epigenetic Alterations in Human Carcinogenesis. Mutat. Res. 2011, 711, 167–173. [Google Scholar] [CrossRef]
- Tunc, O.; Tremellen, K. Oxidative DNA Damage Impairs Global Sperm DNA Methylation in Infertile Men. J. Assist. Reprod. Genet. 2009, 26, 537–544. [Google Scholar] [CrossRef]
- Weitzman, S.A.; Turk, P.W.; Milkowski, D.H.; Kozlowski, K. Free Radical Adducts Induce Alterations in DNA Cytosine Methylation. Proc. Natl. Acad. Sci. USA 1994, 91, 1261–1264. [Google Scholar]
- Turk, P.W.; Laayoun, A.; Smith, S.S.; Weitzman, S.A. DNA Adduct 8-Hydroxyl-2′-Deoxyguanosine (8-Hydroxyguanine) Affects Function of Human DNA Methyltransferase. Carcinogen. 1995, 16, 1253–1255. [Google Scholar] [CrossRef]
- Valinluck, V.; Sowers, L.C. Endogenous Cytosine Damage Products Alter the Site Selectivity of Human DNA Maintenance Methyltransferase DNMT1. Cancer Res. 2007, 67, 946–950. [Google Scholar] [CrossRef]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-Methylcytosine to 5-Hydroxymethylcytosine in Mammalian DNA by MLL Partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef]
- Globisch, D.; Münzel, M.; Müller, M.; Michalakis, S.; Wagner, M.; Koch, S.; Brückl, T.; Biel, M.; Carell, T. Tissue Distribution of 5-Hydroxymethylcytosine and Search for Active Demethylation Intermediates. PLoS One 2010, 5, e15367. [Google Scholar] [Green Version]
- Kriaucionis, S.; Heintz, N. The Nuclear DNA Base 5-Hydroxymethylcytosine Is Present in Purkinje Neurons and the Brain. Science 2009, 324, 929–930. [Google Scholar] [CrossRef]
- Ruzov, A.; Tsenkina, Y.; Serio, A.; Dudnakova, T.; Fletcher, J.; Bai, Y.; Chebotareva, T.; Pells, S.; Hannoun, Z.; Sullivan, G.; Chandran, S.; Hay, D.C.; Bradley, M.; Wilmut, I.; De Sousa, P. Lineage-Specific Distribution of High Levels of Genomic 5-Hydroxymethylcytosine in Mammalian Development. Cell Res. 2011, 21, 1332–1342. [Google Scholar] [CrossRef]
- Bhutani, N.; Burns, D.M.; Blau, H.M. DNA Demethylation Dynamics. Cell 2011, 146, 866–872. [Google Scholar] [CrossRef]
- Fouse, S.D.; Nagarajan, R.O.; Costello, J.F. Genome-Scale DNA Methylation Analysis. Epigenomics 2010, 2, 105–117. [Google Scholar] [CrossRef]
- Jin, S.G.; Kadam, S.; Pfeifer, G.P. Examination of the Specificity of DNA Methylation Profiling Techniques towards 5-Methylcytosine and 5-Hydroxymethylcytosine. Nucleic Acids Res. 2010, 38, e125. [Google Scholar] [CrossRef]
- Valinluck, V.; Tsai, H.-H.; Rogstad, D.K.; Burdzy, A.; Bird, A.; Sowers, L.C. Oxidative Damage to Methyl-CpG Sequences Inhibits the Binding of the Methyl-CpG Binding Domain (MBD) of Methyl-CpG Binding Protein 2 (MeCP2). Nucleic Acids Res. 2004, 32, 4100–4108. [Google Scholar] [CrossRef]
- Afanas’ev, I. New Nucleophilic Mechanisms of Ros-Dependent Epigenetic Modifications: Comparison of Aging and Cancer. Aging Dis. 2014, 5, 52–62. [Google Scholar] [CrossRef]
- Vilkaitis, G.; Merkiene, E.; Serva, S.; Weinhold, E.; Klimasauskas, S. The Mechanism of DNA Cytosine-5 Methylation. Kinetic and Mutational Dissection of Hhai Methyltransferase. J. Biol. Chem. 2001, 276, 20924–20934. [Google Scholar]
- Gerasimaitė, R.; Merkienė, E.; Klimašauskas, S. Direct Observation of Cytosine Flipping and Covalent Catalysis in a DNA Methyltransferase. Nucleic Acids Res. 2011, 39, 3771–3780. [Google Scholar] [CrossRef]
- O’Hagan, H.; Wang, W.; Sen, S. Oxidative Damage Targets Complexes Containing DNA Methyltransferases, SIRT1, and Polycomb Members to Promoter CpG Islands. Cancer Cell 2011, 20, 606–619. [Google Scholar] [CrossRef]
- Fan, W.; Luo, J. SIRT1 Regulates UV-Induced DNA Repair through Deacetylating XPA. Mol. Cell 2010, 39, 247–258. [Google Scholar] [CrossRef]
- Espada, J.; Ballestar, E.; Santoro, R.; Fraga, M.F.; Villar-Garea, A.; Németh, A.; Lopez-Serra, L.; Ropero, S.; Aranda, A.; Orozco, H.; et al. Epigenetic Disruption of Ribosomal RNA Genes and Nucleolar Architecture in DNA Methyltransferase 1 (Dnmt1) Deficient Cells. Nucleic Acids Res. 2007, 35, 2191–2198. [Google Scholar] [CrossRef]
- Viré, E.; Brenner, C.; Deplus, R.; Blanchon, L.; Fraga, M.; Didelot, C.; Morey, L.; Van Eynde, A.; Bernard, D.; Vanderwinden, J.-M.; et al. The Polycomb Group Protein EZH2 Directly Controls DNA Methylation. Nature 2006, 439, 871–874. [Google Scholar]
- Mortusewicz, O.; Schermelleh, L.; Walter, J.; Cardoso, M.C.; Leonhardt, H. Recruitment of DNA Methyltransferase I to DNA Repair Sites. Proc. Natl. Acad. Sci. USA 2005, 102, 8905–8909. [Google Scholar] [CrossRef]
- O’Hagan, H.M.; Mohammad, H.P.; Baylin, S.B. Double Strand Breaks Can Initiate Gene Silencing and SIRT1-Dependent Onset of DNA Methylation in an Exogenous Promoter CpG Island. PLoS Genet. 2008, 4, e1000155. [Google Scholar]
- MacLeod, A.; Rouleau, J.; Szyf, M. Regulation of DNA Methylation by the Ras Signaling Pathway. J. Biol. Chem. 1995, 270, 11327–11337. [Google Scholar] [CrossRef]
- Kang, K.A.; Zhang, R.; Kim, G.Y.; Bae, S.C.; Hyun, J.W. Epigenetic Changes Induced by Oxidative Stress in Colorectal Cancer Cells: Methylation of Tumor Suppressor RUNX3. Tumour Biol. 2012, 33, 403–412. [Google Scholar] [CrossRef]
- Fuks, F.; Burgers, W.A.; Brehm, A.; Hughes-Davies, L.; Kouzarides, T. DNA Methyltransferase Dnmt1 Associates with Histone Deacetylase Activity. Nat. Genet. 2000, 24, 88–91. [Google Scholar] [CrossRef]
- Fuks, F. DNA Methylation and Histone Modifications: Teaming up to Silence Genes. Curr. Opin. Genet. Dev. 2005, 15, 490–495. [Google Scholar] [CrossRef]
- Shao, D.; Fry, J.L.; Han, J.; Hou, X.; Pimentel, D.R.; Matsui, R.; Cohen, R.A.; Bachschmid, M.M. A Redox-Resistant Sirtuin-1 Mutant Protects against Hepatic Metabolic and Oxidant Stress. J. Biol. Chem. 2014, 289, 7293–7306. [Google Scholar]
- Peng, L.; Yuan, Z.; Ling, H.; Fukasawa, K.; Robertson, K.; Olashaw, N.; Koomen, J.; Chen, J.; Lane, W.S.; Seto, E. SIRT1 Deacetylates the DNA Methyltransferase 1 (DNMT1) Protein and Alters Its Activities. Mol. Cell. Biol. 2011, 31, 4720–4734. [Google Scholar] [CrossRef]
- Cedar, H.; Bergman, Y. Linking DNA Methylation and Histone Modification: Patterns and Paradigms. Nat. Rev. Genet. 2009, 10, 295–304. [Google Scholar] [CrossRef]
- Fraga, M.F.; Esteller, M. Epigenetics and Aging: The Targets and the Marks. Trends Genet. 2007, 23, 413–418. [Google Scholar] [CrossRef]
- Sedivy, J.M.; Banumathy, G.; Adams, P.D. Aging by Epigenetics—A Consequence of Chromatin Damage? Exp. Cell Res. 2008, 314, 1909–1917. [Google Scholar] [CrossRef]
- O’Sullivan, R.J.; Karlseder, J. The Great Unravelling: Chromatin as a Modulator of the Aging Process. Trends Biochem. Sci. 2012, 37, 466–476. [Google Scholar] [CrossRef]
- Bollati, V.; Galimberti, D.; Pergoli, L.; Dalla Valle, E.; Barretta, F.; Cortini, F.; Scarpini, E.; Bertazzi, P.A.; Baccarelli, A. DNA Methylation in Repetitive Elements and Alzheimer Disease. Brain. Behav. Immun. 2011, 25, 1078–1083. [Google Scholar] [CrossRef]
- Mastroeni, D.; Grover, A.; Delvaux, E.; Whiteside, C.; Coleman, P.D.; Rogers, J. Epigenetic Changes in Alzheimer’s Disease: Decrements in DNA Methylation. Neurobiol. Aging 2010, 31, 2025–2037. [Google Scholar] [CrossRef]
- Park, S.-Y.; Yoo, E.J.; Cho, N.-Y.; Kim, N.; Kang, G.H. Comparison of CpG Island Hypermethylation and Repetitive DNA Hypomethylation in Premalignant Stages of Gastric Cancer, Stratified for Helicobacter Pylori Infection. J. Pathol. 2009, 219, 410–416. [Google Scholar] [CrossRef]
- Graff, J.R.; Herman, J.G.; Lapidus, R.G.; Chopra, H.; Xu, R.; Jarrard, D.F.; Isaacs, W.B.; Pitha, P.M.; Davidson, N.E.; Baylin, S.B. E-Cadherin Expression Is Silenced by DNA Hypermethylation in Human Breast and Prostate Carcinomas. Cancer Res. 1995, 55, 5195–5199. [Google Scholar]
- Yang, B.; Guo, M.; Herman, J.G.; Clark, D.P. Aberrant Promoter Methylation Profiles of Tumor Suppressor Genes in Hepatocellular Carcinoma. Am. J. Pathol. 2003, 163, 1101–1107. [Google Scholar] [CrossRef]
- Donkena, K.V.; Young, C.Y.F.; Tindall, D.J. Oxidative Stress and DNA Methylation in Prostate Cancer. Obstet. Gynecol. Int. 2010, 2010, 302051. [Google Scholar]
- Gaudet, F.; Hodgson, J.G.; Eden, A.; Jackson-Grusby, L.; Dausman, J.; Gray, J.W.; Leonhardt, H.; Jaenisch, R. Induction of Tumors in Mice by Genomic Hypomethylation. Science 2003, 300, 489–492. [Google Scholar] [CrossRef]
- Eden, A.; Gaudet, F.; Waghmare, A.; Jaenisch, R. Chromosomal Instability and Tumors Promoted by DNA Hypomethylation. Science 2003, 300, 455. [Google Scholar] [CrossRef]
- Hegele, R.A. Drawing the Line in Progeria Syndromes. Lancet 2003, 362, 416–417. [Google Scholar] [CrossRef]
- Eriksson, M.; Brown, W.T.; Gordon, L.B.; Glynn, M.W.; Singer, J.; Scott, L.; Erdos, M.R.; Robbins, C.M.; Moses, T.Y.; Berglund, P.; et al. Recurrent de Novo Point Mutations in Lamin A Cause Hutchinson-Gilford Progeria Syndrome. Nature 2003, 423, 293–298. [Google Scholar]
- Yu, C.E.; Oshima, J.; Fu, Y.H.; Wijsman, E.M.; Hisama, F.; Alisch, R.; Matthews, S.; Nakura, J.; Miki, T.; Ouais, S.; Martin, G.M.; Mulligan, J.; Schellenberg, G.D. Positional Cloning of the Werner’s Syndrome Gene. Science 1996, 272, 258–262. [Google Scholar]
- Liu, B.; Wang, Z.; Zhang, L.; Ghosh, S.; Zheng, H.; Zhou, Z. Depleting the Methyltransferase Suv39h1 Improves DNA Repair and Extends Lifespan in a Progeria Mouse Model. Nat. Commun. 2013, 4, 2–12. [Google Scholar]
- Heyn, H.; Moran, S.; Esteller, M. Aberrant DNA Methylation Profiles in the Premature Aging Disorders Hutchinson-Gilford Progeria and Werner Syndrome. Epigenetics 2013, 8, 28–33. [Google Scholar]
- Hwang, I.K.; Moon, S.M.; Yoo, K.-Y.; Li, H.; Kwon, H.D.; Hwang, H.S.; Choi, S.K.; Lee, B.-H.; Kim, J.D.; Won, M.H. C-Myb Immunoreactivity, Protein and mRNA Levels Significantly Increase in the Aged Hippocampus Proper in Gerbils. Neurochem. Res. 2007, 32, 1091–1097. [Google Scholar] [CrossRef]
- Lee, Y.-H.; Lee, N.-H.; Bhattarai, G.; Hwang, P.-H.; Kim, T.-I.; Jhee, E.-C.; Yi, H.-K. C-Myb Has a Character of Oxidative Stress Resistance in Aged Human Diploid Fibroblasts: Regulates SAPK/JNK and Hsp60 Pathway Consequently. Biogerontology 2010, 11, 267–274. [Google Scholar] [CrossRef]
- Casillas, M.A.; Lopatina, N.; Andrews, L.G.; Tollefsbol, T.O. Transcriptional Control of the DNA Methyltransferases Is Altered in Aging and Neoplastically-Transformed Human Fibroblasts. Mol. Cell. Biochem. 2003, 252, 33–43. [Google Scholar] [CrossRef]
- Lopatina, N.; Haskell, J.F.; Andrews, L.G.; Poole, J.C.; Saldanha, S.; Tollefsbol, T. Differential Maintenance and de Novo Methylating Activity by Three DNA Methyltransferases in Aging and Immortalized Fibroblasts. J. Cell. Biochem. 2002, 84, 324–334. [Google Scholar] [CrossRef]
- Timp, W.; Feinberg, A. Cancer as a Dysregulated Epigenome Allowing Cellular Growth Advantage at the Expense of the Host. Nat. Rev. Cancer 2013, 13, 497–510. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Rang, F.J.; Boonstra, J. Causes and Consequences of Age-Related Changes in DNA Methylation: A Role for ROS? Biology 2014, 3, 403-425. https://doi.org/10.3390/biology3020403
Rang FJ, Boonstra J. Causes and Consequences of Age-Related Changes in DNA Methylation: A Role for ROS? Biology. 2014; 3(2):403-425. https://doi.org/10.3390/biology3020403
Chicago/Turabian StyleRang, Franka J., and Johannes Boonstra. 2014. "Causes and Consequences of Age-Related Changes in DNA Methylation: A Role for ROS?" Biology 3, no. 2: 403-425. https://doi.org/10.3390/biology3020403