Dnmt3b Prefers Germ Line Genes and Centromeric Regions: Lessons from the ICF Syndrome and Cancer and Implications for Diseases

Abstract
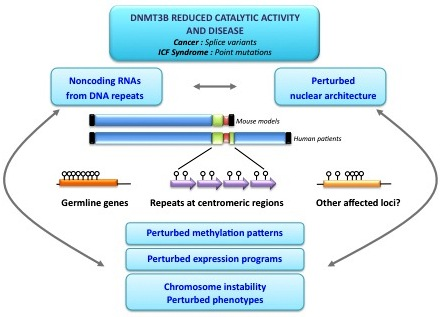
:
{kind=link}
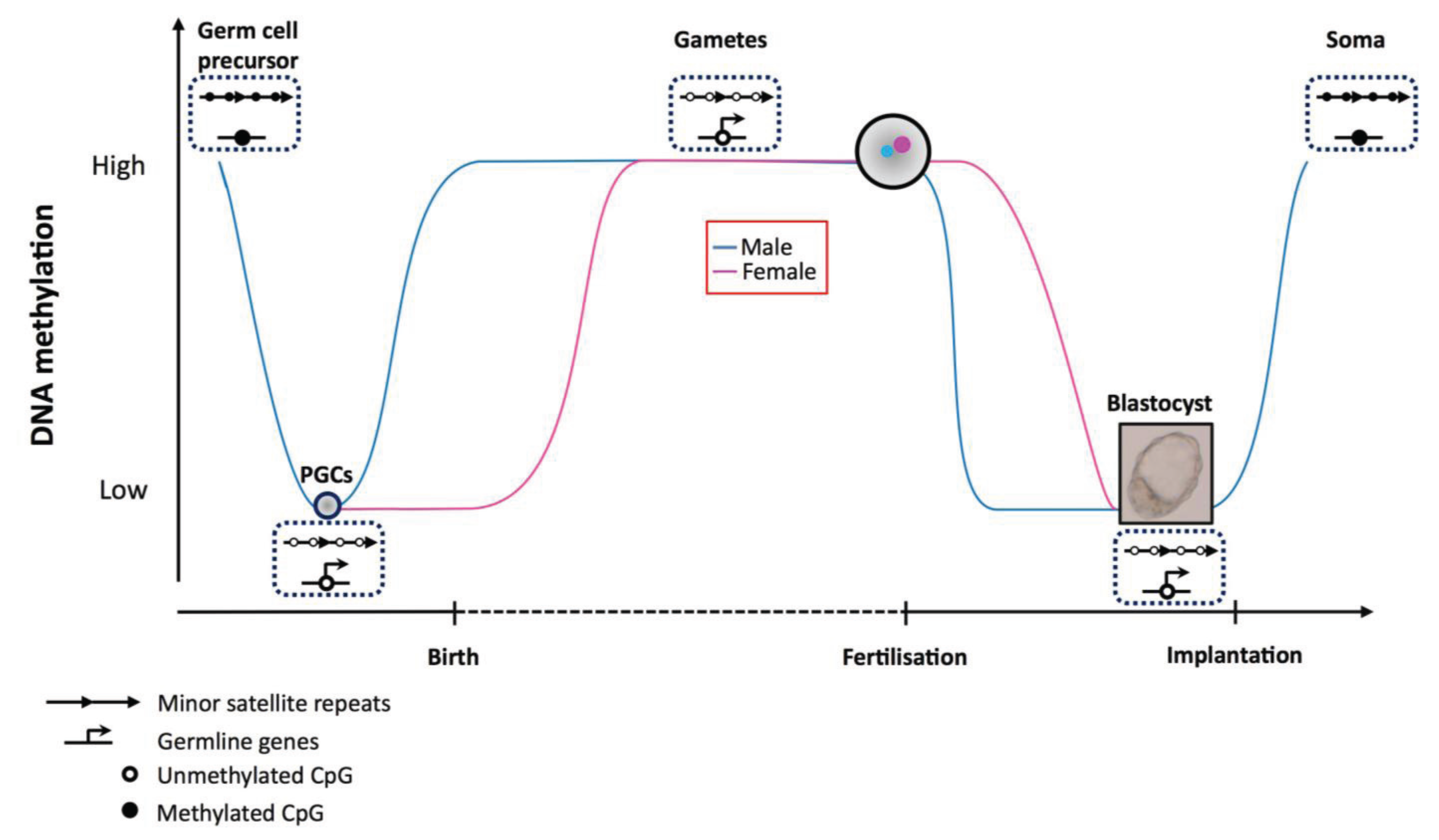{kind=link}
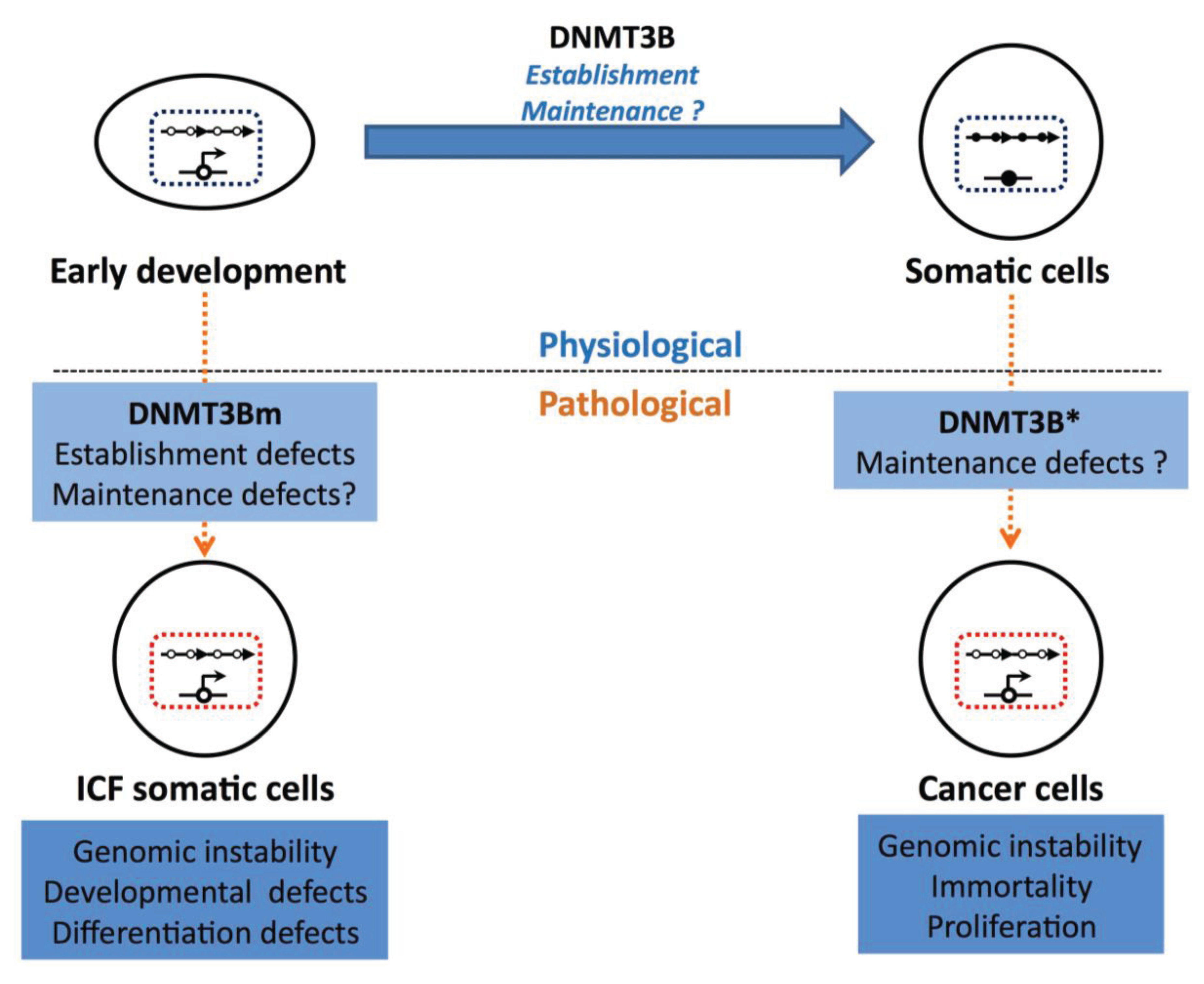{kind=link}
{kind=link}
1. An Introduction to DNA methylation
2. DNMT3B: A de novo Methyltransferase with Many Isoforms
3. Genomic Loci Affected by DNMT3B Loss of Function.
3.1. Centromeric and Pericentromeric DNA Repeats
3.2. Germ Line Genes
3.3. Other DNA Repeats
4. Dnmt3b and Development
4.1. Early Development

4.2. Targeting Dnmt3b during Development
5. DNMT3B and Disease
5.1. DNMT3B Variants in Cancer
5.2. Germ Line Mutations in DNMT3B in the ICF Syndrome
6. Consequences of Hypomethylation at DNMT3B Targets for Disease

6.1. Germ Line Genes and “Meiomitosis”
6.2. Transcription of Satellite Repeats and Chromosomal Instability
6.3. Hypomethylation of Satellite Repeats and Perturbed Nuclear Organization

7. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jones, P.A.; Taylor, S.M.; Wilson, V. DNA modification, differentiation, and transformation. J. Exp. Zool. 1983, 228, 287–295. [Google Scholar] [CrossRef]
- Meissner, A.; Mikkelsen, T.S.; Gu, H.; Wernig, M.; Hanna, J.; Sivachenko, A.; Zhang, X.; Bernstein, B.E.; Nusbaum, C.; Jaffe, D.B.; et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 2010, 454, 766–770. [Google Scholar]
- Weber, M.; Hellmann, I.; Stadler, M.B.; Ramos, L.; Paabo, S.; Rebhan, M.; Schubeler, D. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat. Genet. 2007, 39, 457–466. [Google Scholar] [CrossRef]
- Weber, M.; Schubeler, D. Genomic patterns of DNA methylation: Targets and function of an epigenetic mark. Curr. Opin. Cell Biol. 2007, 19, 273–280. [Google Scholar] [CrossRef]
- Illingworth, R.; Kerr, A.; Desousa, D.; Jorgensen, H.; Ellis, P.; Stalker, J.; Jackson, D.; Clee, C.; Plumb, R.; Rogers, J.; et al. A novel CpG island set identifies tissue-specific methylation at developmental gene loci. PLoS Biol. 2008, 6, e22. [Google Scholar]
- Razin, A.; Cedar, H. DNA methylation and gene expression. Microbiol. Rev. 1991, 55, 451–458. [Google Scholar]
- Bird, A.P.; Wolffe, A.P. Methylation-induced repression—Belts, braces, and chromatin. Cell 1999, 99, 451–454. [Google Scholar] [CrossRef]
- Shukla, S.; Kavak, E.; Gregory, M.; Imashimizu, M.; Shutinoski, B.; Kashlev, M.; Oberdoerffer, P.; Sandberg, R.; Oberdoerffer, S. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature 2011, 479, 74–79. [Google Scholar]
- Maunakea, A.K.; Nagarajan, R.P.; Bilenky, M.; Ballinger, T.J.; D'Souza, C.; Fouse, S.D.; Johnson, B.E.; Hong, C.; Nielsen, C.; Zhao, Y.; et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 2010, 466, 253–257. [Google Scholar] [CrossRef]
- Li, E.; Bestor, T.H.; Jaenisch, R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 1992, 69, 915–926. [Google Scholar] [CrossRef]
- Walsh, C.P.; Chaillet, J.R.; Bestor, T.H. Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nat. Genet. 1998, 20, 116–117. [Google Scholar] [CrossRef]
- Sasaki, H.; Jones, P.A.; Chaillet, J.R.; Ferguson-Smith, A.C.; Barton, S.C.; Reik, W.; Surani, M.A. Parental imprinting: Potentially active chromatin of the repressed maternal allele of the mouse insulin-like growth factor II (IGF2) gene. Genes Dev. 1992, 6, 1843–1856. [Google Scholar] [CrossRef]
- Li, E.; Beard, C.; Jaenisch, R. Role for DNA methylation in genomic imprinting. Nature 1993, 366, 362–365. [Google Scholar] [CrossRef]
- Panning, B.; Jaenisch, R. DNA hypomethylation can activate Xist expression and silence X-linked genes. Genes Dev. 1996, 10, 1991–2002. [Google Scholar] [CrossRef]
- Okano, M.; Xie, S.; Li, E. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat. Genet. 1998, 19, 219–220. [Google Scholar] [CrossRef]
- Bostick, M.; Kim, J.K.; Esteve, P.O.; Clark, A.; Pradhan, S.; Jacobsen, S.E. Uhrf1 plays a role in maintaining DNA methylation in mammalian cells. Science 2007, 317, 1760–1764. [Google Scholar]
- Hermann, A.; Goyal, R.; Jeltsch, A. The Dnmt1 DNA-(cytosine-C5)-methyltransferase methylates DNA processively with high preference for hemimethylated target sites. J. Biol. Chem. 2004, 279, 48350–48359. [Google Scholar] [CrossRef]
- Chedin, F.; Lieber, M.R.; Hsieh, C.L. The DNA methyltransferase-like protein DNMT3L stimulates de novo methylation by Dnmt3a. Proc. Natl. Acad. Sci. USA 2002, 99, 16916–16921. [Google Scholar] [CrossRef]
- Guenatri, M.; Duffie, R.; Iranzo, J.; Fauque, P.; Bourc’his, D. Plasticity in Dnmt3L-dependent and -independent modes of de novo methylation in the developing mouse embryo. Development 2013, 140, 562–572. [Google Scholar] [CrossRef]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef]
- Liang, G.; Chan, M.F.; Tomigahara, Y.; Tsai, Y.C.; Gonzales, F.A.; Li, E.; Laird, P.W.; Jones, P.A. Cooperativity between DNA methyltransferases in the maintenance methylation of repetitive elements. Mol. Cell Biol. 2002, 22, 480–491. [Google Scholar] [CrossRef]
- Chen, T.; Ueda, Y.; Dodge, J.E.; Wang, Z.; Li, E. Establishment and maintenance of genomic methylation patterns in mouse embryonic stem cells by Dnmt3a and Dnmt3b. Mol. Cell. Biol. 2003, 23, 5594–5605. [Google Scholar]
- Walton, E.L.; Francastel, C.; Velasco, G. Maintenance of DNA methylation: Dnmt3b joins the dance. Epigenetics 2011, 6, 1373–1377. [Google Scholar] [CrossRef]
- Chen, T.; Tsujimoto, N.; Li, E. The PWWP domain of Dnmt3a and Dnmt3b is required for directing DNA methylation to the major satellite repeats at pericentric heterochromatin. Mol. Cell Biol. 2004, 24, 9048–9058. [Google Scholar] [CrossRef]
- Bachman, K.E.; Rountree, M.R.; Baylin, S.B. Dnmt3a and Dnmt3b are transcriptional repressors that exhibit unique localization properties to heterochromatin. J. Biol. Chem. 2001, 276, 32282–32287. [Google Scholar] [CrossRef]
- Zhang, Y.; Jurkowska, R.; Soeroes, S.; Rajavelu, A.; Dhayalan, A.; Bock, I.; Rathert, P.; Brandt, O.; Reinhardt, R.; Fischle, W.; et al. Chromatin methylation activity of Dnmt3a and Dnmt3a/3L is guided by interaction of the add domain with the histone H3 tail. Nucl. Acids Res. 2010, 38, 4246–4253. [Google Scholar] [CrossRef]
- Gordon, C.A.; Hartono, S.R.; Chedin, F. Inactive DNMT3B splice variants modulate de novo DNA methylation. PLoS One 2013, 8, e69486. [Google Scholar] [CrossRef]
- Gopalakrishnan, S.; van Emburgh, B.O.; Shan, J.; Su, Z.; Fields, C.R.; Vieweg, J.; Hamazaki, T.; Schwartz, P.H.; Terada, N.; Robertson, K.D. A novel DNMT3B splice variant expressed in tumor and pluripotent cells modulates genomic DNA methylation patterns and displays altered DNA binding. Mol. Cancer Res. 2009, 7, 1622–1634. [Google Scholar] [CrossRef]
- Ostler, K.R.; Davis, E.M.; Payne, S.L.; Gosalia, B.B.; Exposito-Cespedes, J.; le Beau, M.M.; Godley, L.A. Cancer cells express aberrant DNMT3B transcripts encoding truncated proteins. Oncogene 2007, 26, 5553–5563. [Google Scholar] [CrossRef]
- Saito, Y.; Kanai, Y.; Sakamoto, M.; Saito, H.; Ishii, H.; Hirohashi, S. Overexpression of a splice variant of DNA methyltransferase 3b, DNMT3B4, associated with DNA hypomethylation on pericentromeric satellite regions during human hepatocarcinogenesis. Proc. Natl. Acad. Sci. USA 2002, 99, 10060–10065. [Google Scholar] [CrossRef]
- Wang, L.; Wang, J.; Sun, S.; Rodriguez, M.; Yue, P.; Jang, S.J.; Mao, L. A novel DNMT3B subfamily, deltaDNMT3B, is the predominant form of DNMT3B in non-small cell lung cancer. Int. J. Oncol. 2008, 29, 201–207. [Google Scholar]
- Weisenberger, D.J.; Velicescu, M.; Cheng, J.C.; Gonzales, F.A.; Liang, G.; Jones, P.A. Role of the DNA methyltransferase variant Dnmt3b3 in DNA methylation. Mol. Cancer Res. 2004, 2, 62–72. [Google Scholar]
- Weisenberger, D.J.; Velicescu, M.; Preciado-Lopez, M.A.; Gonzales, F.A.; Tsai, Y.C.; Liang, G.; Jones, P.A. Identification and characterization of alternatively spliced variants of DNA methyltransferase 3a in mammalian cells. Gene 2002, 298, 91–99. [Google Scholar] [CrossRef]
- Huntriss, J.; Hinkins, M.; Oliver, B.; Harris, S.E.; Beazley, J.C.; Rutherford, A.J.; Gosden, R.G.; Lanzendorf, S.E.; Picton, H.M. Expression of mRNAs for DNA methyltransferases and methyl-CpG-binding proteins in the human female germ line, preimplantation embryos, and embryonic stem cells. Mol. Reprod. Dev. 2004, 67, 323–336. [Google Scholar] [CrossRef]
- Robertson, K.D.; Uzvolgyi, E.; Liang, G.; Talmadge, C.; Sumegi, J.; Gonzales, F.A.; Jones, P.A. The human DNA methyltransferases (DNMTs) 1, 3a and 3b: Coordinate mRNA expression in normal tissues and overexpression in tumors. Nucl. Acids Res. 1999, 27, 2291–2298. [Google Scholar] [CrossRef]
- Xu, G.L.; Bestor, T.H.; Bourc’his, D.; Hsieh, C.L.; Tommerup, N.; Bugge, M.; Hulten, M.; Qu, X.; Russo, J.J.; Viegas-Pequignot, E. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature 1999, 402, 187–191. [Google Scholar] [CrossRef]
- Guenatri, M.; Bailly, D.; Maison, C.; Almouzni, G. Mouse centric and pericentric satellite repeats form distinct functional heterochromatin. J. Cell. Biol. 2004, 166, 493–505. [Google Scholar] [CrossRef]
- Yamagishi, Y.; Sakuno, T.; Shimura, M.; Watanabe, Y. Heterochromatin links to centromeric protection by recruiting shugoshin. Nature 2008, 455, 251–255. [Google Scholar] [CrossRef]
- Choo, K.H. The Centromere; Oxford University Press Inc.: New York, NY, USA, 1997. [Google Scholar]
- Martens, J.H.; O’Sullivan, R.J.; Braunschweig, U.; Opravil, S.; Radolf, M.; Steinlein, P.; Jenuwein, T. The profile of repeat-associated histone lysine methylation states in the mouse epigenome. EMBO J. 2005, 24, 800–812. [Google Scholar] [CrossRef]
- Vourc’h, C.; Biamonti, G. Transcription of satellite DNAs in mammals. Prog. Mol. Subcell. Biol. 2011, 51, 95–118. [Google Scholar] [CrossRef]
- Choo, K.H. Domain organization at the centromere and neocentromere. Dev. Cell. 2001, 1, 165–177. [Google Scholar]
- Earnshaw, W.C.; Cooke, C.A. Proteins of the inner and outer centromere of mitotic chromosomes. Genome 1989, 31, 541–552. [Google Scholar]
- Eymery, A.; Callanan, M.; Vourc’h, C. The secret message of heterochromatin: New insights into the mechanisms and function of centromeric and pericentric repeat sequence transcription. Int. J. Dev. Biol. 2009, 53, 259–268. [Google Scholar]
- Hall, L.E.; Mitchell, S.E.; O’Neill, R.J. Pericentric and centromeric transcription: A perfect balance required. Chromosom. Res. 2012, 20, 535–546. [Google Scholar] [CrossRef]
- Bouzinba-Segard, H.; Guais, A.; Francastel, C. Accumulation of small murine minor satellite transcripts leads to impaired centromeric architecture and function. Proc. Natl. Acad. Sci. USA 2006, 103, 8709–8714. [Google Scholar] [CrossRef]
- Ferri, F.; Bouzinba-Segard, H.; Velasco, G.; Hubé, F.; Francastel, C. Non-coding murine centromeric transcripts associate with and potentiate Aurora B kinase. Nucl. Acids Res. 2009, 37, 5071–5080. [Google Scholar] [CrossRef]
- Probst, A.V.; Okamoto, I.; Casanova, M.; El Marjou, F.; Le Baccon, P.; Almouzni, G. A strand-specific burst in transcription of pericentric satellites is required for chromocenter formation and early mouse development. Dev. Cell. 2010, 19, 625–638. [Google Scholar] [CrossRef]
- Borgel, J.; Guibert, S.; Li, Y.; Chiba, H.; Schubeler, D.; Sasaki, H.; Forne, T.; Weber, M. Targets and dynamics of promoter DNA methylation during early mouse development. Nat. Genet. 2010, 42, 1093–1100. [Google Scholar] [CrossRef]
- Velasco, G.; Hube, F.; Rollin, J.; Neuillet, D.; Philippe, C.; Bouzinba-Segard, H.; Galvani, A.; Viegas-Pequignot, E.; Francastel, C. Dnmt3b recruitment through E2F6 transcriptional repressor mediates germ-line gene silencing in murine somatic tissues. Proc. Natl. Acad. Sci. USA 2010, 107, 9281–9286. [Google Scholar] [CrossRef]
- De Smet, C.; Lurquin, C.; Lethe, B.; Martelange, V.; Boon, T. DNA methylation is the primary silencing mechanism for a set of germ line- and tumor-specific genes with a CpG-rich promoter. Mol. Cell. Biol. 1999, 19, 7327–7335. [Google Scholar]
- Maatouk, D.M.; Kellam, L.D.; Mann, M.R.; Lei, H.; Li, E.; Bartolomei, M.S.; Resnick, J.L. DNA methylation is a primary mechanism for silencing postmigratory primordial germ cell genes in both germ cell and somatic cell lineages. Development 2006, 133, 3411–3418. [Google Scholar] [CrossRef]
- Hackett, J.A.; Reddington, J.P.; Nestor, C.E.; Dunican, D.S.; Branco, M.R.; Reichmann, J.; Reik, W.; Surani, M.A.; Adams, I.R.; Meehan, R.R. Promoter DNA methylation couples genome-defence mechanisms to epigenetic reprogramming in the mouse germline. Development 2012, 139, 3623–3632. [Google Scholar] [CrossRef]
- Tsien, F.; Sun, B.; Hopkins, N.E.; Vedanarayanan, V.; Figlewicz, D.; Winokur, S.; Ehrlich, M. Methylation of the FSHD syndrome-linked subtelomeric repeat in normal and FSHD cell cultures and tissues. Mol. Genet. Metab. 2001, 74, 322–331. [Google Scholar] [CrossRef]
- Nishiyama, R.; Qi, L.; Tsumagari, K.; Weissbecker, K.; Dubeau, L.; Champagne, M.; Sikka, S.; Nagai, H.; Ehrlich, M. A DNA repeat, NBL2, is hypermethylated in some cancers but hypomethylated in others. Cancer Biol. Ther. 2005, 4, 440–448. [Google Scholar]
- Kondo, T.; Bobek, M.P.; Kuick, R.; Lamb, B.; Zhu, X.; Narayan, A.; Bourc’his, D.; Viegas-Pequignot, E.; Ehrlich, M.; Hanash, S.M. Whole-genome methylation scan in ICF syndrome: Hypomethylation of non-satellite DNA repeats D4Z4 and NBL2. Hum. Mol. Genet. 2000, 9, 597–604. [Google Scholar] [CrossRef]
- Miniou, P.; Bourc’his, D.; Molina Gomes, D.; Jeanpierre, M.; Viegas-Pequignot, E. Undermethylation of Alu sequences in ICF syndrome: Molecular and in situ analysis. Cytogenet. Cell. Genet. 1997, 77, 308–313. [Google Scholar] [CrossRef]
- Yehezkel, S.; Segev, Y.; Viegas-Pequignot, E.; Skorecki, K.; Selig, S. Hypomethylation of subtelomeric regions in ICF syndrome is associated with abnormally short telomeres and enhanced transcription from telomeric regions. Hum. Mol. Genet. 2008, 17, 2776–2789. [Google Scholar] [CrossRef]
- Brock, G.J.; Charlton, J.; Bird, A. Densely methylated sequences that are preferentially localized at telomere-proximal regions of human chromosomes. Gene 1999, 240, 269–277. [Google Scholar]
- Craig, J.M.; Earle, E.; Canham, P.; Wong, L.H.; Anderson, M.; Choo, K.H. Analysis of mammalian proteins involved in chromatin modification reveals new metaphase centromeric proteins and distinct chromosomal distribution patterns. Hum. Mol. Genet. 2003, 12, 3109–3121. [Google Scholar] [CrossRef]
- Ueda, Y.; Okano, M.; Williams, C.; Chen, T.; Georgopoulos, K.; Li, E. Roles for Dnmt3b in mammalian development: A mouse model for the ICF syndrome. Development 2006, 133, 1183–1192. [Google Scholar]
- Marks, H.; Kalkan, T.; Menafra, R.; Denissov, S.; Jones, K.; Hofemeister, H.; Nichols, J.; Kranz, A.; Stewart, A.F.; Smith, A.; et al. The transcriptional and epigenomic foundations of ground state pluripotency. Cell. 2012, 149, 590–604. [Google Scholar] [CrossRef]
- Kriaucionis, S.; Heintz, N. The nuclear DNA base 5-hydroxymethylcytosine is present in purkinje neurons and the brain. Science 2009, 324, 929–930. [Google Scholar]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by mll partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef]
- Seisenberger, S.; Peat, J.R.; Hore, T.A.; Santos, F.; Dean, W.; Reik, W. Reprogramming DNA methylation in the mammalian life cycle: Building and breaking epigenetic barriers. Philos. Trans. R Soc. Lond. B Biol. Sci. 2013, 368, 20110330. [Google Scholar]
- Guibert, S.; Weber, M. Functions of DNA methylation and hydroxymethylation in mammalian development. Curr. Top. Dev. Biol. 2013, 104, 47–83. [Google Scholar] [CrossRef]
- Sakaue, M.; Ohta, H.; Kumaki, Y.; Oda, M.; Sakaide, Y.; Matsuoka, C.; Yamagiwa, A.; Niwa, H.; Wakayama, T.; Okano, M. DNA methylation is dispensable for the growth and survival of the extraembryonic lineages. Curr. Biol. 2010, 20, 1452–1457. [Google Scholar] [CrossRef]
- Tsumura, A.; Hayakawa, T.; Kumaki, Y.; Takebayashi, S.; Sakaue, M.; Matsuoka, C.; Shimotohno, K.; Ishikawa, F.; Li, E.; Ueda, H.R.; et al. Maintenance of self-renewal ability of mouse embryonic stem cells in the absence of DNA methyltransferases Dnmt1, Dnmt3a and Dnmt3b. Genes Cells 2006, 11, 805–814. [Google Scholar]
- Jackson-Grusby, L.; Beard, C.; Possemato, R.; Tudor, M.; Fambrough, D.; Csankovszki, G.; Dausman, J.; Lee, P.; Wilson, C.; Lander, E.; et al. Loss of genomic methylation causes p53-dependent apoptosis and epigenetic deregulation. Nat. Genet. 2001, 27, 31–39. [Google Scholar] [CrossRef]
- Senner, C.E.; Krueger, F.; Oxley, D.; Andrews, S.; Hemberger, M. DNA methylation profiles define stem cell identity and reveal a tight embryonic-extraembryonic lineage boundary. Stem Cells 2012, 30, 2732–2745. [Google Scholar]
- Veillard, A.C.; Marks, H.; Bernardo, A.S.; Jouneau, L.; Laloe, D.; Boulanger, L.; Kaan, A.; Brochard, V.; Tosolini, M.; Pedersen, R.; et al. Stable methylation at promoters distinguishes epiblast stem cells from embryonic stem cells and the in vivo epiblast. Stem Cells Dev. 2014, 23, 2014–2029. [Google Scholar]
- Habibi, E.; Brinkman, A.B.; Arand, J.; Kroeze, L.I.; Kerstens, H.H.; Matarese, F.; Lepikhov, K.; Gut, M.; Brun-Heath, I.; Hubner, N.C.; et al. Whole-genome bisulfite sequencing of two distinct interconvertible DNA methylomes of mouse embryonic stem cells. Cell. Stem Cell. 2013, 13, 360–369. [Google Scholar] [CrossRef]
- Leitch, H.G.; McEwen, K.R.; Turp, A.; Encheva, V.; Carroll, T.; Grabole, N.; Mansfield, W.; Nashun, B.; Knezovich, J.G.; Smith, A.; et al. Naive pluripotency is associated with global DNA hypomethylation. Nat. Struct. Mol. Biol. 2013, 20, 311–316. [Google Scholar]
- Arand, J.; Spieler, D.; Karius, T.; Branco, M.R.; Meilinger, D.; Meissner, A.; Jenuwein, T.; Xu, G.; Leonhardt, H.; Wolf, V.; et al. In vivo control of CpG and non- CpG DNA methylation by DNA methyltransferases. PLoS Genet 2012, 8, e1002750. [Google Scholar]
- Zvetkova, I.; Apedaile, A.; Ramsahoye, B.; Mermoud, J.E.; Crompton, L.A.; John, R.; Feil, R.; Brockdorff, N. Global hypomethylation of the genome in XX embryonic stem cells. Nat. Genet. 2005, 37, 1274–1279. [Google Scholar] [CrossRef]
- Grabole, N.; Tischler, J.; Hackett, J.A.; Kim, S.; Tang, F.; Leitch, H.G.; Magnusdottir, E.; Surani, M.A. Prdm14 promotes germline fate and naive pluripotency by repressing FGF signalling and DNA methylation. EMBO Rep. 2013, 14, 629–637. [Google Scholar] [CrossRef]
- Guibert, S.; Forne, T.; Weber, M. Global profiling of DNA methylation erasure in mouse primordial germ cells. Genome Res. 2012, 22, 633–641. [Google Scholar] [CrossRef]
- Hajkova, P.; Erhardt, S.; Lane, N.; Haaf, T.; El-Maarri, O.; Reik, W.; Walter, J.; Surani, M.A. Epigenetic reprogramming in mouse primordial germ cells. Mech. Dev. 2002, 117, 15–23. [Google Scholar]
- Lees-Murdock, D.J.; de Felici, M.; Walsh, C.P. Methylation dynamics of repetitive DNA elements in the mouse germ cell lineage. Genomics 2003, 82, 230–237. [Google Scholar] [CrossRef]
- La Salle, S.; Mertineit, C.; Taketo, T.; Moens, P.B.; Bestor, T.H.; Trasler, J.M. Windows for sex-specific methylation marked by DNA methyltransferase expression profiles in mouse germ cells. Dev. Biol. 2004, 268, 403–415. [Google Scholar]
- Yamagata, K.; Yamazaki, T.; Miki, H.; Ogonuki, N.; Inoue, K.; Ogura, A.; Baba, T. Centromeric DNA hypomethylation as an epigenetic signature discriminates between germ and somatic cell lineages. Dev. Biol. 2007, 312, 419–426. [Google Scholar] [CrossRef]
- Riggs, A.D.; Xiong, Z. Methylation and epigenetic fidelity. Proc. Natl. Acad. Sci. USA 2004, 101, 4–5. [Google Scholar] [CrossRef]
- Brandeis, M.; Frank, D.; Keshet, I.; SIegfried, Z.; Mendelsohn, M.; Nemes, A.; Temper, V.; Razin, A.; Cedar, H. Sp1 elements protect a CpG island from de novo methylation. Nature 1994, 371, 435–438. [Google Scholar]
- Macleod, D.; Charlton, J.; Cullins, J.; Bird, A.P. Sp1 sites in the mouse Aprt gene promoter are required to prevent methylation of the CpG island. Genes Dev. 1994, 8, 2282–2292. [Google Scholar]
- Lienert, F.; Wirbelauer, C.; Som, I.; Dean, A.; Mohn, F.; Schubeler, D. Identification of genetic elements that autonomously determine DNA methylation states. Nat. Genet. 2011, 43, 1091–1097. [Google Scholar]
- Wienholz, B.L.; Kareta, M.S.; Moarefi, A.H.; Gordon, C.A.; Ginno, P.A.; Chedin, F. Dnmt3l modulates significant and distinct flanking sequence preference for DNA methylation by Dnmt3a and Dnmt3b in vivo. PLoS Genet. 2010, 6, 1001106. [Google Scholar]
- Hervouet, E.; Vallette, F.M.; Cartron, P.F. Dnmt3/transcription factor interactions as crucial players in targeted DNA methylation. Epigenetics 2009, 4, 487–499. [Google Scholar]
- Maeda, I.; Okamura, D.; Tokitake, Y.; Ikeda, M.; Kawaguchi, H.; Mise, N.; Abe, K.; Noce, T.; Okuda, A.; Matsui, Y. Max is a repressor of germ cell-related gene expression in mouse embryonic stem cells. Nat. Commun. 2013, 4, 1754. [Google Scholar]
- Gopalakrishnan, S.; Sullivan, B.A.; Trazzi, S.; Della Valle, G.; Robertson, K.D. DNMT3B interacts with constitutive centromere protein CENP-C to modulate DNA methylation and the histone code at centromeric regions. Hum. Mol. Genet. 2009, 18, 3178–3193. [Google Scholar] [CrossRef]
- Dennis, K.; Fan, T.; Geiman, T.; Yan, Q.; Muegge, K. Lsh, a member of the SNF2 family, is required for genome-wide methylation. Genes Dev. 2001, 15, 2940–2944. [Google Scholar]
- Geiman, T.M.; Tessarollo, L.; Anver, M.R.; Kopp, J.B.; Ward, J.M.; Muegge, K. Lsh, a SNF2 family member, is required for normal murine development. Biochim. Biophys. Acta 2001, 1526, 211–220. [Google Scholar]
- Huang, J.; Fan, T.; Yan, Q.; Zhu, H.; Fox, S.; Issaq, H.J.; Best, L.; Gangi, L.; Munroe, D.; Muegge, K. Lsh, an epigenetic guardian of repetitive elements. Nucl. Acids Res. 2004, 32, 5019–5028. [Google Scholar]
- Myant, K.; Termanis, A.; Sundaram, A.Y.; Boe, T.; Li, C.; Merusi, C.; Burrage, J.; de Las Heras, J.I.; Stancheva, I. Lsh and G9a/GLP complex are required for developmentally programmed DNA methylation. Genome Res. 2011, 21, 83–94. [Google Scholar]
- Tao, Y.; Xi, S.; Shan, J.; Maunakea, A.; Che, A.; Briones, V.; Lee, E.Y.; Geiman, T.; Huang, J.; Stephens, R.; et al. Lsh, chromatin remodeling family member, modulates genome-wide cytosine methylation patterns at nonrepeat sequences. Proc. Natl. Acad. Sci. USA 2011, 108, 5626–5631. [Google Scholar] [CrossRef]
- Xi, S.; Geiman, T.M.; Briones, V.; Guang Tao, Y.; Xu, H.; Muegge, K. Lsh participates in DNA methylation and silencing of stem cell genes. Stem Cells 2009, 27, 2691–2702. [Google Scholar] [CrossRef]
- Dunican, D.S.; Cruickshanks, H.A.; Suzuki, M.; Semple, C.A.; Davey, T.; Arceci, R.J.; Greally, J.; Adams, I.R.; Meehan, R.R. Lsh regulates LTR retrotransposon repression independently of Dnmt3b function. Genome Biol. 2013, 14, R146. [Google Scholar] [CrossRef]
- Myant, K.; Stancheva, I. Lsh cooperates with DNA methyltransferases to repress transcription. Mol. Cell. Biol. 2008, 28, 215–226. [Google Scholar] [CrossRef]
- Aravin, A.A.; Sachidanandam, R.; Girard, A.; Fejes-Toth, K.; Hannon, G.J. Developmentally regulated piRNA clusters implicate MILI in transposon control. Science 2007, 316, 744–747. [Google Scholar] [CrossRef]
- Schmitz, K.M.; Mayer, C.; Postepska, A.; Grummt, I. Interaction of noncoding RNA with the rdna promoter mediates recruitment of Dnmt3b and silencing of rRNA genes. Genes Dev. 2010, 24, 2264–2269. [Google Scholar]
- Volpe, T.A.; Kidner, C.; Hall, I.M.; Teng, G.; Grewal, S.I.; Martienssen, R.A. Regulation of heterochromatic silencing and histone h3 lysine-9 methylation by RNAi. Science 2002, 297, 1833–1837. [Google Scholar] [CrossRef]
- Scarano, M.I.; Strazzullo, M.; Matarazzo, M.R.; D’Esposito, M. DNA methylation 40 years later: Its role in human health and disease. J. Cell. Physiol. 2005, 204, 21–35. [Google Scholar]
- Wilson, A.S.; Power, B.E.; Molloy, P.L. DNA hypomethylation and human diseases. Biochim. Biophys. Acta. 2007, 1775, 138–162. [Google Scholar]
- Robertson, K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005, 6, 597–610. [Google Scholar]
- Gronbaek, K.; Hother, C.; Jones, P.A. Epigenetic changes in cancer. APMIS 2007, 115, 1039–1059. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Vogelstein, B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983, 301, 89–92. [Google Scholar] [CrossRef]
- Ehrlich, M. DNA hypomethylation in cancer cells. Epigenomics 2009, 1, 239–259. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Vogelstein, B. Hypomethylation of Ras oncogenes in primary human cancers. Biochem. Biophys. Res. Commun. 1983, 111, 47–54. [Google Scholar] [CrossRef]
- Scanlan, M.J.; Gure, A.O.; Jungbluth, A.A.; Old, L.J.; Chen, Y.T. Cancer/testis antigens: An expanding family of targets for cancer immunotherapy. Immunol. Rev. 2002, 188, 22–32. [Google Scholar] [CrossRef]
- Sincic, N.; Herceg, Z. DNA methylation and cancer: Ghosts and angels above the genes. Curr. Opin. Oncol. 2011, 23, 69–76. [Google Scholar] [CrossRef]
- Vasanthakumar, A.; Lepore, J.B.; Zegarek, M.H.; Kocherginsky, M.; Singh, M.; Davis, E.M.; Link, P.A.; Anastasi, J.; Le Beau, M.M.; Karpf, A.R.; et al. Dnmt3b is a haploinsufficient tumor suppressor gene in myc-induced lymphomagenesis. Blood 2013, 121, 2059–2063. [Google Scholar] [CrossRef]
- Van Emburgh, B.O.; Robertson, K.D. Modulation of Dnmt3b function in vitro by interactions with Dnmt3l, Dnmt3a and Dnmt3b splice variants. Nucl. Acids Res. 2011, 39, 4984–5002. [Google Scholar] [CrossRef]
- Almeida, L.G.; Sakabe, N.J.; de Oliveira, A.R.; Silva, M.C.; Mundstein, A.S.; Cohen, T.; Chen, Y.T.; Chua, R.; Gurung, S.; Gnjatic, S.; et al. CTdatabase: a knowledge-base of high-throughput and curated data on cancer-testis antigens. Nucl. Acids Res. 2009, 37, D816–D819. [Google Scholar] [CrossRef]
- Hansen, R.S.; Wijmenga, C.; Luo, P.; Stanek, A.M.; Canfield, T.K.; Weemaes, C.M.; Gartler, S.M. The Dnmt3b DNA methyltransferase gene is mutated in the icf immunodeficiency syndrome. Proc. Natl. Acad. Sci. USA 1999, 96, 14412–14417. [Google Scholar]
- Fryns, J.P.; Azou, M.; Jaeken, J.; Eggermont, E.; Pedersen, J.C.; van den Berghe, H. Centromeric instability of chromosomes 1, 9, and 16 associated with combined immunodeficiency. Hum. Genet. 1981, 57, 108–110. [Google Scholar]
- Tiepolo, L.; Maraschio, P.; Gimelli, G.; Cuoco, C.; Gargani, G.F.; Romano, C. Multibranched chromosomes 1, 9, and 16 in a patient with combined IGA and IGE deficiency. Hum. Genet. 1979, 51, 127–137. [Google Scholar] [CrossRef]
- Ehrlich, M.; Buchanan, K.L.; Tsien, F.; Jiang, G.; Sun, B.; Uicker, W.; Weemaes, C.M.; Smeets, D.; Sperling, K.; Belohradsky, B.H.; et al. DNA methyltransferase 3B mutations linked to the ICF syndrome cause dysregulation of lymphogenesis genes. Hum. Mol. Genet. 2001, 10, 2917–2931. [Google Scholar] [CrossRef]
- Hagleitner, M.M.; Lankester, A.; Maraschio, P.; Hulten, M.; Fryns, J.P.; Schuetz, C.; Gimelli, G.; Davies, E.G.; Gennery, A.; Belohradsky, B.H.; et al. Clinical spectrum of immunodeficiency, centromeric instability and facial dysmorphism (icf syndrome). J. Med. Genet. 2008, 45, 93–99. [Google Scholar]
- Carpenter, N.J.; Filipovich, A.; Blaese, R.M.; Carey, T.L.; Berkel, A.I. Variable immunodeficiency with abnormal condensation of the heterochromatin of chromosomes 1, 9, and 16. J. Pediatr. 1988, 112, 757–760. [Google Scholar] [CrossRef]
- Maraschio, P.; Zuffardi, O.; Dalla Fior, T.; Tiepolo, L. Immunodeficiency, centromeric heterochromatin instability of chromosomes 1, 9, and 16, and facial anomalies: The ICF syndrome. J. Med. Genet. 1988, 25, 173–180. [Google Scholar] [CrossRef]
- Tuck-Muller, C.M.; Narayan, A.; Tsien, F.; Smeets, D.F.; Sawyer, J.; Fiala, E.S.; Sohn, O.S.; Ehrlich, M. DNA hypomethylation and unusual chromosome instability in cell lines from ICF syndrome patients. Cytogenet. Cell. Genet. 2000, 89, 121–128. [Google Scholar] [CrossRef]
- Jeanpierre, M.; Turleau, C.; Aurias, A.; Prieur, M.; Ledeist, F.; Fischer, A.; Viegas-Pequignot, E. An embryonic-like methylation pattern of classical satellite DNA is observed in icf syndrome. Hum. Mol. Genet. 1993, 2, 731–735. [Google Scholar] [CrossRef]
- Ji, W.; Hernandez, R.; Zhang, X.Y.; Qu, G.Z.; Frady, A.; Varela, M.; Ehrlich, M. DNA demethylation and pericentromeric rearrangements of chromosome 1. Mutat. Res. 1997, 379, 33–41. [Google Scholar]
- Jiang, Y.L.; Rigolet, M.; Bourc’his, D.; Nigon, F.; Bokesoy, I.; Fryns, J.P.; Hulten, M.; Jonveaux, P.; Maraschio, P.; Megarbane, A.; et al. Dnmt3b mutations and DNA methylation defect define two types of ICF syndrome. Hum. Mutat. 2005, 25, 56–63. [Google Scholar]
- Wijmenga, C.; Hansen, R.S.; Gimelli, G.; Bjorck, E.J.; Davies, E.G.; Valentine, D.; Belohradsky, B.H.; van Dongen, J.J.; Smeets, D.F.; van den Heuvel, L.P.; et al. Genetic variation in ICF syndrome: Evidence for genetic heterogeneity. Hum. Mutat. 2000, 16, 509–517. [Google Scholar]
- Gowher, H.; Jeltsch, A. Molecular enzymology of the catalytic domains of the Dnmt3a and Dnmt3b DNA methyltransferases. J. Biol. Chem. 2002, 277, 20409–20414. [Google Scholar]
- Moarefi, A.H.; Chedin, F. ICF syndrome mutations cause a broad spectrum of biochemical defects in Dnmt3b-mediated de novo DNA methylation. J. Mol. Biol. 2011, 409, 758–772. [Google Scholar] [CrossRef]
- De Greef, J.C.; Wang, J.; Balog, J.; den Dunnen, J.T.; Frants, R.R.; Straasheijm, K.R.; Aytekin, C.; van der Burg, M.; Duprez, L.; Ferster, A.; et al. Mutations in ZBTB24 are associated with immunodeficiency, centromeric instability, and facial anomalies syndrome type 2. Am. J. Hum. Genet. 2011, 88, 796–804. [Google Scholar]
- Lee, S.U.; Maeda, T. Pok/zbtb proteins: An emerging family of proteins that regulate lymphoid development and function. Immunol. Rev. 2012, 247, 107–119. [Google Scholar] [CrossRef]
- Weemaes, C.M.; van Tol, M.J.; Wang, J.; van Ostaijen-Ten Dam, M.M.; van Eggermond, M.C.; Thijssen, P.E.; Aytekin, C.; Brunetti-Pierri, N.; van der Burg, M.; Graham Davies, E.; et al. Heterogeneous clinical presentation in icf syndrome: Correlation with underlying gene defects. Eur. J. Hum. Genet. 2013, 21, 1219–1225. [Google Scholar] [CrossRef]
- Velasco, G.; Walton, E.L.; Sterlin, D.; Hedouin, S.; Nitta, H.; Yuya, I.; Fouyssac, F.; Megarbane, A.; Sasaki, H.; Picard, C.; et al. Germline genes hypomethylation and expression define a molecular signature in peripheral blood of ICF patients: Implications for diagnosis and etiology. Orphanet. J. Rare Dis. 2014, 9, 56. [Google Scholar] [CrossRef] [Green Version]
- Nitta, H.; Unoki, M.; Ichiyanagi, K.; Kosho, T.; Shigemura, T.; Takahashi, H.; Velasco, G.; Francastel, C.; Picard, C.; Kubota, T.; et al. Three novel ZBTB24 mutations identified in japanese and cape verdean type 2 ICF syndrome patients. J. Hum. Genet. 2013, 58, 455–460. [Google Scholar] [CrossRef]
- Van den Brand, M.; Flucke, U.E.; Bult, P.; Weemaes, C.M.; van Deuren, M. Angiosarcoma in a patient with immunodeficiency, centromeric region instability, facial anomalies (ICF) syndrome. Am. J. Med. Genet. 2011, 155A, 622–625. [Google Scholar]
- Narayan, A.; Tuck-Muller, C.; Weissbecker, K.; Smeets, D.; Ehrlich, M. Hypersensitivity to radiation-induced non-apoptotic and apoptotic death in cell lines from patients with the ICF chromosome instability syndrome. Mutat. Res. 2000, 456, 1–15. [Google Scholar]
- Wang, J.; Emadali, A.; le Bescont, A.; Callanan, M.; Rousseaux, S.; Khochbin, S. Induced malignant genome reprogramming in somatic cells by testis-specific factors. Biochim. Biophys. Acta. 2011, 1809, 221–225. [Google Scholar]
- Rousseaux, S.; Wang, J.; Khochbin, S. Cancer hallmarks sustained by ectopic activations of placenta/male germline genes. Cell. Cycle 2013, 12, 2331–2332. [Google Scholar] [CrossRef]
- Rousseaux, S.; Debernardi, A.; Jacquiau, B.; Vitte, A.L.; Vesin, A.; Nagy-Mignotte, H.; Moro-Sibilot, D.; Brichon, P.Y.; Lantuejoul, S.; Hainaut, P.; et al. Ectopic activation of germline and placental genes identifies aggressive metastasis-prone lung cancers. Sci. Transl. Med. 2013, 5, 186ra166. [Google Scholar]
- Simpson, A.J.; Caballero, O.L.; Jungbluth, A.; Chen, Y.T.; Old, L.J. Cancer/testis antigens, gametogenesis and cancer. Nat. Rev. Cancer 2005, 5, 615–625. [Google Scholar] [CrossRef]
- Janic, A.; Mendizabal, L.; Llamazares, S.; Rossell, D.; Gonzalez, C. Ectopic expression of germline genes drives malignant brain tumor growth in Drosophila. Science 2010, 330, 1824–1827. [Google Scholar]
- Rudert, F.; Bronner, S.; Garnier, J.M.; Dolle, P. Transcripts from opposite strands of gamma satellite DNA are differentially expressed during mouse development. Mamm. Genome 1995, 6, 76–83. [Google Scholar] [CrossRef]
- Efroni, S.; Duttagupta, R.; Cheng, J.; Dehghani, H.; Hoeppner, D.J.; Dash, C.; Bazett-Jones, D.P.; le Grice, S.; McKay, R.D.; Buetow, K.H.; et al. Global transcription in pluripotent embryonic stem cells. Cell Stem Cell 2008, 2, 437–447. [Google Scholar] [CrossRef]
- Lu, J.; Gilbert, D.M. Proliferation-dependent and cell cycle regulated transcription of mouse pericentric heterochromatin. J. Cell Biol. 2007, 179, 411–421. [Google Scholar] [CrossRef]
- Eymery, A.; Horard, B.; el Atifi-Borel, M.; Fourel, G.; Berger, F.; Vitte, A.L.; van den Broeck, A.; Brambilla, E.; Fournier, A.; Callanan, M.; et al. A transcriptomic analysis of human centromeric and pericentric sequences in normal and tumor cells. Nucl. Acids Res. 2009, 37, 6340–6354. [Google Scholar] [CrossRef] [Green Version]
- Ting, D.T.; Lipson, D.; Paul, S.; Brannigan, B.W.; Akhavanfard, S.; Coffman, E.J.; Contino, G.; Deshpande, V.; Iafrate, A.J.; Letovsky, S.; et al. Aberrant overexpression of satellite repeats in pancreatic and other epithelial cancers. Science 2011, 331, 593–596. [Google Scholar]
- Alexiadis, V.; Ballestas, M.E.; Sanchez, C.; Winokur, S.; Vedanarayanan, V.; Warren, M.; Ehrlich, M. RNApol-ChIP analysis of transcription from FSHD-linked tandem repeats and satellite DNA. Biochim. Biophys. Acta 2007, 1769, 29–40. [Google Scholar]
- Enukashvily, N.I.; Donev, R.; Waisertreiger, I.S.; Podgornaya, O.I. Human chromosome 1 satellite 3 DNA is decondensed, demethylated and transcribed in senescent cells and in A431 epithelial carcinoma cells. Cytogenet. Genome Res. 2007, 118, 42–54. [Google Scholar] [CrossRef]
- Bergmann, J.H.; Jakubsche, J.N.; Martins, N.M.; Kagansky, A.; Nakano, M.; Kimura, H.; Kelly, D.A.; Turner, B.M.; Masumoto, H.; Larionov, V.; et al. Epigenetic engineering: Histone H3K9 acetylation is compatible with kinetochore structure and function. J. Cell Sci. 2012, 125, 411–421. [Google Scholar] [CrossRef]
- Zhu, Q.; Pao, G.M.; Huynh, A.M.; Suh, H.; Tonnu, N.; Nederlof, P.M.; Gage, F.H.; Verma, I.M. BRCA1 tumour suppression occurs via heterochromatin-mediated silencing. Nature 2011, 477, 179–184. [Google Scholar] [CrossRef]
- Jaco, I.; Canela, A.; Vera, E.; Blasco, M.A. Centromere mitotic recombination in mammalian cells. J. Cell Biol. 2008, 181, 885–892. [Google Scholar] [CrossRef]
- Francastel, C.; Schubeler, D.; Martin, D.I.; Groudine, M. Nuclear compartmentalization and gene activity. Nat. Rev. Mol. Cell Biol. 2000, 1, 137–143. [Google Scholar] [CrossRef]
- Fisher, A.G.; Merkenschlager, M. Gene silencing, cell fate and nuclear organisation. Curr. Opin. Genet. Dev. 2002, 12, 193–197. [Google Scholar] [CrossRef]
- Luciani, J.J.; Depetris, D.; Usson, Y.; Metzler-Guillemain, C.; Mignon-Ravix, C.; Mitchell, M.J.; Megarbane, A.; Sarda, P.; Sirma, H.; Moncla, A.; et al. PML nuclear bodies are highly organised DNA-protein structures with a function in heterochromatin remodelling at the G2 phase. J. Cell. Sci. 2006, 119, 2518–2531. [Google Scholar] [CrossRef]
- Jefferson, A.; Colella, S.; Moralli, D.; Wilson, N.; Yusuf, M.; Gimelli, G.; Ragoussis, J.; Volpi, E.V. Altered intra-nuclear organisation of heterochromatin and genes in ICF syndrome. PLoS One 2010, 5, e11364. [Google Scholar] [CrossRef]
- Ehrlich, M.; Tsien, F.; Herrera, D.; Blackman, V.; Roggenbuck, J.; Tuck-Muller, C.M. High frequencies of ICF syndrome-like pericentromeric heterochromatin decondensation and breakage in chromosome 1 in a chorionic villus sample. J. Med. Genet. 2001, 38, 882–884. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Walton, E.L.; Francastel, C.; Velasco, G. Dnmt3b Prefers Germ Line Genes and Centromeric Regions: Lessons from the ICF Syndrome and Cancer and Implications for Diseases. Biology 2014, 3, 578-605. https://doi.org/10.3390/biology3030578
Walton EL, Francastel C, Velasco G. Dnmt3b Prefers Germ Line Genes and Centromeric Regions: Lessons from the ICF Syndrome and Cancer and Implications for Diseases. Biology. 2014; 3(3):578-605. https://doi.org/10.3390/biology3030578
Chicago/Turabian StyleWalton, Emma L., Claire Francastel, and Guillaume Velasco. 2014. "Dnmt3b Prefers Germ Line Genes and Centromeric Regions: Lessons from the ICF Syndrome and Cancer and Implications for Diseases" Biology 3, no. 3: 578-605. https://doi.org/10.3390/biology3030578