Discerning Primary and Secondary Factors Responsible for Clinical Fatigue in Multisystem Diseases

{kind=link}
Abstract
:1. Introduction
2. Skeletal Muscle Adaptations in Chronic Multi-System Diseases
2.1. Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS)
2.2. Multiple Sclerosis (MS) (Aka, Encephalomyelitis Disseminate)
2.3. Cancers
2.4. Cardiovascular Disorders and Heart Failure
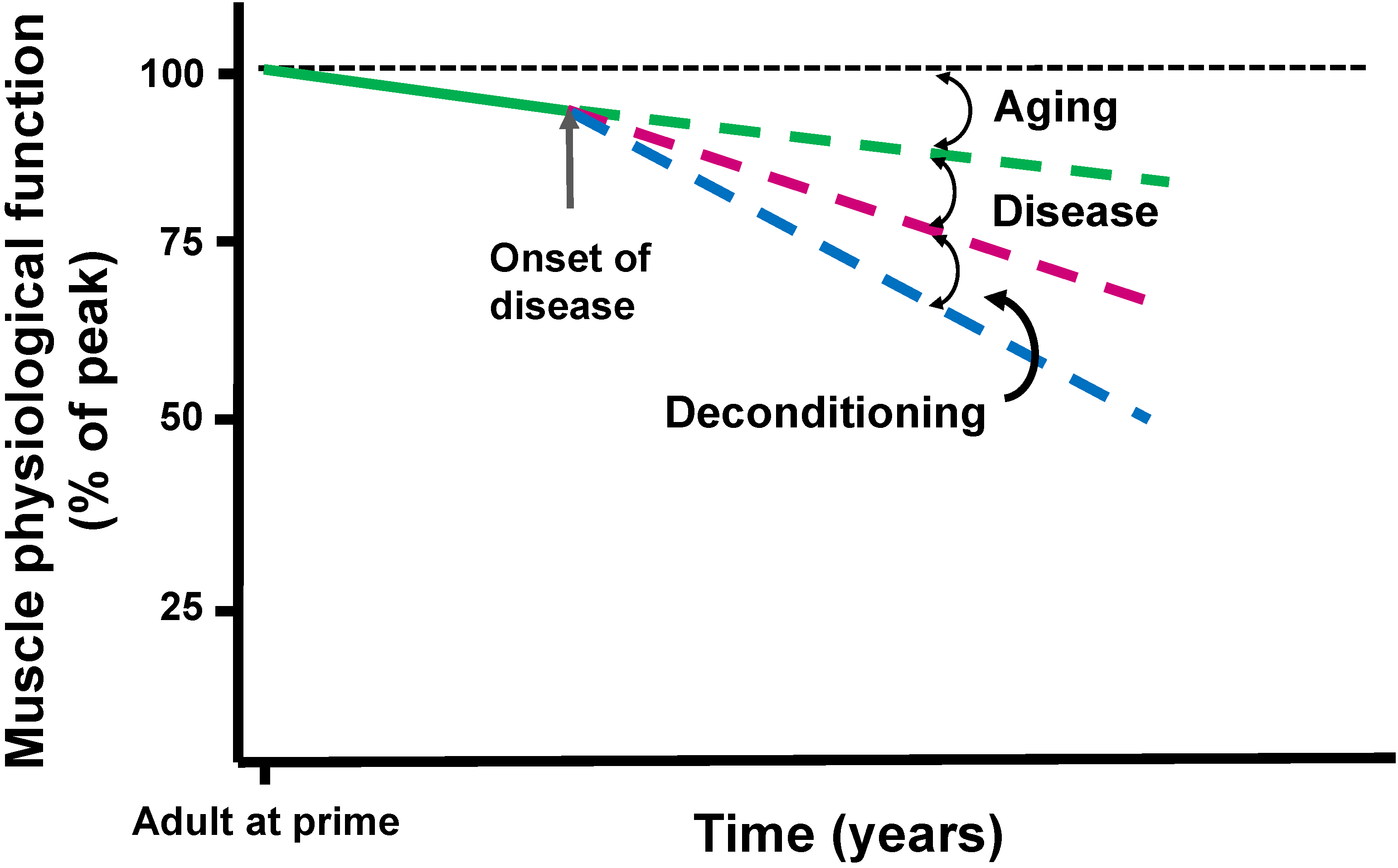
3. Disease and Muscle Deconditioning

Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ferriolli, E.; Skipworth, R.J.E.; Hendry, P.; Scott, A.; Stensteth, J.; Dahele, M.; Wall, L.; Greig, C.; Fallon, M.; Strasser, F.; et al. Physical activity monitoring: A responsive and meaningful patient-centered outcome for surgery, chemotherapy, or radiotherapy? J. Pain Symptom Manag. 2012, 43, 1025–1035. [Google Scholar] [CrossRef]
- Toth, M.J.; Gottlieb, S.S.; Goran, M.I.; Fisher, M.L.; Poehlman, E.T. Daily energy expenditure in free-living heart failure patients. Am. J. Physiol. Endocrinol. Metab. 1997, 272, E469–E475. [Google Scholar]
- Pitta, F.; Troosters, T.; Spruit, M.A.; Probst, V.S.; Decramer, M.; Gosselink, R. Characteristics of physical activities in daily life in chronic obstructive pulmonary disease. Am. J. Respir Crit Care Med. 2005, 171, 972–977. [Google Scholar] [CrossRef]
- Edwards, R.H. Human muscle function and fatigue. Ciba Found. Symp. 1981, 82, 1–18. [Google Scholar]
- Kent-Braun, J.A.; Fitts, R.H.; Christie, A. Skeletal muscle fatigue. Compr. Physiol. 2012, 2, 997–1044. [Google Scholar]
- Carruthers, B.M.; van de Sande, M.I.; De Meirleir, K.L.; Klimas, N.G.; Broderick, G.; Mitchell, T.; Staines, D.; Powles, A.C.; Speight, N.; Vallings, R.; et al. Myalgic encephalomyelitis: International Consensus Criteria. J. Intern. Med. 2011, 270, 327–338. [Google Scholar] [CrossRef]
- Holmes, G.P.; Kaplan, J.E.; Gantz, N.M.; Komaroff, A.L.; Schonberger, L.B.; Straus, S.E.; Jones, J.F.; Dubois, R.E.; Cunningham-Rundles, C.; Pahwa, S.; et al. Chronic fatigue syndrome: A working case definition. Ann. Intern. Med. 1988, 108, 387–389. [Google Scholar] [CrossRef]
- Morris, G.; Maes, M. A neuro-immune model of Myalgic Encephalomyelitis/Chronic fatigue syndrome. Metab. Brain Dis. 2013, 28, 523–540. [Google Scholar] [CrossRef]
- Morris, G.; Maes, M. Myalgic encephalomyelitis/chronic fatigue syndrome and encephalomyelitis disseminata/multiple sclerosis show remarkable levels of similarity in phenomenology and neuroimmune characteristics. BMC Med. 2013, 11, 205. [Google Scholar] [CrossRef]
- Bierl, C.; Nisenbaum, R.; Hoaglin, D.C.; Randall, B.; Jones, A.B.; Unger, E.R.; Reeves, W.C. Regional distribution of fatiguing illnesses in the United States: A pilot study. Popul. Health Metr. 2004, 2, 1. [Google Scholar] [CrossRef]
- Sommer, C.; Hauser, W.; Burgmer, M.; Engelhardt, R.; Gerhold, K.; Petzke, F.; Schmidt-Wilcke, T.; Spath, M.; Tolle, T.; Uceyler, N.; et al. Etiology and pathophysiology of fibromyalgia syndrome. Schmerz 2012, 26, 259–267. [Google Scholar] [CrossRef]
- Klimas, N.G.; Koneru, A.O. Chronic fatigue syndrome: Inflammation, immune function, and neuroendocrine interactions. Curr. Rheumatol. Rep. 2007, 9, 482–487. [Google Scholar] [CrossRef]
- Barnden, L.R.; Crouch, B.; Kwiatek, R.; Burnet, R.; Mernone, A.; Chryssidis, S.; Scroop, G.; Del Fante, P. A brain MRI study of chronic fatigue syndrome: Evidence of brainstem dysfunction and altered homeostasis. NMR Biomed. 2011, 24, 1302–1312. [Google Scholar] [CrossRef]
- Pall, M.L. Elevated, sustained peroxynitrite levels as the cause of chronic fatigue syndrome. Med. Hypotheses 2000, 54, 115–125. [Google Scholar] [CrossRef]
- Pall, M.L. Novel Disease Paradigm Produces Explanations for a Whole Group of Illnesses; Washington State University: Pullman, WA, USA, 2009. [Google Scholar]
- Booth, N.E.; Myhill, S.; McLaren-Howard, J. Mitochondrial dysfunction and the pathophysiology of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). Int. J. Clin. Exp. Med. 2012, 5, 208. [Google Scholar]
- Myhill, S.; Booth, N.E.; McLaren-Howard, J. Targeting mitochondrial dysfunction in the treatment of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS) - a clinical audit. Int. J. Clin. Exp. Med. 2013, 6, 1–15. [Google Scholar]
- Morris, G.; Maes, M. Increased nuclear factor-kappaB and loss of p53 are key mechanisms in Myalgic Encephalomyelitis/chronic fatigue syndrome (ME/CFS). Med. Hypotheses 2012, 79, 607–613. [Google Scholar] [CrossRef]
- Carlson, B.E.; Vigoreaux, J.O.; Maughan, D.W. Diffusion coefficients of endogenous cytosolic proteins from rabbit skinned muscle fibers. Biophys. J. 2014, 106, 780–792. [Google Scholar] [CrossRef]
- Massie, B.M.; Conway, M.; Rajagopalan, B.; Yonge, R.; Frostick, S.; Ledingham, J.; Sleight, P.; Radda, G. Skeletal muscle metabolism during exercise under ischemic conditions in congestive heart failure: Evidence for abnormalities unrelated to blood flow. Circulation 1988, 78, 320–326. [Google Scholar] [CrossRef]
- Newton, D.J.; Kennedy, G.; Chan, K.K.; Lang, C.C.; Belch, J.J.; Khan, F. Large and small artery endothelial dysfunction in chronic fatigue syndrome. Int. J. Cardiol. 2012, 154, 335–336. [Google Scholar] [CrossRef]
- Lemle, M.D. Hypothesis: Chronic fatigue syndrome is caused by dysregulation of hydrogen sulfide metabolism. Med. Hypotheses 2009, 72, 108–109. [Google Scholar] [CrossRef]
- Kobayashi, Y.M.; Rader, E.P.; Crawford, R.W.; Iyengar, N.K.; Thedens, D.R.; Faulkner, J.A.; Parikh, S.V.; Weiss, R.M.; Chamberlain, J.S.; Moore, S.A.; et al. Sarcolemma-localized nNOS is required to maintain activity after mild exercise. Nature 2008, 456, 511–515. [Google Scholar] [CrossRef]
- Lane, R.J.; Barrett, M.C.; Taylor, D.J.; Kemp, G.J.; Lodi, R. Heterogeneity in chronic fatigue syndrome: Evidence from magnetic resonance spectroscopy of muscle. Neuromuscul. Disord. 1998, 8, 204–209. [Google Scholar] [CrossRef]
- Wong, R.; Lopaschuk, G.; Zhu, G.; Walker, D.; Catellier, D.; Burton, D.; Teo, K.; Collins-Nakai, R.; Montague, T. Skeletal muscle metabolism in the chronic fatigue syndrome. In vivo assessment by 31P nuclear magnetic resonance spectroscopy. Chest 1992, 102, 1716–1722. [Google Scholar]
- McCully, K.K.; Natelson, B.H.; Iotti, S.; Sisto, S.; Leigh, J.S., Jr. Reduced oxidative muscle metabolism in chronic fatigue syndrome. Muscle Nerve 1996, 19, 621–625. [Google Scholar] [CrossRef]
- Connolly, S.; Smith, D.G.; Doyle, D.; Fowler, C.J. Chronic fatigue: Electromyographic and neruopathological evaluation. J. Neuro 1993, 240, 435–438. [Google Scholar]
- Fulle, S.; Belia, S.; Vecchiet, J.; Morabito, C.; Vecchiet, L.; Fano, G. Modification of the functional capacity of sarcoplasmic reticulum membranes in patients suffering from chronic fatigue syndrome. Neuromuscul. Disord. 2003, 13, 479–484. [Google Scholar] [CrossRef]
- Light, A.R.; Bateman, L.; Jo, D.; Hughen, R.W.; Vanhaitsma, T.A.; White, A.T.; Light, K.C. Gene expression alterations at baseline and following moderate exercise in patients with Chronic Fatigue Syndrome and Fibromyalgia Syndrome. J. Intern. Med. 2012, 271, 64–81. [Google Scholar] [CrossRef]
- Light, A.R.; White, A.T.; Hughen, R.W.; Light, K.C. Moderate exercise increases expression for sensory, adrenergic, and immune genes in chronic fatigue syndrome patients but not in normal subjects. J. Pain 2009, 10, 1099–1112. [Google Scholar] [CrossRef]
- Pall, M.L. Chronic fatigue syndrome/myalgic encephalitis. Br. J. Gen. Pract. 2002, 52, 762; author reply 763–764. [Google Scholar]
- Shor, S. Pathogenesis of Chronic Fatigue Syndrome, a Multisystem Hypothesis. J. Chronic Fatigue Syndr. 2003, 11, 51–68. [Google Scholar] [CrossRef]
- McCully, K.K.; Natelson, B.H. Impaired oxygen delivery to muscle in chronic fatigue syndrome. Clin. Sci. (Lond) 1999, 97, 603–608; discussion 611–613. [Google Scholar]
- McCully, K.K.; Smith, S.; Rajaei, S.; Leigh, J.S., Jr.; Natelson, B.H. Muscle metabolism with blood flow restriction in chronic fatigue syndrome. J. Appl. Physiol. (1985) 2004, 96, 871–878. [Google Scholar] [CrossRef]
- VanNess, J.M.; Snell, C.R.; Stevens, S.R. Diminished Cardiopulmonary Capacity during Post-Exertional Malaise. J. Chronic Dis. Syndr. 2007, 14, 77–85. [Google Scholar] [CrossRef]
- Carruthers, B.M.; van de Sande, M.I.; De Meirleir, K.L.; Klimas, N.G.; Broderick, G.; Mitchell, T.; Staines, D.; Powles, A.C.; Speight, N.; Vallings, R.; et al. Myalgic Encephalohyeltitis—Adult & Paediatric: International Concensus Primer for Medical Practitioners; Carruthers & van de Sande: Vancouver, BC, Canada, 2012. [Google Scholar]
- LaManca, J.J.; Sisto, S.A.; DeLuca, J.; Johnson, S.K.; Lange, G.; Pareja, J.; Cook, S.; Natelson, B.H. Influence of exhaustive treadmill exercise on cognitive functioning in chronic fatigue syndrome. Am. J. Med. 1998, 105, 59S–65S. [Google Scholar] [CrossRef]
- Blackwood, S.K.; MacHale, S.M.; Power, M.J.; Goodwin, G.M.; Lawrie, S.M. Effects of exercise on cognitive and motor function in chronic fatigue syndrome and depression. J. Neurol. Neurosurg. Psychiatry 1998, 65, 541–546. [Google Scholar] [CrossRef]
- Cook, D.B.; Nagelkirk, P.R.; Peckerman, A.; Poluri, A.; Mores, J.; Natelson, B.H. Exercise and cognitive performance in chronic fatigue syndrome. Med. Sci. Sports Exerc. 2005, 37, 1460–1467. [Google Scholar] [CrossRef]
- Yoshiuchi, K.; Cook, D.B.; Ohashi, K.; Kumano, H.; Kuboki, T.; Yamamoto, Y.; Natelson, B.H. A real-time assessment of the effect of exercise in chronic fatigue syndrome. Physiol. Behav. 2007, 92, 963–968. [Google Scholar] [CrossRef]
- VanNess, J.M.; Stevens, S.R.; Bateman, L.; Stiles, T.L.; Snell, C.R. Postexertional malaise in women with chronic fatigue syndrome. J. Womens Health (Larchmt) 2010, 19, 239–244. [Google Scholar] [CrossRef]
- Gonsette, R.E. Neurodegeneration in multiple sclerosis: The role of oxidative stress and excitotoxicity. J. Neurol. Sci. 2008, 274, 48–53. [Google Scholar] [CrossRef]
- van Horssen, J.; Schreibelt, G.; Drexhage, J.; Hazes, T.; Dijkstra, C.D.; van der Valk, P.; de Vries, H.E. Severe oxidative damage in multiple sclerosis lesions coincides with enhanced antioxidant enzyme expression. Free Radic. Biol. Med. 2008, 45, 1729–1737. [Google Scholar]
- Lu, F.; Selak, M.; O'Connor, J.; Croul, S.; Lorenzana, C.; Butunoi, C.; Kalman, B. Oxidative damage to mitochondrial DNA and activity of mitochondrial enzymes in chronic active lesions of multiple sclerosis. J. Neurol. Sci. 2000, 177, 95–103. [Google Scholar] [CrossRef]
- Mahad, D.J.; Ziabreva, I.; Campbell, G.; Lax, N.; White, K.; Hanson, P.S.; Lassmann, H.; Turnbull, D.M. Mitochondrial changes within axons in multiple sclerosis. Brain 2009, 132, 1161–1174. [Google Scholar] [CrossRef]
- Higgins, G.C.; Beart, P.M.; Shin, Y.S.; Chen, M.J.; Cheung, N.S.; Nagley, P. Oxidative stress: Emerging mitochondrial and cellular themes and variations in neuronal injury. J. Alzheimers Dis. 2010, 20, S453–S473. [Google Scholar]
- Adhya, S.; Johnson, G.; Herbert, J.; Jaggi, H.; Babb, J.S.; Grossman, R.I.; Inglese, M. Pattern of hemodynamic impairment in multiple sclerosis: Dynamic susceptibility contrast perfusion MR imaging at 3.0 T. Neuroimage 2006, 33, 1029–1035. [Google Scholar] [CrossRef]
- Brooks, D.J.; Leenders, K.L.; Head, G.; Marshall, J.; Legg, N.J.; Jones, T. Studies on regional cerebral oxygen utilisation and cognitive function in multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 1984, 47, 1182–1191. [Google Scholar] [CrossRef]
- Rashid, W.; Parkes, L.M.; Ingle, G.T.; Chard, D.T.; Toosy, A.T.; Altmann, D.R.; Symms, M.R.; Tofts, P.S.; Thompson, A.J.; Miller, D.H. Abnormalities of cerebral perfusion in multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 2004, 75, 1288–1293. [Google Scholar] [CrossRef]
- Debouverie, M.; Pittion, S. Fatigue and episodic exhaustion as a feature of multiple sclerosis. Rev. Neurol. (Paris) 2006, 162, 295–297. [Google Scholar] [CrossRef]
- Lenman, A.J.; Tulley, F.M.; Vrbova, G.; Dimitrijevic, M.R.; Towle, J.A. Muscle fatigue in some neurological disorders. Muscle Nerve 1989, 12, 938–942. [Google Scholar] [CrossRef]
- Sharma, K.R.; Kent-Braun, J.; Mynhier, M.A.; Weiner, M.W.; Miller, R.G. Evidence of an abnormal intramuscular component of fatigue in multiple sclerosis. Muscle Nerve 1995, 18, 1403–1411. [Google Scholar] [CrossRef]
- Petajan, J.H.; White, A.T. Motor-evoked potentials in response to fatiguing grip exercise in multiple sclerosis patients. Clin. Neurophysiol. 2000, 111, 2188–2195. [Google Scholar] [CrossRef]
- Ng, A.V.; Miller, R.G.; Gelinas, D.; Kent-Braun, J.A. Functional relationships of central and peripheral muscle alterations in multiple sclerosis. Muscle Nerve 2004, 29, 843–852. [Google Scholar] [CrossRef]
- McArdle, W.D.; Katch, F.I.; Katch, V.L. Excercise Physiology, 7th ed.; Wolters Kluwer/Lippincott Williams & Wilkins Health: Philadelphia, PA, USA, 2010. [Google Scholar]
- Ng, A.V.; Kent-Braun, J.A. Quantitation of lower physical activity in persons with multiple sclerosis. Med. Sci. Sports Exerc. 1997, 29, 517–523. [Google Scholar] [CrossRef]
- Vogelzang, N.J.; Breibart, W.; Cella, D.; Curt, G.A.; Groopman, J.E.; Horning, S.J.; Itri, L.M.; Johnson, D.H.; Scherr, S.L.; Portenoy, R.K. Patient, caregiver, and oncologist perceptions of cancer-related fatigue: Results of a tripart assessment survey. The Fatigue Coalition. Semin. Hematol. 1997, 34, 4–12. [Google Scholar]
- Butt, Z.; Rosenbloom, S.K.; Abernethy, A.P.; Beaumont, J.L.; Paul, D.; Hamptom, D.; Jacobsen, P.B.; Syrjala, K.L.; Von Roenn, J.H.; Cella, D. Fatigue is the most important symptom for advanced cancer patients who have had chemotherapy. J. Natl. Compr. Cancer Netw. 2008, 6, 448–455. [Google Scholar]
- Cella, D.; Davis, K.; Breitbart, W.; Curt, G.; Fatigue Coalition. Cancer-related fatigue: Prevalence of proposed diagnostic criteria in a United States sample of cancer survivors. J. Clin. Oncol. 2001, 19, 3385–3391. [Google Scholar]
- Braithwaite, D.; Satariano, W.A.; Sternfeld, B.; Hiatt, R.A.; Ganz, P.A.; Kerlikowske, K.; Moore, D.H.; Slattery, M.L.; Tammemagi, M.; Castillo, A.; et al. Long-term prognostic role of functional limitations among women with breast cancer. J. Natl. Compr. Cancer Netw. 2010, 102, 1468–1477. [Google Scholar] [CrossRef]
- Brunelli, A.; Pompili, C.; Berardi, R.; Mazzanti, P.; Onofri, A.; Salati, M.; Cascinu, S.; Sabbatini, A. Performance at preoperative stair-climbing test is associated with prognosis after pulmonary resection in Stage I non-small cell lung cancers. Ann. Thorac. Surg. 2012, 93, 1796–1800. [Google Scholar] [CrossRef]
- Jones, L.W.; Hornsby, W.E.; Goetzinger, A.; Forbes, L.M.; Sherrard, E.L.; Quist, M.; Lane, A.T.; West, M.; Eves, N.D.; Gradison, M.; et al. Prognostic significance of functional capacity and exercise behavior in patients with metastatic non-small cell lung cancer. Lung Cancer 2012, 76, 248–252. [Google Scholar] [CrossRef]
- Miller, K.L.; Kocak, Z.; Kahn, D.; Zhou, S.-M.; Baydush, A.; Hollis, D.; Folz, R.J.; Tisch, A.; Clough, R.; Yu, X.; et al. Preliminary report of the 6-minute walk test as a predictor of radiation-induced pulmonary toxicity. Int. J. Radiat. Oncol. Biol. Phys. 2005, 62, 1009–1013. [Google Scholar] [CrossRef]
- Jatoi, A.; Hillman, S.; Stella, P.; Mailliard, J.; Sloan, J.; Vanone, S.; Cannon, M.; Kutteh, L.; Kanard, A.; Jett, J. Daily activities: Exploring their spectrum and prognostic impact in older, chemotherapy-treated lung cancer patients. Support. Care Cancer 2003, 11, 460–464. [Google Scholar] [CrossRef]
- Fearon, K.C.; Glass, D.J.; Guttridge, D.C. Cancer cachexia: Mediators, signaling, and metabolic pathways. Cell Metab. 2012, 16, 153–166. [Google Scholar] [CrossRef]
- Toth, M.J.; Miller, M.S.; Callahan, D.M.; Sweeny, A.P.; Nunez, I.; Grunberg, S.M.; Der-Torossian, H.; Couch, M.E.; Dittus, K. Molecular mechanisms underlying skeletal muscle weakness in human cancer: Reduced myosin-actin cross-bridge formation and kinetics. J. Appl. Physiol. 2013, 114, 858–868. [Google Scholar] [CrossRef]
- Acharyya, S.; Ladner, K.J.; Nelsen, L.L.; Damrauer, J.; Reiser, P.J.; Swoap, S.; Guttridge, D.C. Cancer cachexia is regulated by selective targeting of skeletal muscle gene products. J. Clin. Invest. 2004, 114, 370–378. [Google Scholar] [CrossRef]
- Eley, H.L.; Skipworth, R.J.E.; Deans, D.A.C.; Fearon, K.C.H.; Tisdale, M.J. Increased expression of phosphorylated forms of RNA-dependent protein kinase and eukaryotic initiation factor 2[alpha] may signal skeletal muscle atrophy in weight-losing cancer patients. Br. J. Cancer 2007, 98, 443–449. [Google Scholar] [CrossRef]
- Cosper, P.F.; Leinwand, L.A. Myosin heavy chain is not selectively decreased in murine cancer cachexia. Int. J. Cancer 2012, 130, 2722–2727. [Google Scholar] [CrossRef]
- Weber, M.-A.; Krakowski-Roosen, H.; Schröder, L.; Kinscherf, R.; Krix, M.; Kopp-Schneider, A.; Essig, M.; Bachert, P.; Kauczor, H.-U.; Hildebrandt, W. Morphology, metabolism, microcirculation, and strength of skeletal muscles in cancer-related cachexia. Acta Oncol. 2009, 48, 116–124. [Google Scholar] [CrossRef]
- Stephens, N.A.; Gray, C.; MacDonald, A.J.; Tan, B.H.; Gallagher, I.J.; Skipworth, R.J.E.; Ross, J.A.; Fearon, K.C.H.; Greig, C.A. Sexual dimorphism modulates the impact of cancer cachexia on lower limb muscle mass and function. Clin. Nutr. 2012, 31, 499–505. [Google Scholar] [CrossRef]
- Aulino, P.; Berardi, E.; Cardillo, V.; Rizzuto, E.; Perniconi, B.; Ramina, C.; Padula, F.; Spugnini, E.; Baldi, A.; Faiola, F.; et al. Molecular, cellular and physiological characterization of the cancer cachexia-inducing C26 colon carcinoma in mouse. BMC Cancer 2010, 10, 363. [Google Scholar] [CrossRef]
- Murphy, K.T.; Chee, A.; Trieu, J.; Naim, T.; Lynch, G.S. Importance of functional and metabolic impairments in the characterization of the C-26 murine model of cancer cachexia. Dis. Model. Mech. 2012, 5, 533–545. [Google Scholar] [CrossRef]
- Toledo, M.; Busquets, S.; Sirisi, S.; Serpe, R.; Orpi, M.; Coutinho, J.; Martinez, R.; Lopez-Soriano, F.J.; Argiles, J.M. Cancer cachexia: Physical activity and muscle force in tumor bearing rats. Oncol. Rep. 2011, 25, 183–193. [Google Scholar]
- Der-Torossian, H.; Couch, M.E.; Dittus, K.; Toth, M.J. Skeletal muscle adaptations to cancer and its treatment: Their fundamental basis and contribution to functional disability. Crit. Rev. Eukaryot. Gene Expr. 2013, 23, 283–297. [Google Scholar] [CrossRef]
- Julienne, C.; Dumas, J.-.F.; Goupille, C.; Pinault, M.; Berri, C.; Collin, A.; Tesseraud, S.; Couet, C.; Servais, S. Cancer cachexia is associated with a decrease in skeletal muscle mitochondrial oxidative capacities without alteration of ATP production efficiency. J. Cachexia Sarcopenia Muscle 2012, 3, 265–275. [Google Scholar]
- Tzika, A.A.; Fontes-Oliveria, C.C.; Shestova, A.A.; Constantinou, C.; Psychogios, N.; Righi, V.; Mintzopoulos, D.; Busquets, S.; Lopez-Soriano, F.J.; Milote, S.; et al. Skeletal muscle mitochondrial uncoupling in a murine cancer cachexia model. Int. J. Oncol. 2013, 43, 886–894. [Google Scholar]
- Gilliam, L.A.A.; Fisher-Wellman, K.H.; Lin, C.-T.; Maples, J.M.; Cathey, B.L.; Neufer, P.D. The anticancer agent doxorubicin disrupts mitochondrial energy metabolism and redox balance in skeletal muscle. Free Radic. Biol. Med. 2013, 65, 988–996. [Google Scholar]
- Pinto, B.M.; Trunzo, J.J.; Reiss, P.; Shiu, S.Y. Exercise participation after diagnosis of breast cancer: Trends and effects on mood and quality of life. Psychooncology 2002, 11, 389–400. [Google Scholar] [CrossRef]
- Courneya, K.S.; McKenzie, D.C.; Mackey, J.R.; Gelmon, K.; Friedenreich, C.M.; Yasui, Y.; Reid, R.D.; Cook, D.; Jespersen, D.; Proulx, C.; et al. Effects of Exercise Dose and Type during Breast Cancer Chemotherapy: Multicenter Randomized Trial. J. Natl. Cancer Inst. 2013, 105, 1821–1832. [Google Scholar] [CrossRef]
- Jones, L.W.; Alfano, C.M. Exercise-oncology research: Past, present, and future. Acta Oncol. 2013, 52, 195–215. [Google Scholar] [CrossRef]
- Coats, A.J.S. The “muscle hypothesis” of chronic heart failure. J. Mol. Cell. Cardiol. 1996, 28, 2255–2262. [Google Scholar] [CrossRef]
- Zizola, C.; Schulze, P.C. Metabolic and structural impairment of skeletal muscle in heart failure. Heart Fail. Rev. 2013, 18, 623–630. [Google Scholar] [CrossRef]
- Mancini, D.M.; Coyle, E.; Coggan, A.; Beltz, J.; Ferraro, N.; Montain, S.; Wilson, J.R. Contribution of intrinsic skeletal muscle changes to 31P-NMR skeletal muscle metabolic abnormalities in patients with chronic heart failure. Circulation 1989, 80, 1338–1346. [Google Scholar] [CrossRef]
- Sullivan, M.J.; Green, H.J.; Cobb, F.R. Skeletal muscle biochemistry and histology in ambulatory patients with long-term heart failure. Circulation 1990, 81, 518–527. [Google Scholar] [CrossRef]
- Callahan, D.M.; Toth, M.J. Skeletal muscle protein metabolism in human heart failure. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 66–71. [Google Scholar] [CrossRef]
- Harrington, D.; Anker, S.; Chua, T.P.; Webb-Peploe, K.M.; Ponikowski, P.P.; Poole-Wilson, P.A.; Coats, A.J.S. Skeletal muscle function and its relation to exercise tolerance in chronic heart failure. J. Am. Coll. Cardiol. 1997, 30, 1758–1764. [Google Scholar] [CrossRef]
- Vescovo, G.; Serafini, F.; Facchin, L.; Tenderini, P.; Carraro, U.; Dalla Libera, L.; Catani, C.; Ambrosio, G.B. Specific changes in skeletal muscle myosin heavy chain composition in cardiac failure: Differences compared with disuse atrophy as assessed on microbiopsies by high resolution electrophoresis. Heart 1996, 76, 337–343. [Google Scholar] [CrossRef]
- Drexler, H.; Riede, U.; Munzel, T.; Konig, H.; Funke, E.; Just, H. Alterations in skeletal muscle in chronic heart failure. Circulation 1992, 85, 1751–1759. [Google Scholar] [CrossRef]
- Minotti, J.R.; Pillay, P.; Chang, L.; Wells, L.; Massie, B.M. Neurophysiological assessment of skeletal muscle fatigue in patients with congestive heart failure. Circulation 1992, 86, 903–908. [Google Scholar] [CrossRef]
- Miller, M.S.; VanBuren, P.; LeWinter, M.M.; Braddock, J.M.; Ades, P.A.; Maughan, D.W.; Palmer, B.M.; Toth, M.J. Chronic heart failure decreases cross-bridge kinetics in single skeletal muscle fibres from humans. J. Physiol. 2010, 588, 4039–4053. [Google Scholar] [CrossRef]
- Miller, M.S.; VanBuren, P.; LeWinter, M.M.; Lecker, S.H.; Selby, D.E.; Palmer, B.M.; Maughan, D.W.; Ades, P.A.; Toth, M.J. Mechanisms underlying skeletal muscle weakness in human heart failure: Alterations in single fiber myosin protein content and function. Circ. Heart Fail. 2009, 2, 700–706. [Google Scholar] [CrossRef]
- Rehn, T.A.; Munkvik, M.; Lunde, P.K.; Sjaastad, I.; Sejersted, O.M. Intrinsic skeletal muscle alterations in chronic heart failure patients: A disease-specific myopathy or a result of deconditioning? Heart Fail. Rev. 2012, 17, 421–436. [Google Scholar]
- Mettauer, B.; Zoll, J.; Sanchez, H.; Lampert, E.; Ribera, F.; Veksler, V.; Bigard, X.; Mateo, P.; Epailly, E.; Lonsdorfer, J.; et al. Oxidative capacity of skeletal muscle in heart failure patients versus sedentary or active controls subjects. J. Am. Coll. Cardiol. 2001, 38, 947–954. [Google Scholar]
- Chati, Z.; Zannad, F.; Jeandel, C.; Lherbier, B.; Escanye, J.-M.; Robert, J.; Aliot, E. Physical deconditioning may be a mechanism for the skeletal muscle energy phosphate metabolism abnormalities in chronic heart failure. Am. Heart J. 1996, 131, 560–566. [Google Scholar] [CrossRef]
- Williams, A.D.; Selig, S.; Hare, D.L.; Hayes, A.; Krum, H.; Patterson, J.; Geerling, R.H.; Toia, D.; Carey, M.F. Reduced exercise tolerance in CHF may be related to factors other than impaired skeletal muscle oxidative capacity. J. Card Fail. 2004, 10, 141–148. [Google Scholar] [CrossRef]
- Toth, M.J.; Shaw, A.O.; Miller, M.S.; VanBuren, P.; LeWinter, M.M.; Maughan, D.; Ades, P.A. Reduced knee extensor function in heart failure is not explained by inactivity. Int. J. Cardiol. 2010, 143, 276–282. [Google Scholar] [CrossRef]
- Pollak, K.A.; Swenson, J.D.; Vanhaitsma, T.A.; Hughen, R.W.; Jo, D.; Light, K.C.; Schweinhardt, P.; Amann, M.; Light, A.R. Exogenously applied muscle metabolites synergistically evoke sensations of muscle fatigue and pain in human subjects. Exp. Physiol. 2014, 99, 368–380. [Google Scholar] [CrossRef]
- White, P.D.; Goldsmith, K.A.; Johnson, A.L.; Potts, L.; Walwyn, R.; DeCesare, J.C.; Baber, H.L.; Burgess, M.; Clark, L.V.; Cox, D.L.; et al. Comparison of adaptive pacing therapy, cognitive behaviour therapy, graded exercise therapy, and specialist medical care for chronic fatigue syndrome (PACE): A randomised trial. Lancet 2011, 377, 823–836. [Google Scholar] [CrossRef]
- Twisk, F.N.; Maes, M. A review on cognitive behavorial therapy (CBT) and graded exercise therapy (GET) in myalgic encephalomyelitis (ME)/chronic fatigue syndrome (CFS): CBT/GET is not only ineffective and not evidence-based, but also potentially harmful for many patients with ME/CFS. Neuro Endocrinol. Lett. 2009, 30, 284–299. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Maughan, D.; Toth, M. Discerning Primary and Secondary Factors Responsible for Clinical Fatigue in Multisystem Diseases. Biology 2014, 3, 606-622. https://doi.org/10.3390/biology3030606
Maughan D, Toth M. Discerning Primary and Secondary Factors Responsible for Clinical Fatigue in Multisystem Diseases. Biology. 2014; 3(3):606-622. https://doi.org/10.3390/biology3030606
Chicago/Turabian StyleMaughan, David, and Michael Toth. 2014. "Discerning Primary and Secondary Factors Responsible for Clinical Fatigue in Multisystem Diseases" Biology 3, no. 3: 606-622. https://doi.org/10.3390/biology3030606