The Structural and Functional Coordination of Glycolytic Enzymes in Muscle: Evidence of a Metabolon?

Abstract
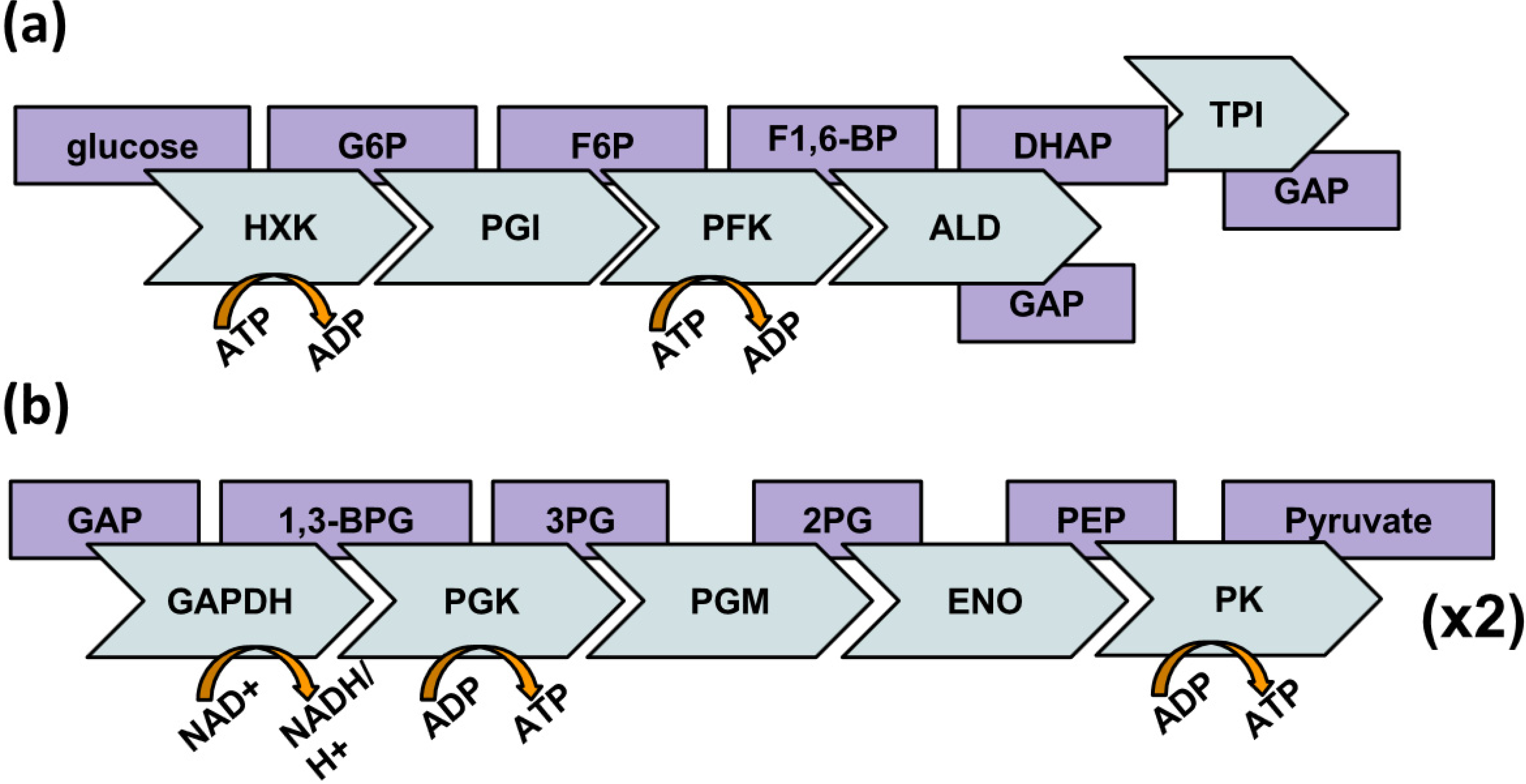
:1. Introduction
2. Behavior of Glycolytic Enzymes

2.1. Moonlighting Functions
2.2. Implications of a Glycolytic Enzyme Complex
2.3. Muscle: An Ordered Model for Metabolism
3. A New Hypothesis on Glycolytic Enzyme Complexes

{kind=link}
{kind=link}
{kind=link}
| Ligands | PFK | ALD | TPI | GAPDH | PGK | PGM | ENO | PK | F-actin |
|---|---|---|---|---|---|---|---|---|---|
| PFK |   | ▪ |  | ▪ |  | | ▪ | | |
| ALD | | ▪ | ▪ | | ▪ | ▪ | | ▪ | |
| TPI | ▪ | ▪ | ▪ | | ▪ | ▪ | ▪ | ▪ | |
| GAPTH | | | ▪ | ▪ | | ▪ | ▪ | ▪ | |
| PGK | ▪ | ▪ | | | ▪ | ▪ | | ▪ | ▪ |
| PGM | | ▪ | ▪ | ▪ | ▪ | ▪ | | ▪ | ▪ |
| ENO | | | ▪ |  | | | ▪ | | ▪ |
| PK | ▪ | ▪ | ▪ | ▪ | ▪ | | ▪ | |
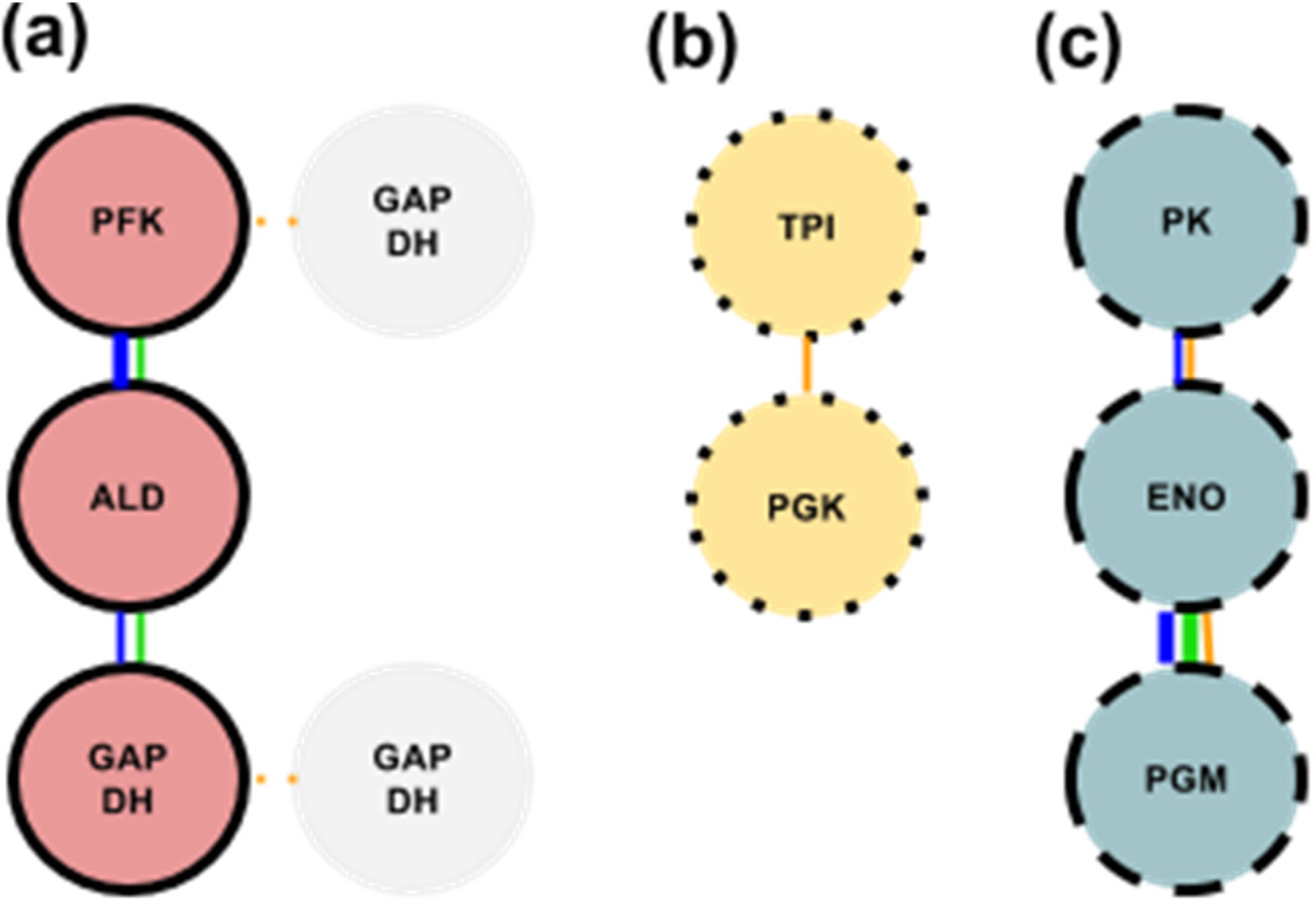
3.1. Glycolytic Subcomplex-1 (GlySCx1): PFK, ALD, & GAPDH

3.2. Glycolytic Subcomplex-2 (GlySCx2): TPI & PGK
3.3. Glycolytic Subcomplex-3 (GlySCx3): PGM, ENO, & PK
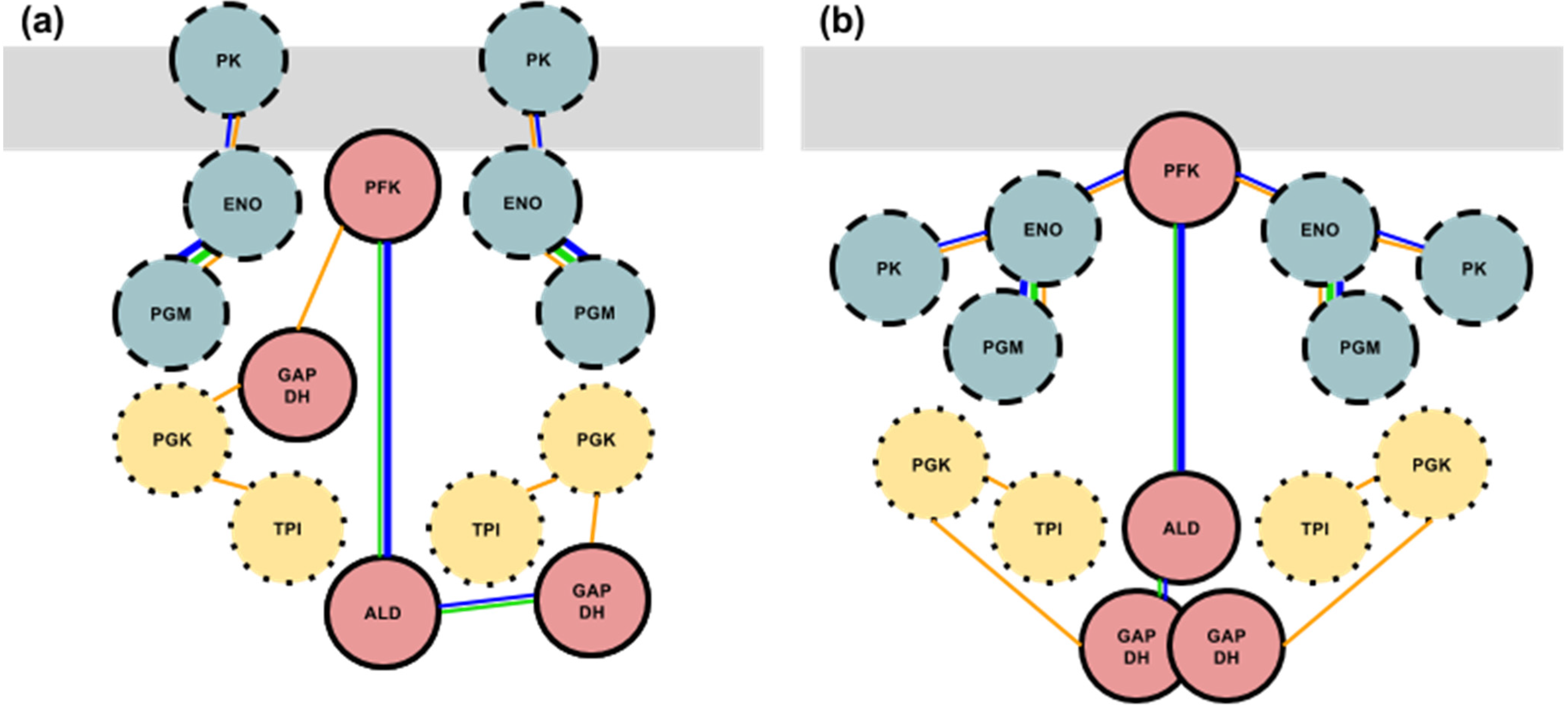
3.4. Formation of the Glycolytic Complex

4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Srere, P.A.; Mathews, C.K. Purification of multienzyme complexes. Methods Enzymol. 1990, 182, 539–551. [Google Scholar]
- Mendes, P.; Kell, D.B.; Westerhoff, H.V. A series of cases in which metabolic channelling can decrease the pool size at constant net flux in a simple dynamic channel. Biochem. Soc. Trans. 1995, 23, 287S. [Google Scholar]
- Saks, V.; Beraud, N.; Wallimann, T. Metabolic compartmentation—A system level property of muscle cells: Real problems of diffusion in living cells. Int. J. Mol. Sci. 2008, 9, 751–767. [Google Scholar]
- Seufferheld, M.J.; Caetano-Anolles, G. Phylogenomics supports a cellularly structured urancestor. J. Mol. Microbiol. Biotechnol. 2013, 23, 178–191. [Google Scholar] [CrossRef]
- Sachs, J.N.; Engelman, D.M. Introduction to the membrane protein reviews: The interplay of structure, dynamics, and environment in membrane protein function. Annu. Rev. Biochem. 2006, 75, 707–712. [Google Scholar] [CrossRef]
- Lieleg, O.; Claessens, M.M.A.E.; Bausch, A.R. Structure and dynamics of cross-linked actin networks. Soft Matter 2010, 6, 218–225. [Google Scholar] [CrossRef]
- Cunningham, J.; Estrella, V.; Lloyd, M.; Gillies, R.; Frieden, B.R.; Gatenby, R. Intracellular electric field and pH optimize protein localization and movement. PLoS One 2012, 7, e36894. [Google Scholar] [CrossRef]
- Opperdoes, F.R.; Borst, P. Localization of nine glycolytic enzymes in a microbody-like organelle in Trypanosoma brucei: The glycosome. FEBS Lett. 1977, 80, 360–364. [Google Scholar] [CrossRef]
- Michels, P.A.; Bringaud, F.; Herman, M.; Hannaert, V. Metabolic functions of glycosomes in trypanosomatids. Biochim. Biophys. Acta 2006, 1763, 1463–1477. [Google Scholar] [CrossRef]
- Dovey, H.F.; Parsons, M.; Wang, C.C. Biogenesis of glycosomes of Trypanosoma brucei: An in vitro model of 3-phosphoglycerate kinase import. Proc. Natl. Acad. Sci. USA 1988, 85, 2598–2602. [Google Scholar] [CrossRef]
- Clarke, F.M.; Masters, C.J. On the association of glycolytic enzymes with structural proteins of skeletal muscle. Biochim. Biophys. Acta 1975, 381, 37–46. [Google Scholar] [CrossRef]
- Kurganov, B.I.; Sugrobova, N.P.; Mil'man, L.S. Supramolecular organization of glycolytic enzymes. J. Theor. Biol. 1985, 116, 509–526. [Google Scholar] [CrossRef]
- Boiteux, A.; Hess, B. Design of glycolysis. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1981, 293, 5–22. [Google Scholar] [CrossRef]
- Srere, P.A. Complexes of sequential metabolic enzymes. Annu. Rev. Biochem. 1987, 56, 89–124. [Google Scholar] [CrossRef]
- Brooks, S.P.; Storey, K.B. Where is the glycolytic complex? A critical evaluation of present data from muscle tissue. FEBS Lett. 1991, 278, 135–138. [Google Scholar] [CrossRef]
- Herman, M.; Perez-Morga, D.; Schtickzelle, N.; Michels, P.A. Turnover of glycosomes during life-cycle differentiation of Trypanosoma brucei. Autophagy 2008, 4, 294–308. [Google Scholar] [CrossRef]
- Nemat-Gorgani, M.; Wilson, J.E. Ambiquitous behavior—A biological phenomenon of general significance? Curr. Top. Cell Regul. 1980, 16, 45–54. [Google Scholar]
- Sagrista, M.L.; Bozal, J. Lactate and malate dehydrogenase binding to the microsomal fraction from chicken liver. Biochimie 1987, 69, 1207–1215. [Google Scholar] [CrossRef]
- Knull, H.R.; Fillmore, S.J. Glycolytic enzyme levels in synaptosomes. Comp. Biochem. Physiol. B 1985, 81, 349–351. [Google Scholar]
- Foemmel, R.S.; Gray, R.H.; Bernstein, I.A. Intracellular localization of fructose 1,6-bisphosphate aldolase. J. Biol. Chem. 1975, 250, 1892–1897. [Google Scholar]
- Farrow, S.M.; Jones, C.T. Membrane-associated pyruvate kinase in developing guinea-pig liver. Biochem. J. 1986, 235, 103–110. [Google Scholar]
- Gutowicz, J.; Terlecki, G. The association of glycolytic enzymes with cellular and model membranes. Cell Mol. Biol. Lett. 2003, 8, 667–680. [Google Scholar]
- Xu, K.Y.; Zweier, J.L.; Becker, L.C. Functional coupling between glycolysis and sarcoplasmic reticulum Ca2+ transport. Circ. Res. 1995, 77, 88–97. [Google Scholar] [CrossRef]
- Xu, K.Y.; Becker, L.C. Ultrastructural localization of glycolytic enzymes on sarcoplasmic reticulum vesticles. J. Histochem. Cytochem. 1998, 46, 419–427. [Google Scholar] [CrossRef]
- Jackson, S.A.; Thomson, M.J.; Clegg, J.S. Glycolysis compared in intact, permeabilized and sonicated L-929 cells. FEBS Lett. 1990, 262, 212–214. [Google Scholar] [CrossRef]
- Hudder, A.; Nathanson, L.; Deutscher, M.P. Organization of mammalian cytoplasm. Mol. Cell Biol. 2003, 23, 9318–9326. [Google Scholar] [CrossRef]
- Maughan, D.W.; Henkin, J.A.; Vigoreaux, J.O. Concentrations of glycolytic enzymes and other cytosolic proteins in the diffusible fraction of a vertebrate muscle proteome. Mol. Cell Proteomics 2005, 4, 1541–1549. [Google Scholar]
- Weiss, J.N.; Korge, P. The cytoplasm: No longer a well-mixed bag. Circ. Res. 2001, 89, 108–110. [Google Scholar]
- Gomez-Arreaza, A.; Acosta, H.; Quinones, W.; Concepcion, J.L.; Michels, P.A.; Avilan, L. Extracellular functions of glycolytic enzymes of parasites: Unpredicted use of ancient proteins. Mol. Biochem. Parasitol. 2014, 193, 75–81. [Google Scholar] [CrossRef]
- Zaffagnini, M.; Fermani, S.; Costa, A.; Lemaire, S.D.; Trost, P. Plant cytoplasmic GAPDH: Redox post-translational modifications and moonlighting properties. Front. Plant Sci. 2013, 4, 450. [Google Scholar] [CrossRef]
- Sirover, M.A. New nuclear functions of the glycolytic protein, glyceraldehyde-3-phosphate dehydrogenase, in mammalian cells. J. Cell Biochem. 2005, 95, 45–52. [Google Scholar] [CrossRef]
- Hara, M.R.; Agrawal, N.; Kim, S.F.; Cascio, M.B.; Fujimuro, M.; Ozeki, Y.; Takahashi, M.; Cheah, J.H.; Tankou, S.K.; Hester, L.D.; et al. S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat. Cell Biol. 2005, 7, 665–674. [Google Scholar] [CrossRef]
- Sen, N.; Hara, M.R.; Kornberg, M.D.; Cascio, M.B.; Bae, B.I.; Shahani, N.; Thomas, B.; Dawson, T.M.; Dawson, V.L.; Snyder, S.H.; et al. Nitric oxide-induced nuclear GAPDH activates p300/CBP and mediates apoptosis. Nat. Cell Biol. 2008, 10, 866–873. [Google Scholar]
- Rawat, P.; Kumar, S.; Sheokand, N.; Raje, C.I.; Raje, M. The multifunctional glycolytic protein glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is a novel macrophage lactoferrin receptor. Biochem. Cell Biol. 2012, 90, 329–338. [Google Scholar] [CrossRef]
- Norris, V.; Amar, P.; Legent, G.; Ripoll, C.; Thellier, M.; Ovadi, J. Sensor potency of the moonlighting enzyme-decorated cytoskeleton: The cytoskeleton as a metabolic sensor. BMC Biochem 2013, 14, 3. [Google Scholar]
- Sriram, G.; Martinez, J.A.; McCabe, E.R.; Liao, J.C.; Dipple, K.M. Single-gene disorders: What role could moonlighting enzymes play? Am. J. Hum. Genet. 2005, 76, 911–924. [Google Scholar] [CrossRef]
- Jeffery, C.J. Proteins with neomorphic moonlighting functions in disease. IUBMB Life 2011, 63, 489–494. [Google Scholar] [CrossRef]
- Keller, M.A.; Turchyn, A.V.; Ralser, M. Non-enzymatic glycolysis and pentose phosphate pathway-like reactions in a plausible Archean ocean. Mol. Syst. Biol. 2014, 10, 725. [Google Scholar] [CrossRef] [Green Version]
- Skulachev, V.P.; Bogachev, A.V.; Kasparinsky, F.O. Principles of Bioenergetics; Springer: New York, NY, USA, 2012. [Google Scholar]
- Chebotareva, N.A. Effect of molecular crowding on the enzymes of glycogenolysis. Biochemistry (Mosc) 2007, 72, 1478–1490. [Google Scholar] [CrossRef]
- Cascante, M.; Sorribas, A.; Canela, E.I. Enzyme-enzyme interactions and metabolite channelling: Alternative mechanisms and their evolutionary significance. Biochem. J. 1994, 298, 313–320. [Google Scholar]
- Ellis, R.J.; Minton Allen, P. Protein aggregation in crowded environments. Biol. Chem. 2006, 387, 485. [Google Scholar] [CrossRef]
- Shearwin, K.; Nanhua, C.; Masters, C. Interactions between glycolytic enzymes and cytoskeletal structure—The influence of ionic strength and molecular crowding. Biochem. Int. 1990, 21, 53–60. [Google Scholar]
- Dhar, A.; Samiotakis, A.; Ebbinghaus, S.; Nienhaus, L.; Homouz, D.; Gruebele, M.; Cheung, M.S. Structure, function, and folding of phosphoglycerate kinase are strongly perturbed by macromolecular crowding. Proc. Natl. Acad. Sci. USA 2010, 107, 17586–17591. [Google Scholar] [CrossRef]
- Easterby, J.S. The analysis of metabolite channelling in multienzyme complexes and multifunctional proteins. Biochem. J. 1989, 264, 605–607. [Google Scholar]
- Shearer, G.; Lee, J.C.; Koo, J.A.; Kohl, D.H. Quantitative estimation of channeling from early glycolytic intermediates to CO in intact Escherichia coli. FEBS J. 2005, 272, 3260–3269. [Google Scholar] [CrossRef]
- Srivastava, D.K.; Bernhard, S.A. Biophysical Chemistry of Metabolic Reaction Sequences in Concentrated Enzyme Solution and in the Cell. Annu. Rev. Biophys. Biophys. Chem. 1987, 16, 175–204. [Google Scholar] [CrossRef]
- Albe, K.R.; Butler, M.H.; Wright, B.E. Cellular concentrations of enzymes and their substrates. J. Theor. Biol. 1990, 143, 163–195. [Google Scholar]
- Ehrenborg, E.; Krook, A. Regulation of skeletal muscle physiology and metabolism by peroxisome proliferator-activated receptor delta. Pharmacol. Rev. 2009, 61, 373–393. [Google Scholar] [CrossRef]
- Stump, C.S.; Henriksen, E.J.; Wei, Y.; Sowers, J.R. The metabolic syndrome: Role of skeletal muscle metabolism. Ann. Med. 2006, 38, 389–402. [Google Scholar] [CrossRef]
- Storlien, L.; Oakes, N.D.; Kelley, D.E. Metabolic flexibility. Proc. Nutr. Soc. 2004, 63, 363–368. [Google Scholar] [CrossRef]
- Galgani, J.E.; Moro, C.; Ravussin, E. Metabolic flexibility and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E1009–E1017. [Google Scholar] [CrossRef]
- Sinacore, D.R.; Gulve, E.A. The role of skeletal muscle in glucose transport, glucose homeostasis, and insulin resistance: Implications for physical therapy. Phys. Ther. 1993, 73, 878–891. [Google Scholar]
- Hittel, D.S.; Hathout, Y.; Hoffman, E.P.; Houmard, J.A. Proteome analysis of skeletal muscle from obese and morbidly obese women. Diabetes 2005, 54, 1283–1288. [Google Scholar] [CrossRef]
- Giebelstein, J.; Poschmann, G.; Hojlund, K.; Schechinger, W.; Dietrich, J.W.; Levin, K.; Beck-Nielsen, H.; Podwojski, K.; Stuhler, K.; Meyer, H.E.; et al. The proteomic signature of insulin-resistant human skeletal muscle reveals increased glycolytic and decreased mitochondrial enzymes. Diabetologia 2012, 55, 1114–1127. [Google Scholar] [Green Version]
- Myhill, S.; Booth, N.E.; McLaren-Howard, J. Chronic fatigue syndrome and mitochondrial dysfunction. Int. J. Clin. Exp. Med. 2009, 2, 1–16. [Google Scholar]
- Booth, N.E.; Myhill, S.; McLaren-Howard, J. Mitochondrial dysfunction and the pathophysiology of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). Int. J. Clin. Exp. Med. 2012, 5, 208–220. [Google Scholar]
- Carlson, B.E.; Vigoreaux, J.O.; Maughan, D.W. Diffusion coefficients of endogenous cytosolic proteins from rabbit skinned muscle fibers. Biophys. J. 2014, 106, 780–792. [Google Scholar] [CrossRef]
- Boateng, S.Y.; Goldspink, P.H. Assembly and maintenance of the sarcomere night and day. Cardiovasc. Res. 2008, 77, 667–675. [Google Scholar] [CrossRef]
- Kinsey, S.T.; Locke, B.R.; Dillaman, R.M. Molecules in motion: Influences of diffusion on metabolic structure and function in skeletal muscle. J. Exp. Biol. 2011, 214, 263–274. [Google Scholar] [CrossRef]
- Harris, S.J.; Winzor, D.J. Equilibrium partition studies of the myofibrillar interactions of glycolytic enzymes. Arch. Biochem. Biophys. 1989, 275, 185–191. [Google Scholar] [CrossRef]
- Pryme, I.F.; Hesketh, J.E. The Cytoskeleton: Role in Cell Physiology; JAI PRESS: Greenwich, CT, USA, 1996; Volume 2. [Google Scholar]
- Hsu, S.C.; Molday, R.S. Glycolytic enzymes and a GLUT-1 glucose transporter in the outer segments of rod and cone photoreceptor cells. J. Biol. Chem. 1991, 266, 21745–21752. [Google Scholar]
- Preller, A.; Wilson, J.E. Localization of the type III isozyme of hexokinase at the nuclear periphery. Arch. Biochem. Biophys. 1992, 294, 482–492. [Google Scholar] [CrossRef]
- Pedley, K.C.; Jones, G.E.; Magnani, M.; Rist, R.J.; Naftalin, R.J. Direct observation of hexokinase translocation in stimulated macrophages. Biochem. J. 1993, 291, 515–522. [Google Scholar]
- Araiza-Olivera, D.; Chiquete-Felix, N.; Rosas-Lemus, M.; Sampedro, J.G.; Pena, A.; Mujica, A.; Uribe-Carvajal, S. A glycolytic metabolon in Saccharomyces cerevisiae is stabilized by F-actin. FEBS J. 2013, 280, 3887–3905. [Google Scholar] [CrossRef]
- Aflalo, C.; Azoulay, H. Binding of rat brain hexokinase to recombinant yeast mitochondria: Effect of environmental factors and the source of porin. J. Bioenerg. Biomembr. 1998, 30, 245–255. [Google Scholar] [CrossRef]
- Golestani, A.; Nemat-Gorgani, M. Hexokinase “binding sites” of normal and tumoral human brain mitochondria. Mol. Cell Biochem. 2000, 215, 115–121. [Google Scholar] [CrossRef]
- Balasubramanian, R.; Karve, A.; Kandasamy, M.; Meagher, R.B.; Moore, B. A role for F-actin in hexokinase-mediated glucose signaling. Plant Physiol. 2007, 145, 1423–1434. [Google Scholar] [CrossRef]
- Balasubramanian, R.; Karve, A.; Moore, B.D. Actin-based cellular framework for glucose signaling by Arabidopsis hexokinase1. Plant Signal Behav. 2008, 3, 322–324. [Google Scholar] [CrossRef]
- Sullivan, D.T.; MacIntyre, R.; Fuda, N.; Fiori, J.; Barrilla, J.; Ramizel, L. Analysis of glycolytic enzyme co-localization in Drosophila flight muscle. J. Exp. Biol. 2003, 206, 2031–2038. [Google Scholar] [CrossRef]
- Parra, J.; Pette, D. Effects of low-frequency stimulation on soluble and structure-bound activities of hexokinase and phosphofructokinase in rat fast-twitch muscle. Biochim. Biophys. Acta 1995, 1251, 154–160. [Google Scholar] [CrossRef]
- Lushchak, V.I.; Bagnyukova, T.V.; Storey, J.M.; Storey, K.B. Influence of exercise on the activity and the distribution between free and bound forms of glycolytic and associated enzymes in tissues of horse mackerel. Braz. J. Med. Biol. Res. 2001, 34, 1055–1064. [Google Scholar] [CrossRef]
- Commichau, F.M.; Rothe, F.M.; Herzberg, C.; Wagner, E.; Hellwig, D.; Lehnik-Habrink, M.; Hammer, E.; Volker, U.; Stulke, J. Novel activities of glycolytic enzymes in Bacillus subtilis: Interactions with essential proteins involved in mRNA processing. Mol. Cell Proteomics 2009, 8, 1350–1360. [Google Scholar] [CrossRef]
- Kowalski, W.; Gizak, A.; Rakus, D. Phosphoglycerate mutase in mammalian striated muscles: Subcellular localization and binding partners. FEBS Lett. 2009, 583, 1841–1845. [Google Scholar] [CrossRef]
- Wojtera-Kwiczor, J.; Gross, F.; Leffers, H.M.; Kang, M.; Schneider, M.; Scheibe, R. Transfer of a Redox-Signal through the Cytosol by Redox-Dependent Microcompartmentation of Glycolytic Enzymes at Mitochondria and Actin Cytoskeleton. Front. Plant Sci. 2012, 3, 284. [Google Scholar]
- Schmitz, H.D.; Bereiter-Hahn, J. Glyceraldehyde-3-phosphate dehydrogenase associates with actin filaments in serum deprived NIH 3T3 cells only. Cell Biol. Int. 2002, 26, 155–164. [Google Scholar] [CrossRef]
- Waingeh, V.F.; Gustafson, C.D.; Kozliak, E.I.; Lowe, S.L.; Knull, H.R.; Thomasson, K.A. Glycolytic enzyme interactions with yeast and skeletal muscle F-actin. Biophys. J. 2006, 90, 1371–1384. [Google Scholar] [CrossRef]
- Minaschek, G.; Groschel-Stewart, U.; Blum, S.; Bereiter-Hahn, J. Microcompartmentation of glycolytic enzymes in cultured cells. Eur. J. Cell Biol. 1992, 58, 418–428. [Google Scholar]
- Puchulu-Campanella, E.; Chu, H.; Anstee, D.J.; Galan, J.A.; Tao, W.A.; Low, P.S. Identification of the components of a glycolytic enzyme metabolon on the human red blood cell membrane. J. Biol. Chem. 2013, 288, 848–858. [Google Scholar]
- Schmitz, J.P.; Groenendaal, W.; Wessels, B.; Wiseman, R.W.; Hilbers, P.A.; Nicolay, K.; Prompers, J.J.; Jeneson, J.A.; van Riel, N.A. Combined in vivo and in silico investigations of activation of glycolysis in contracting skeletal muscle. Am. J. Physiol. Cell Physiol. 2013, 304, C180–C193. [Google Scholar] [CrossRef]
- Melnikow, E.; Xu, S.; Liu, J.; Bell, A.J.; Ghedin, E.; Unnasch, T.R.; Lustigman, S. A potential role for the interaction of Wolbachia surface proteins with the Brugia malayi glycolytic enzymes and cytoskeleton in maintenance of endosymbiosis. PLoS Negl. Trop. Dis. 2013, 7, e2151. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Morris, A.J.; Tolan, D.R.; Pagliaro, L. The molecular nature of the F-actin binding activity of aldolase revealed with site-directed mutants. J. Biol. Chem. 1996, 271, 6861–6865. [Google Scholar]
- Schindler, R.; Weichselsdorfer, E.; Wagner, O.; Bereiter-Hahn, J. Aldolase-localization in cultured cells: Cell-type and substrate-specific regulation of cytoskeletal associations. Biochem. Cell Biol. 2001, 79, 719–728. [Google Scholar] [CrossRef]
- Cao, F.; Yanagihara, N.; Burke, J.M. Progressive association of a “soluble” glycolytic enzyme with the detergent-insoluble cytoskeleton during in vitro morphogenesis of MDCK epithelial cells. Cell Motil. Cytoskeleton 1999, 44, 133–142. [Google Scholar] [CrossRef]
- Holtgräwe, D.; Scholz, A.; Altmann, B.; Scheibe, R. Cytoskeleton-associated, carbohydrate-metabolizing enzymes in maize identified by yeast two-hybrid screening. Physiol. Plant. 2005, 125, 141–156. [Google Scholar] [CrossRef]
- Forlemu, N.Y.; Njabon, E.N.; Carlson, K.L.; Schmidt, E.S.; Waingeh, V.F.; Thomasson, K.A. Ionic strength dependence of F-actin and glycolytic enzyme associations: A Brownian dynamics simulations approach. Proteins 2011, 79, 2813–2827. [Google Scholar] [CrossRef]
- Forlemu, N.Y.; Waingeh, V.F.; Ouporov, I.V.; Lowe, S.L.; Thomasson, K.A. Theoretical study of interactions between muscle aldolase and F-actin: Insight into different species. Biopolymers 2007, 85, 60–71. [Google Scholar] [CrossRef]
- Ouporov, I.V.; Knull, H.R.; Huber, A.; Thomasson, K.A. Brownian Dynamics Simulations of Aldolase Binding Glyceraldehyde 3-Phosphate Dehydrogenase and the Possibility of Substrate Channeling. Biophys. J. 2001, 80, 2527–2535. [Google Scholar] [CrossRef]
- Ouporov, I.V.; Knull, H.R.; Lowe, S.L.; Thomasson, K.A. Interactions of glyceraldehyde-3-phosphate dehydrogenase with G- and F-actin predicted by Brownian dynamics. J. Mol. Recognit. 2001, 14, 29–41. [Google Scholar] [CrossRef]
- Ouporov, I.V.; Knull, H.R.; Thomasson, K.A. Brownian Dynamics Simulations of Interactions between Aldolase and G- or F-Actin. Biophys. J. 1999, 76, 17–27. [Google Scholar] [CrossRef]
- O'Reilly, G.; Clarke, F. Identification of an actin binding region in aldolase. FEBS Lett. 1993, 321, 69–72. [Google Scholar] [CrossRef]
- Gustafson, C. Glycolytic Enzyme Interactions with Wild Type and Mutant Saccharomyces Cervisiae Actin: Comparison with Skeletal Muscle Actin. M.S. thesis, University of North Dakota, Grand Forks, ND, USA, 1996. [Google Scholar]
- Marcondes, M.C.; Sola-Penna, M.; Torres Rda, S.; Zancan, P. Muscle-type 6-phosphofructo-1-kinase and aldolase associate conferring catalytic advantages for both enzymes. IUBMB Life 2011, 63, 435–445. [Google Scholar] [CrossRef]
- Rais, B.; Ortega, F.; Puigjaner, J.; Comin, B.; Orosz, F.; Ovadi, J.; Cascante, M. Quantitative characterization of homo- and heteroassociations of muscle phosphofructokinase with aldolase. Biochim. Biophys. Acta 2000, 1479, 303–314. [Google Scholar] [CrossRef]
- Real-Hohn, A.; Zancan, P.; Da Silva, D.; Martins, E.R.; Salgado, L.T.; Mermelstein, C.S.; Gomes, A.M.; Sola-Penna, M. Filamentous actin and its associated binding proteins are the stimulatory site for 6-phosphofructo-1-kinase association within the membrane of human erythrocytes. Biochimie 2010, 92, 538–544. [Google Scholar]
- Romagnoli, S.; Faleri, C.; Bini, L.; Baskin, T.I.; Cresti, M. Cytosolic proteins from tobacco pollen tubes that crosslink microtubules and actin filaments in vitro are metabolic enzymes. Cytoskeleton (Hoboken) 2010, 67, 745–754. [Google Scholar] [CrossRef]
- Coelho, R.G.; Calaca Ide, C.; Celestrini Dde, M.; Correia, A.H.; Costa, M.A.; Sola-Penna, M. Clotrimazole disrupts glycolysis in human breast cancer without affecting non-tumoral tissues. Mol. Genet. Metab. 2011, 103, 394–398. [Google Scholar] [CrossRef]
- Da Silva, D.; Ausina, P.; Alencar, E.M.; Coelho, W.S.; Zancan, P.; Sola-Penna, M. Metformin reverses hexokinase and phosphofructokinase downregulation and intracellular distribution in the heart of diabetic mice. IUBMB Life 2012, 64, 766–774. [Google Scholar]
- Coelho, W.S.; Costa, K.C.; Sola-Penna, M. Serotonin stimulates mouse skeletal muscle 6-phosphofructo-1-kinase through tyrosine-phosphorylation of the enzyme altering its intracellular localization. Mol. Genet. Metab. 2007, 92, 364–370. [Google Scholar] [CrossRef]
- Wassenberg, D.; Wuhrer, M.; Beaucamp, N.; Schurig, H.; Wozny, M.; Reusch, D.; Fabry, S.; Jaenicke, R. Local variability of the phosphoglycerate kinase-triosephosphate isomerase fusion protein from Thermotoga maritima MSB 8. Biol. Chem. 2001, 382, 693–697. [Google Scholar] [CrossRef]
- Lowe, S.L.; Adrian, C.; Ouporov, I.V.; Waingeh, V.F.; Thomasson, K.A. Brownian dynamics simulations of glycolytic enzyme subsets with F-actin. Biopolymers 2003, 70, 456–470. [Google Scholar] [CrossRef]
- Merkulova, T.; Lucas, M.; Jabet, C.; Lamande, N.; Rouzeau, J.D.; Gros, F.; Lazar, M.; Keller, A. Biochemical characterization of the mouse muscle-specific enolase: Developmental changes in electrophoretic variants and selective binding to other proteins. Biochem. J. 1997, 323, 791–800. [Google Scholar]
- Lehotzky, A.; Telegdi, M.; Liliom, K.; Ovadi, J. Interaction of phosphofructokinase with tubulin and microtubules. Quantitative evaluation of the mutual effects. J. Biol. Chem. 1993, 268, 10888–10894. [Google Scholar]
- Mitchell, B.F.; Pedersen, L.B.; Feely, M.; Rosenbaum, J.L.; Mitchell, D.R. Atp production in Chlamydomonas reinhardtii flagella by glycolytic enzymes. Mol. Biol. Cell 2005, 16, 4509–4518. [Google Scholar] [CrossRef]
- Volker, K.W.; Knull, H. A glycolytic enzyme binding domain on tubulin. Arch. Biochem. Biophys. 1997, 338, 237–243. [Google Scholar] [CrossRef]
- Volker, K.W.; Reinitz, C.A.; Knull, H.R. Glycolytic enzymes and assembly of microtubule networks. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 1995, 112, 503–514. [Google Scholar] [CrossRef]
- Keller, A.; Peltzer, J.; Carpentier, G.; Horvath, I.; Olah, J.; Duchesnay, A.; Orosz, F.; Ovadi, J. Interactions of enolase isoforms with tubulin and microtubules during myogenesis. Biochim. Biophys. Acta 2007, 1770, 919–926. [Google Scholar] [CrossRef]
- Foucault, G.; Vacher, M.; Merkulova, T.; Keller, A.; Arrio-Dupont, M. Presence of enolase in the M-band of skeletal muscle and possible indirect interaction with the cytosolic muscle isoform of creatine kinase. Biochem. J. 1999, 338, 115–121. [Google Scholar] [CrossRef]
- Hakobyan, D.; Nazaryan, K. Molecular dynamics study of interaction and substrate channeling between neuron-specific enolase and B-type phosphoglycerate mutase. Proteins 2010, 78, 1691–1704. [Google Scholar]
- Hakobyan, D.; Nazaryan, K. Molecular dynamics simulation of interactions in glycolytic enzymes. Biochemistry (Mosc) 2006, 71, 370–375. [Google Scholar] [CrossRef]
- Hakobyan, D.; Nazaryan, K. Investigation of interaction between enolase and phosphoglycerate mutase using molecular dynamics simulation. J. Biomol. Struct. Dyn. 2006, 23, 625–634. [Google Scholar] [CrossRef]
- Engel, M.; Mazurek, S.; Eigenbrodt, E.; Welter, C. Phosphoglycerate mutase-derived polypeptide inhibits glycolytic flux and induces cell growth arrest in tumor cell lines. J. Biol. Chem. 2004, 279, 35803–35812. [Google Scholar] [CrossRef]
- Kholodenko, B.N.; Sakamoto, N.; Puigjaner, J.; Westerhoff, H.V.; Cascante, M. Strong control on the transit time in metabolic channelling. FEBS Lett. 1996, 389, 123–125. [Google Scholar] [CrossRef]
- Stephan, P.; Clarke, F.; Morton, D. The indirect binding of triose-phosphate isomerase to myofibrils to form a glycolytic enzyme mini-complex. Biochim. Biophys. Acta 1986, 873, 127–135. [Google Scholar] [CrossRef]
- Ritterson Lew, C.; Tolan, D.R. Targeting of several glycolytic enzymes using RNA interference reveals aldolase affects cancer cell proliferation through a non-glycolytic mechanism. J. Biol. Chem. 2012, 287, 42554–42563. [Google Scholar] [CrossRef]
- Wojtas, K.; Slepecky, N.; von Kalm, L.; Sullivan, D. Flight muscle function in Drosophila requires colocalization of glycolytic enzymes. Mol. Biol. Cell 1997, 8, 1665–1675. [Google Scholar] [CrossRef]
- Vigoreaux, J. Nature’s Versatile Engine: Insect Flight Muscle Inside and Out. In Molecular Biology Intelligence Unit; Springer: London, UK, 2006; pp. 188–195. [Google Scholar]
- Eanes, W.F.; Merritt, T.J.; Flowers, J.M.; Kumagai, S.; Sezgin, E.; Zhu, C.T. Flux control and excess capacity in the enzymes of glycolysis and their relationship to flight metabolism in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 2006, 103, 19413–19418. [Google Scholar] [CrossRef]
- Duffy, J.B. GAL4 system in Drosophila: A fly geneticist’s Swiss army knife. Genesis 2002, 34, 1–15. [Google Scholar] [CrossRef]
- Boles, E.; Zimmermann, F.K. Saccharomyces cerevisiae phosphoglucose isomerase and fructose bisphosphate aldolase can be replaced functionally by the corresponding enzymes of Escherichia coli and Drosophila melanogaster. Curr. Genet. 1993, 23, 187–191. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Menard, L.; Maughan, D.; Vigoreaux, J. The Structural and Functional Coordination of Glycolytic Enzymes in Muscle: Evidence of a Metabolon? Biology 2014, 3, 623-644. https://doi.org/10.3390/biology3030623
Menard L, Maughan D, Vigoreaux J. The Structural and Functional Coordination of Glycolytic Enzymes in Muscle: Evidence of a Metabolon? Biology. 2014; 3(3):623-644. https://doi.org/10.3390/biology3030623
Chicago/Turabian StyleMenard, Lynda, David Maughan, and Jim Vigoreaux. 2014. "The Structural and Functional Coordination of Glycolytic Enzymes in Muscle: Evidence of a Metabolon?" Biology 3, no. 3: 623-644. https://doi.org/10.3390/biology3030623