The Human ABCG1 Transporter Mobilizes Plasma Membrane and Late Endosomal Non-Sphingomyelin-Associated-Cholesterol for Efflux and Esterification


Abstract
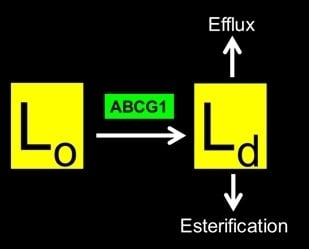
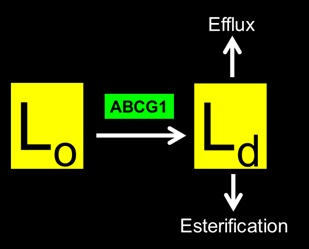
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental Section
3. Results and Discussion
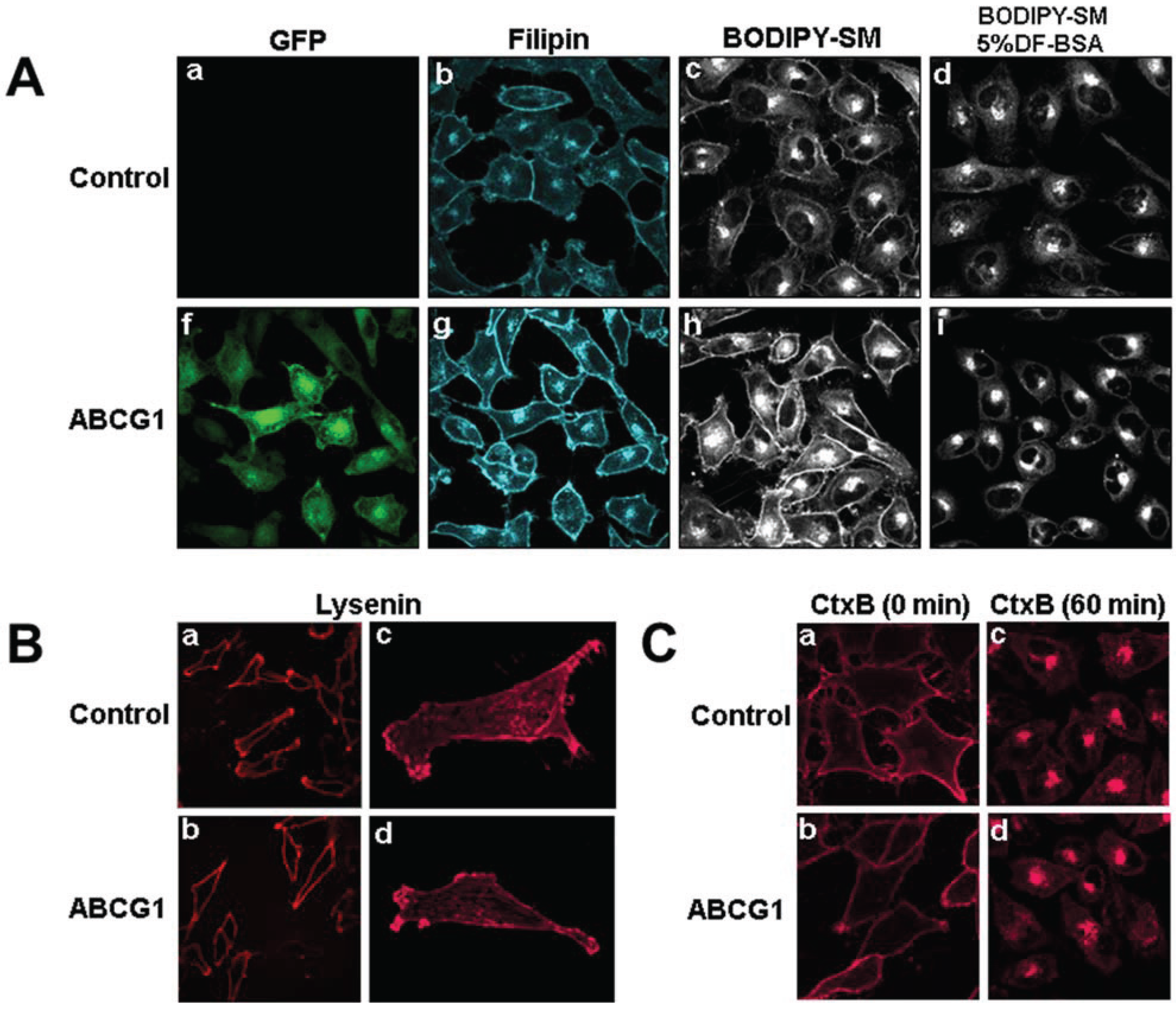
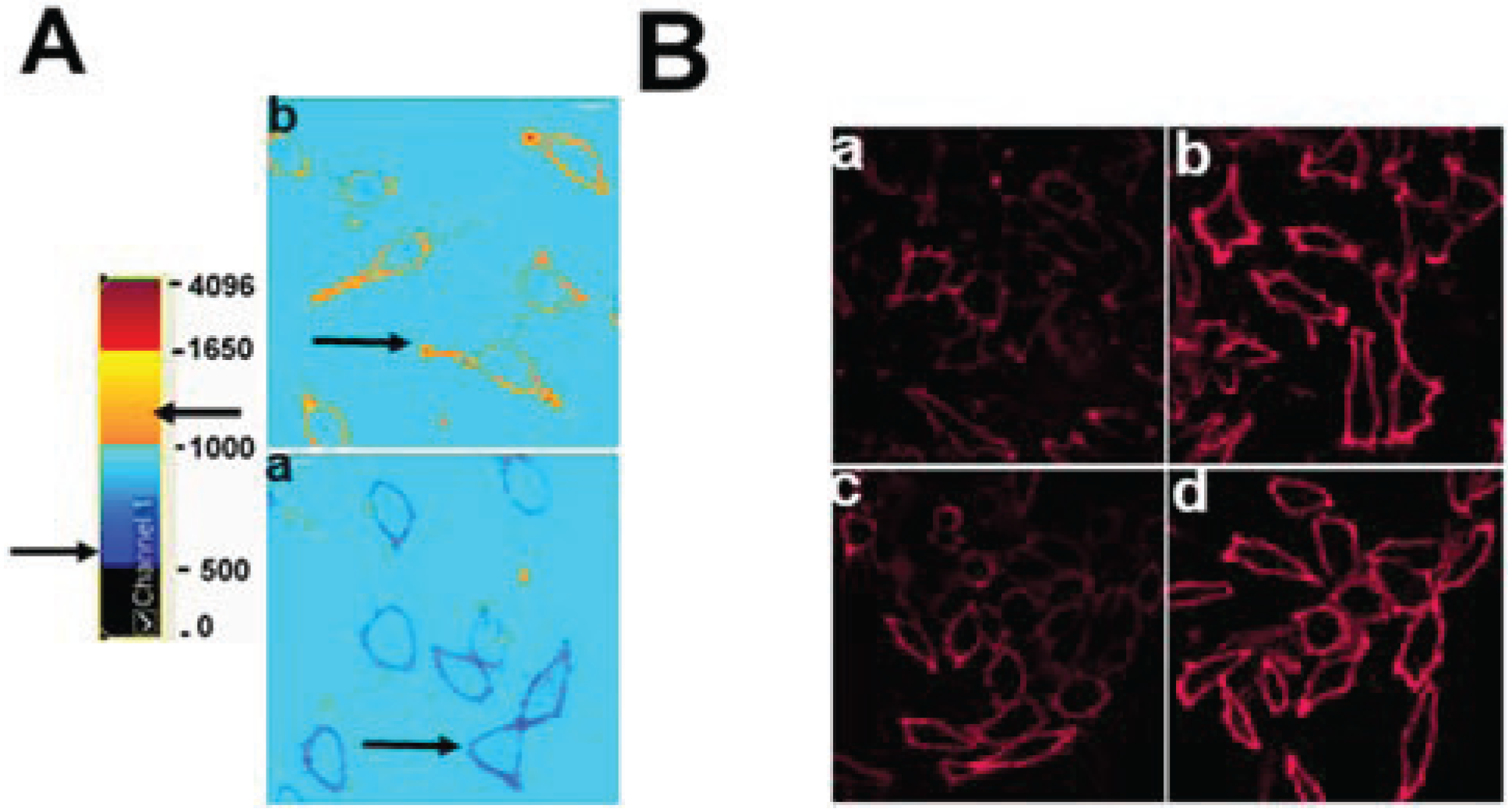
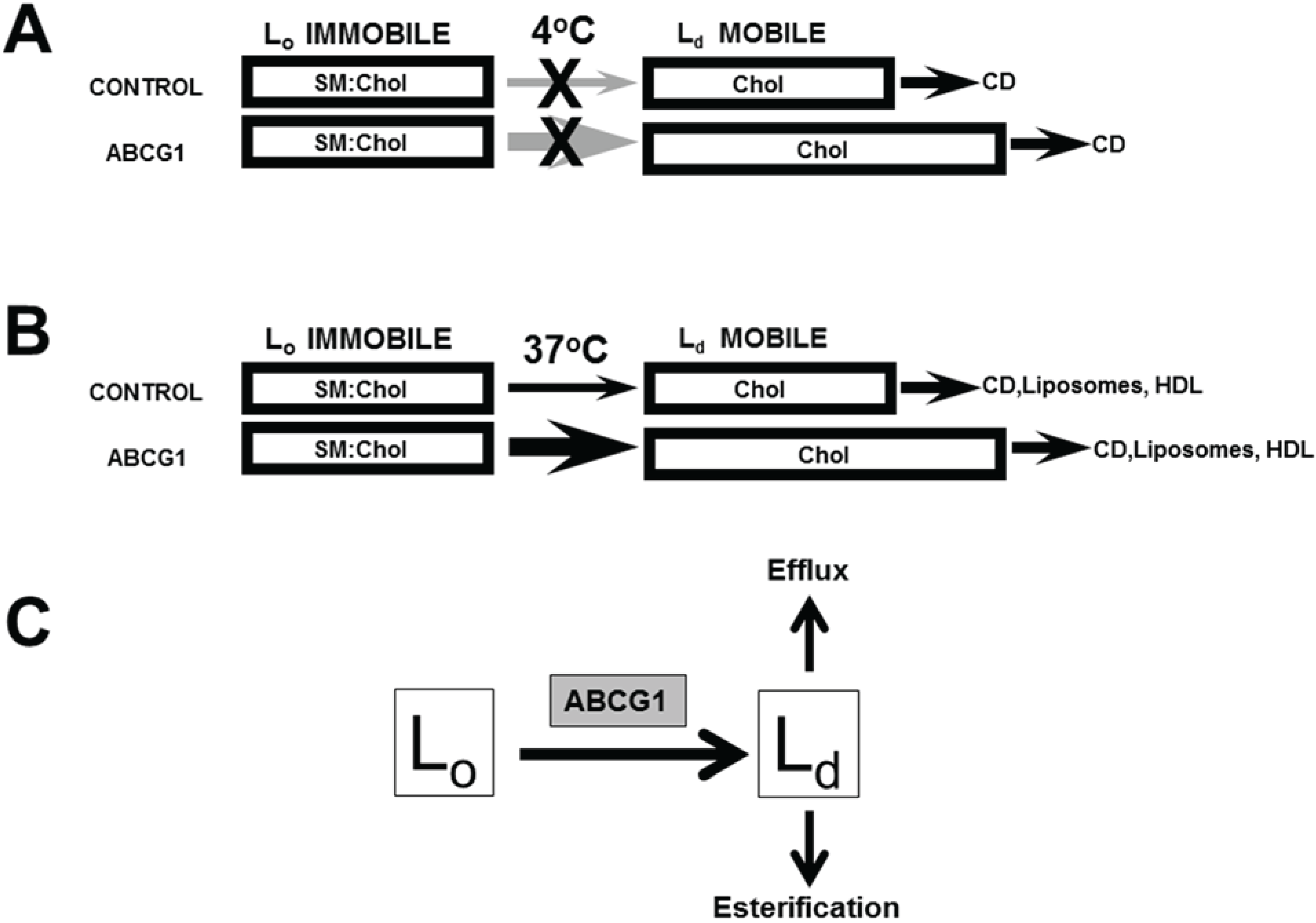
3.1. Cholesterol at the PM and LE of ABCG1-Cells Markedly Increases Incorporation of Exogenous Fluorescent SM, a Marker for Non-SM-Associated Cholesterol

3.2. ABCG1-GFP Increases PM and LE Non-SM-Associated Detergent-Soluble Cholesterol as Well as PM Non-SM-Associated Detergent-Resistant Cholesterol

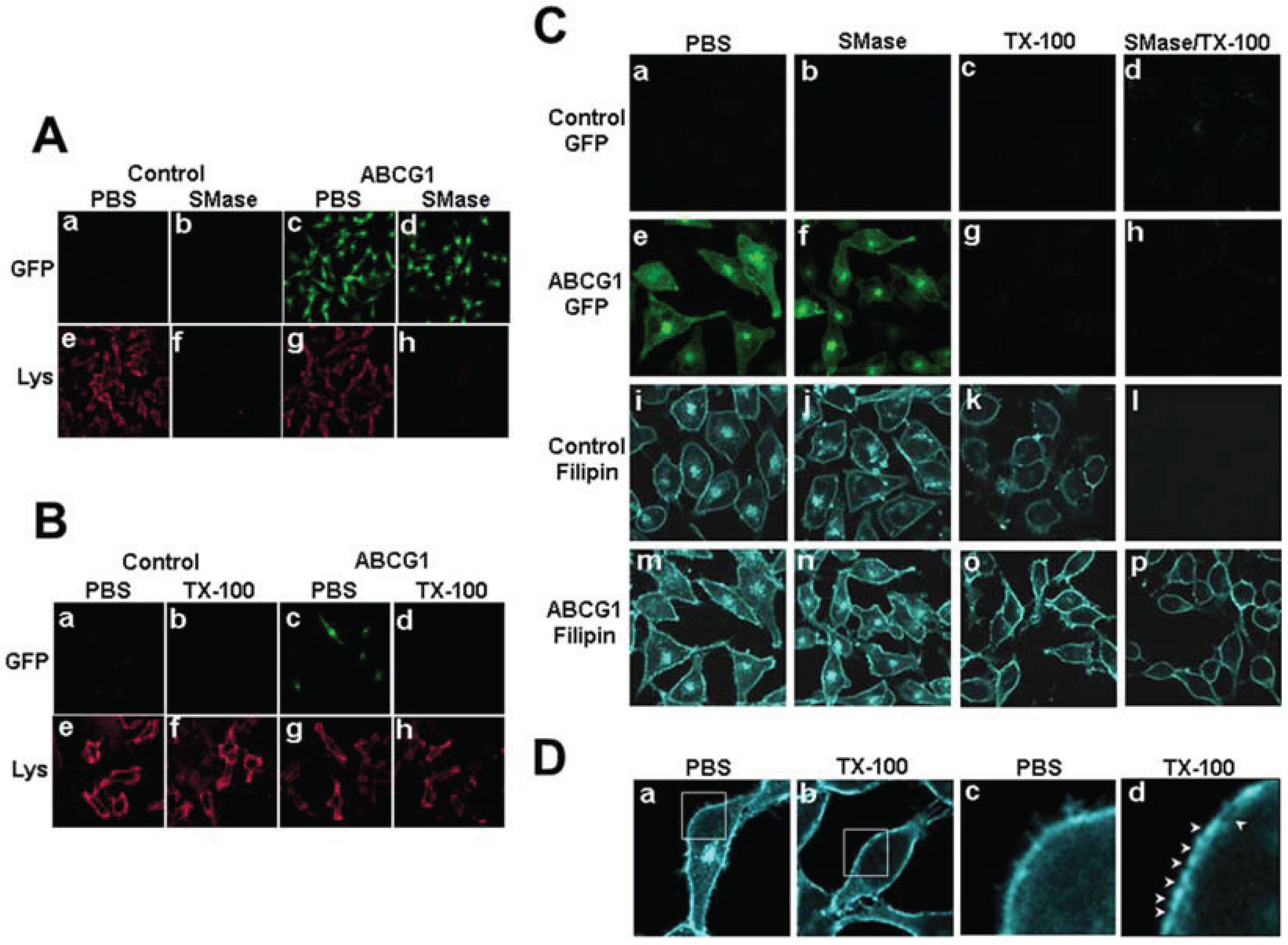
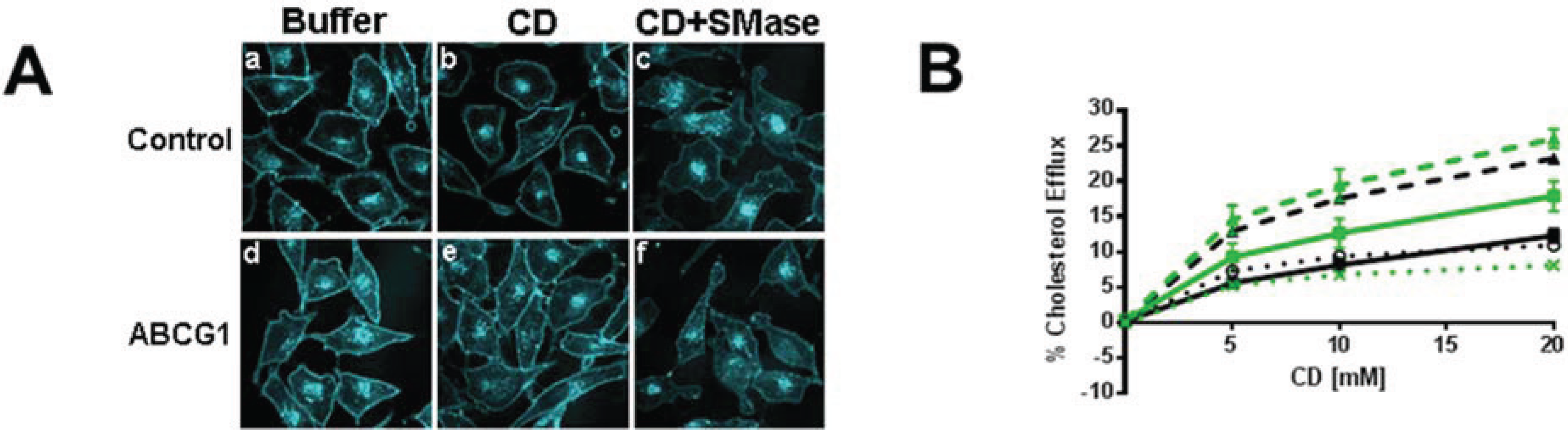
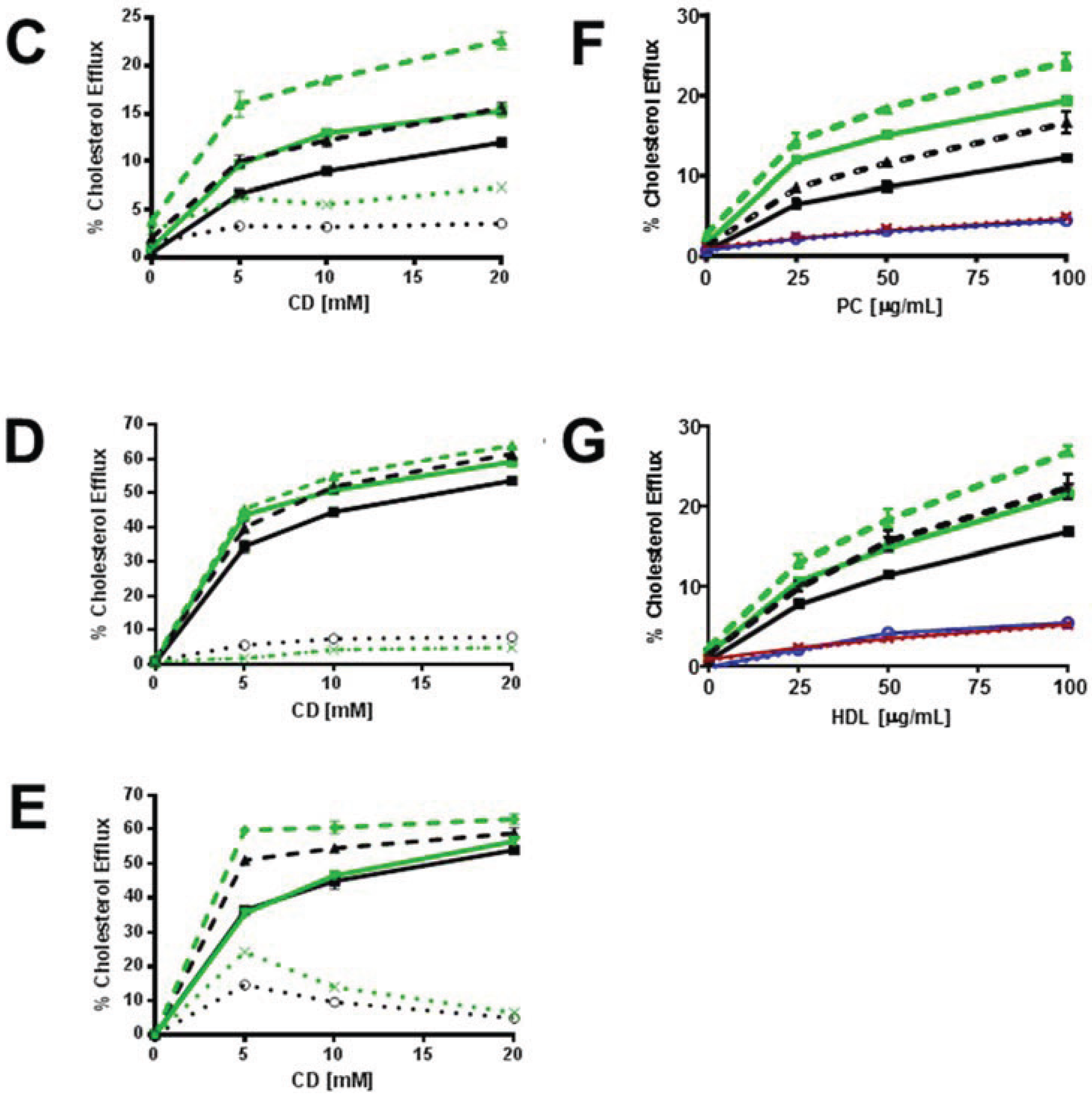
3.2.1. ABCG1-GFP Expression Increases PM and LE Pools of Non-SM-Associated Cholesterol Available for Efflux to CD
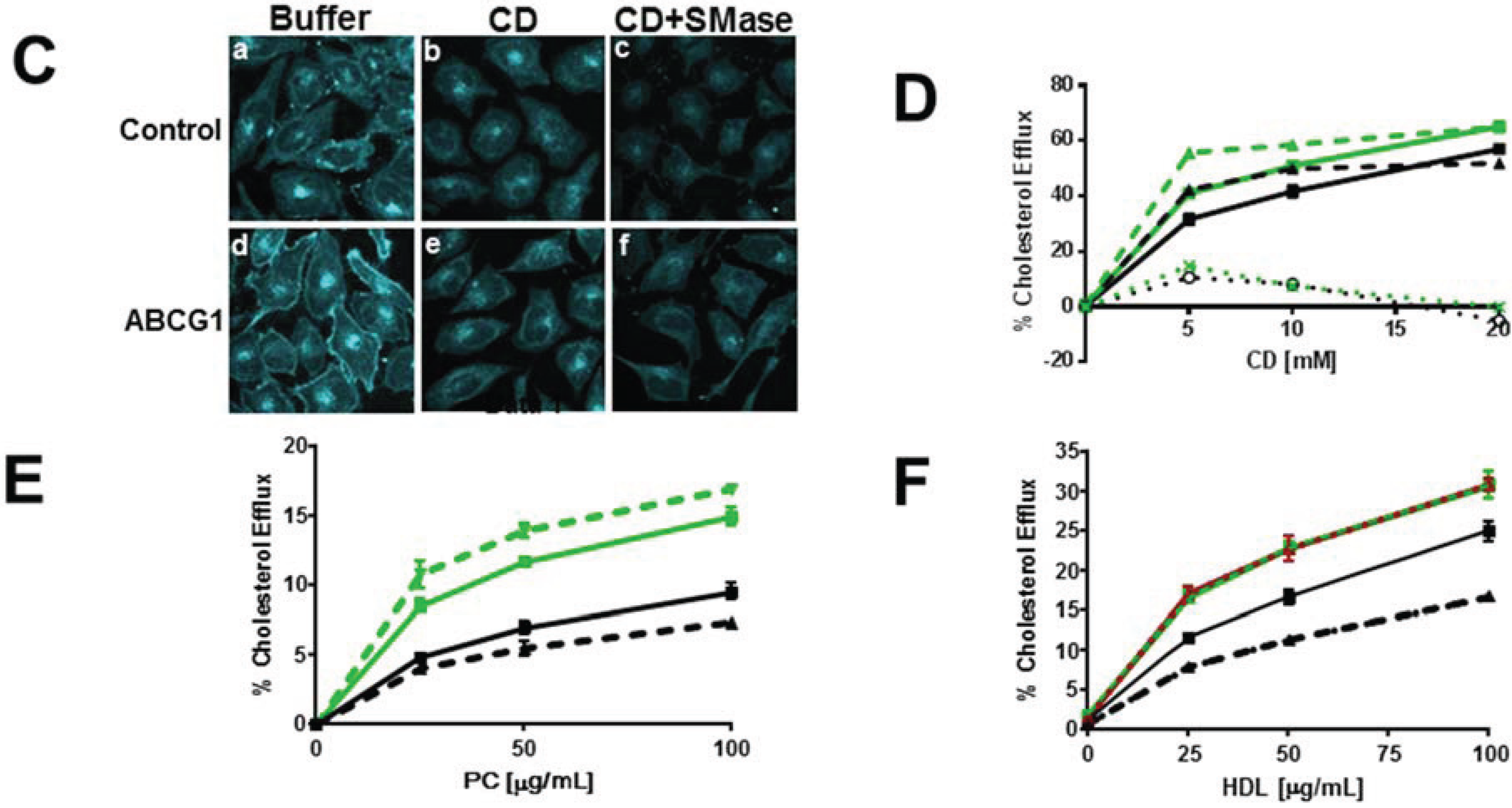
3.2.2. SMase Hydrolysis of Cellular SM Increases ABCG1-Mediated Cellular Cholesterol Efflux to Extracellular Lipid Acceptors

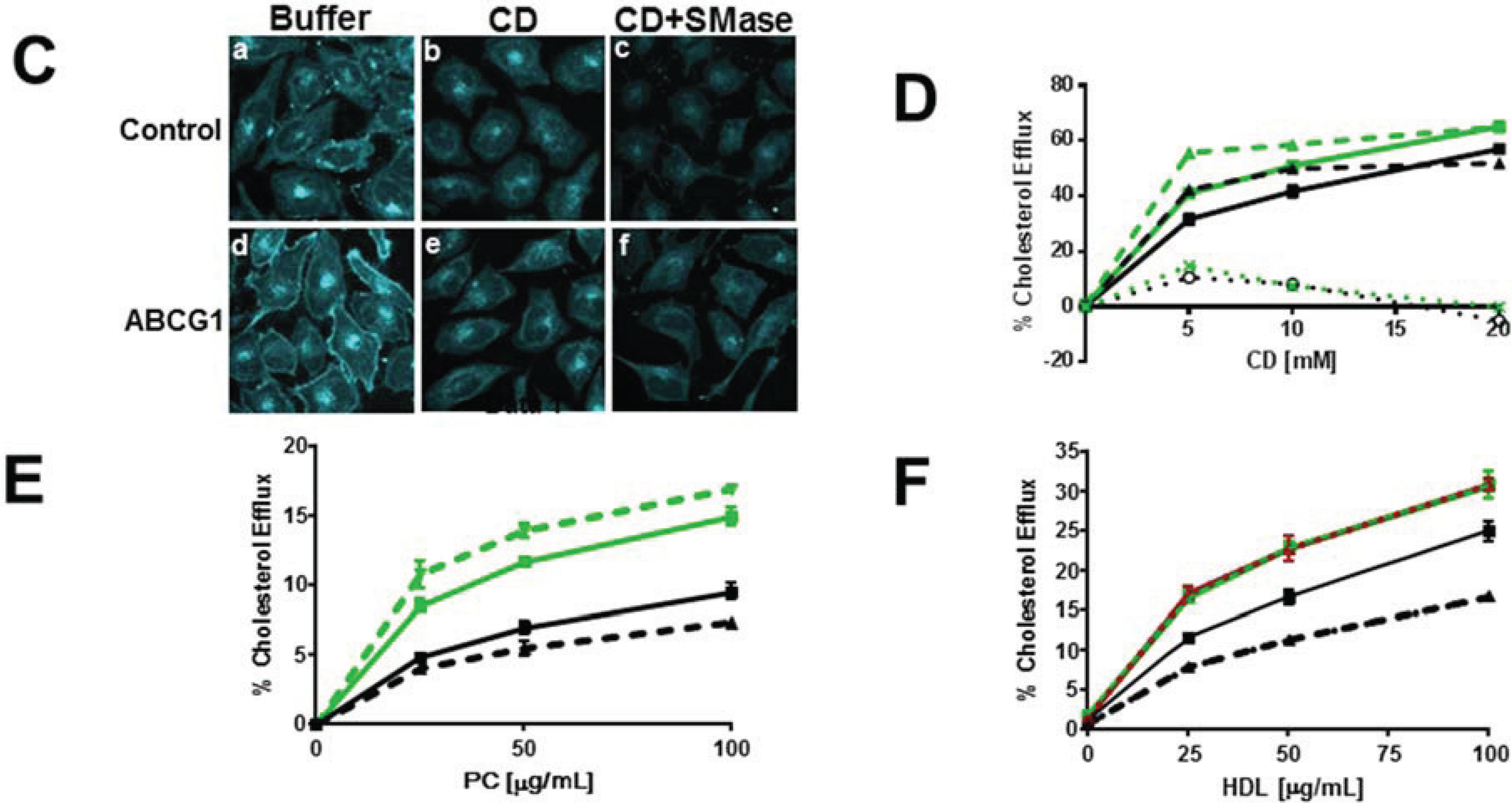
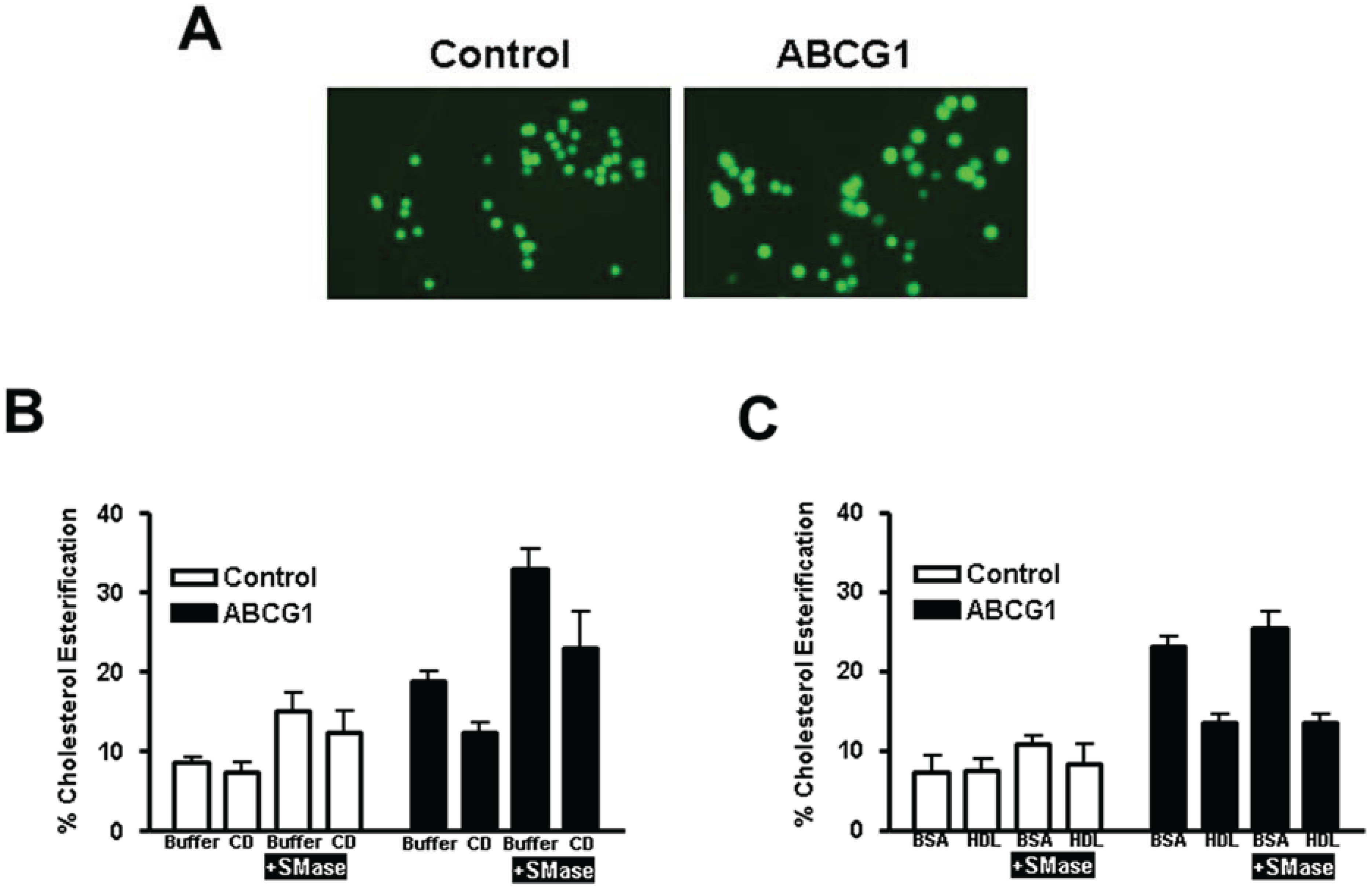
3.2.3. ABCG1-Mediated Increased Cellular Cholesterol Esterification is Enhanced by SMase-Mediated Cholesterol Mobilization and is Reduced by CD and HDL



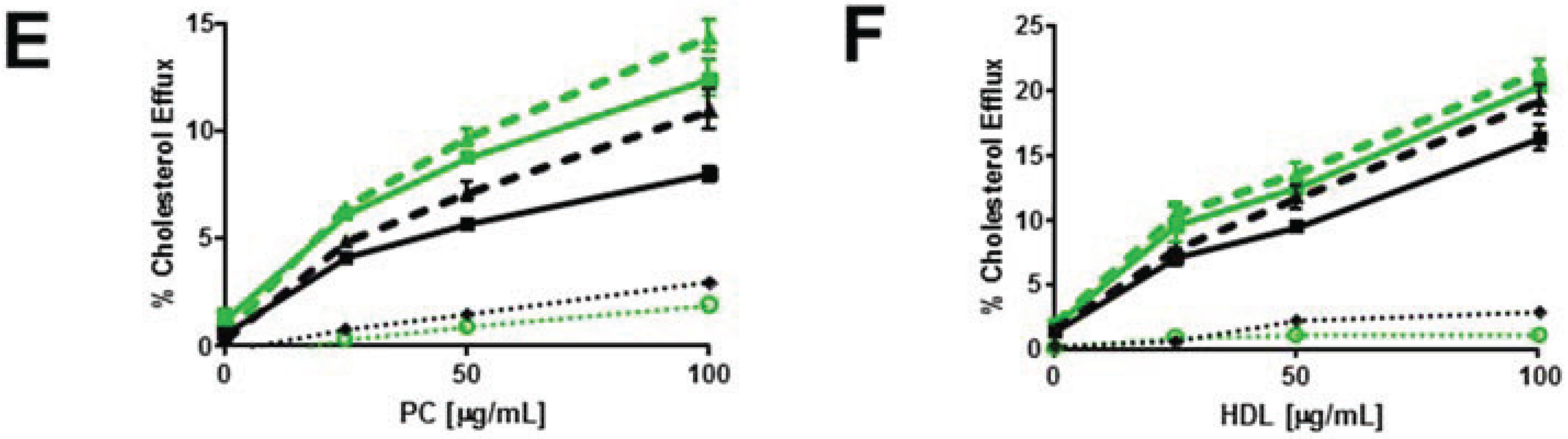
3.3. Enrichment of Membrane SM with Exogenous SM Increases ABCG1-Mediated Cellular Cholesterol Efflux
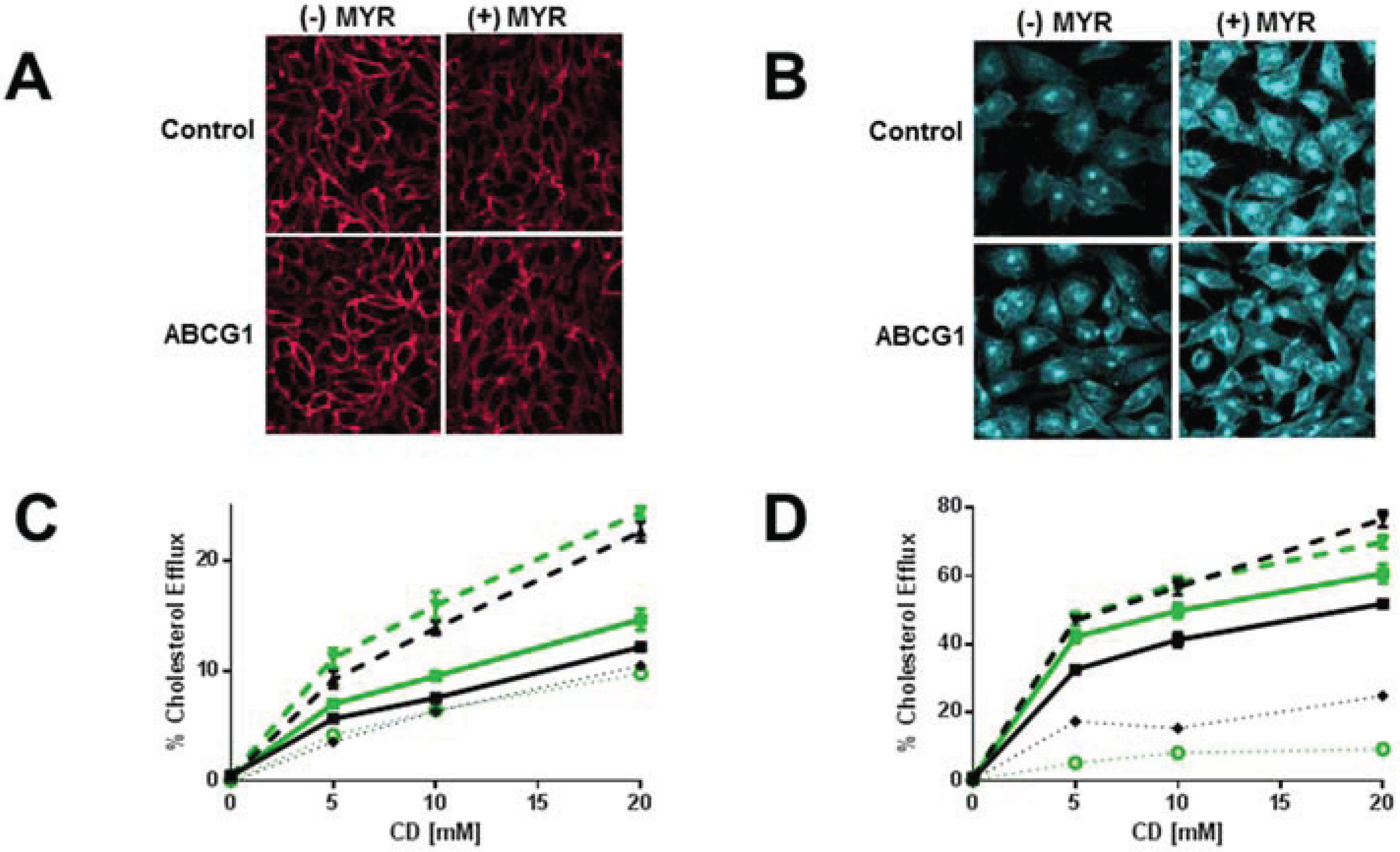
3.4. Myriocin-Mediated Reduction of Cellular SM Increases Cellular Cholesterol Efflux


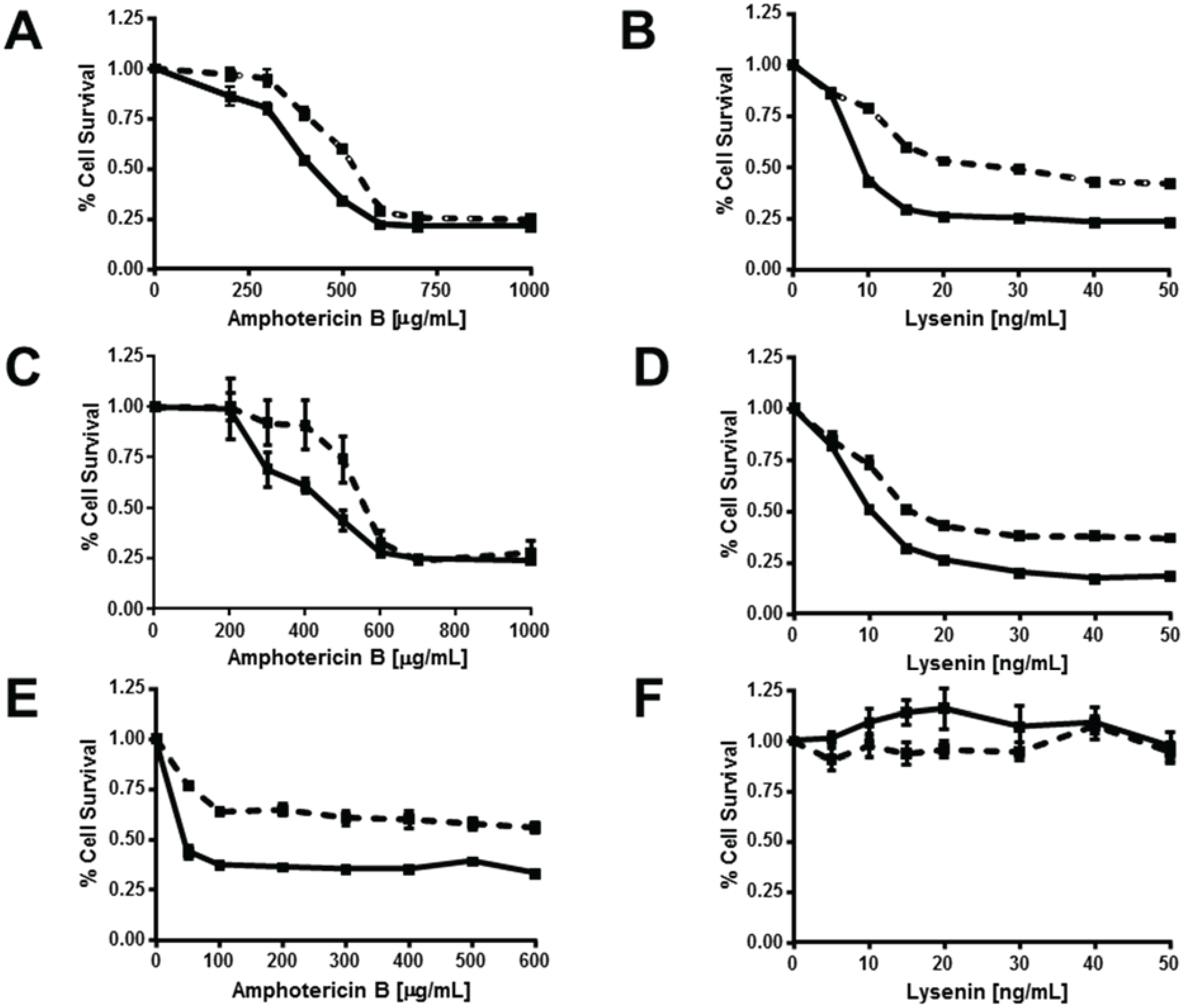
3.5. ABCG1 Confers Resistance to both Cholesterol-Dependent and Sphingomyelin-Dependent Toxin-Induced Cytolysis

4. Conclusions

Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Neufeld, E.B.; O’Brien, K.; Walts, A.D.; Stonik, J.A.; Demosky, S.J.; Malide, D.; Combs, C.A.; Remaley, A.T. Cellular localization and trafficking of the human ABCG1 transporter. Biology (Basel) 2014, 3, 781–800. [Google Scholar]
- Vaughan, A.M.; Oram, J.F. ABCG1 redistributes cell cholesterol to domains removable by high density lipoprotein but not by lipid-depleted apolipoproteins. J. Biol. Chem. 2005, 280, 30150–30157. [Google Scholar] [CrossRef] [PubMed]
- Pagler, T.A.; Wang, M.; Mondal, M.; Murphy, A.J.; Westerterp, M.; Moore, K.J.; Maxfield, F.R.; Tall, A.R. Deletion of ABCA1 and ABCG1 impairs macrophage migration because of increased Rac1 signaling. Circ. Res. 2011, 108, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, H.; Kimura, Y.; Kioka, N.; Matsuo, M.; Ueda, K. Atpase activity of human ABCG1 is stimulated by cholesterol and sphingomyelin. J. Lipid Res. 2013, 54, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Sano, O.; Kobayashi, A.; Nagao, K.; Kumagai, K.; Kioka, N.; Hanada, K.; Ueda, K.; Matsuo, M. Sphingomyelin-dependence of cholesterol efflux mediated by ABCG1. J. Lipid Res. 2007, 48, 2377–2384. [Google Scholar] [CrossRef] [PubMed]
- Simons, K.; Ikonen, E. How cells handle cholesterol. Science 2000, 290, 1721–1726. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Feigenson, G.W. A microscopic interaction model of maximum solubility of cholesterol in lipid bilayers. Biophys. J. 1999, 76, 2142–2157. [Google Scholar] [CrossRef] [PubMed]
- Eggeling, C.; Ringemann, C.; Medda, R.; Schwarzmann, G.; Sandhoff, K.; Polyakova, S.; Belov, V.N.; Hein, B.; von Middendorff, C.; Schonle, A.; et al. Direct observation of the nanoscale dynamics of membrane lipids in a living cell. Nature 2009, 457, 1159–1162. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, E.B.; Cooney, A.M.; Pitha, J.; Dawidowicz, E.A.; Dwyer, N.K.; Pentchev, P.G.; Blanchette-Mackie, E.J. Intracellular trafficking of cholesterol monitored with a cyclodextrin. J. Biol. Chem. 1996, 271, 21604–21613. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, S.A.; Gunther, G.; Tricerri, M.A.; Gratton, E. Methyl-beta-cyclodextrins preferentially remove cholesterol from the liquid disordered phase in giant unilamellar vesicles. J. Membr. Biol. 2011, 241, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lopez, C.A.; de Vries, A.H.; Marrink, S.J. Computational microscopy of cyclodextrin mediated cholesterol extraction from lipid model membranes. Sci. Rep. 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Schuck, S.; Honsho, M.; Ekroos, K.; Shevchenko, A.; Simons, K. Resistance of cell membranes to different detergents. PNAS 2003, 100, 5795–5800. [Google Scholar] [CrossRef] [PubMed]
- Okwu, A.K.; Xu, X.X.; Shiratori, Y.; Tabas, I. Regulation of the threshold for lipoprotein-induced acyl-coA:Cholesterol o-acyltransferase stimulation in macrophages by cellular sphingomyelin content. J. Lipid Res. 1994, 35, 644–655. [Google Scholar] [PubMed]
- Feng, B.; Yao, P.M.; Li, Y.; Devlin, C.M.; Zhang, D.; Harding, H.P.; Sweeney, M.; Rong, J.X.; Kuriakose, G.; Fisher, E.A.; et al. The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat. Cell Biol. 2003, 5, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Ihrke, G.; Neufeld, E.B.; Meads, T.; Shanks, M.R.; Cassio, D.; Laurent, M.; Schroer, T.A.; Pagano, R.E.; Hubbard, A.L. Wif-B cells: An in vitro model for studies of hepatocyte polarity. J. Cell Biol. 1993, 123, 1761–1775. [Google Scholar] [CrossRef] [PubMed]
- Ishitsuka, R.; Yamaji-Hasegawa, A.; Makino, A.; Hirabayashi, Y.; Kobayashi, T. A lipid-specific toxin reveals heterogeneity of sphingomyelin-containing membranes. Biophys. J. 2004, 86, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, E.B.; Remaley, A.T.; Demosky, S.J.; Stonik, J.A.; Cooney, A.M.; Comly, M.; Dwyer, N.K.; Zhang, M.; Blanchette-Mackie, J.; Santamarina-Fojo, S.; et al. Cellular localization and trafficking of the human ABCA1 transporter. J. Biol. Chem. 2001, 276, 27584–27590. [Google Scholar] [CrossRef] [PubMed]
- Brewer, H.B., Jr.; Ronan, R.; Meng, M.; Bishop, C. Isolation and characterization of apolipoproteins A-I, A-II, And A-IV. Methods enzymol. 1986, 128, 223–246. [Google Scholar] [PubMed]
- Remaley, A.T.; Thomas, F.; Stonik, J.A.; Demosky, S.J.; Bark, S.E.; Neufeld, E.B.; Bocharov, A.V.; Vishnyakova, T.G.; Patterson, A.P.; Eggerman, T.L.; et al. Synthetic amphipathic helical peptides promote lipid efflux from cells by an ABCA1-dependent and an ABCA1-independent pathway. J. Lipid Res. 2003, 44, 828–836. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, N.L.; Andemariam, B.; Underwood, K.W.; Panchalingam, K.; Sternberg, D.; Kielian, M.; Liscum, L. Analysis of a chinese hamster ovary cell mutant with defective mobilization of cholesterol from the plasma membrane to the endoplasmic reticulum. J. Lipid Res. 1997, 38, 1973–1987. [Google Scholar] [PubMed]
- Hanada, K.; Hara, T.; Fukasawa, M.; Yamaji, A.; Umeda, M.; Nishijima, M. Mammalian cell mutants resistant to a sphingomyelin-directed cytolysin. Genetic and biochemical evidence for complex formation of the LCB1 protein with the LCB2 protein for serine palmitoyltransferase. J. Biol. Chem. 1998, 273, 33787–33794. [Google Scholar] [CrossRef] [PubMed]
- Puri, V.; Watanabe, R.; Dominguez, M.; Sun, X.; Wheatley, C.L.; Marks, D.L.; Pagano, R.E. Cholesterol modulates membrane traffic along the endocytic pathway in sphingolipid-storage diseases. Nat. Cell Biol. 1999, 1, 386–388. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, E.B.; Stonik, J.A.; Demosky, S.J., Jr.; Knapper, C.L.; Combs, C.A.; Cooney, A.; Comly, M.; Dwyer, N.; Blanchette-Mackie, J.; Remaley, A.T.; et al. The ABCA1 transporter modulates late endocytic trafficking: Insights from the correction of the genetic defect in tangier disease. J. Biol. Chem. 2004, 279, 15571–15578. [Google Scholar] [CrossRef] [PubMed]
- Hao, M.; Mukherjee, S.; Maxfield, F.R. Cholesterol depletion induces large scale domain segregation in living cell membranes. PNAS 2001, 98, 13072–13077. [Google Scholar] [CrossRef] [PubMed]
- Pagano, R.E.; Watanabe, R.; Wheatley, C.; Chen, C.S. Use of N-[5-(5,7-dimethyl boron dipyrromethene difluoride-sphingomyelin to study membrane traffic along the endocytic pathway. Chem. Physic. Lipids 1999, 102, 55–63. [Google Scholar] [CrossRef]
- Pagano, R.E.; Martin, O.C.; Kang, H.C.; Haugland, R.P. A novel fluorescent ceramide analogue for studying membrane traffic in animal cells: Accumulation at the golgi apparatus results in altered spectral properties of the sphingolipid precursor. J. Cell Biol. 1991, 113, 1267–1279. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.S.; Martin, O.C.; Pagano, R.E. Changes in the spectral properties of a plasma membrane lipid analog during the first seconds of endocytosis in living cells. Biophys. J. 1997, 72, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Merritt, E.A.; Sarfaty, S.; van den Akker, F.; L’Hoir, C.; Martial, J.A.; Hol, W.G. Crystal structure of cholera toxin B-pentamer bound to receptor GM1 pentasaccharide. Pro. Sci. 1994, 3, 166–175. [Google Scholar] [CrossRef]
- Kiyokawa, E.; Baba, T.; Otsuka, N.; Makino, A.; Ohno, S.; Kobayashi, T. Spatial and functional heterogeneity of sphingolipid-rich membrane domains. J. Biol. Chem. 2005, 280, 24072–24084. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Lan, D.; Chen, W.; Matsuura, F.; Tall, A.R. ATP-binding cassette transporters G1 and G4 mediate cellular cholesterol efflux to high-density lipoproteins. PNAS 2004, 101, 9774–9779. [Google Scholar] [CrossRef] [PubMed]
- Sankaranarayanan, S.; Oram, J.F.; Asztalos, B.F.; Vaughan, A.M.; Lund-Katz, S.; Adorni, M.P.; Phillips, M.C.; Rothblat, G.H. Effects of acceptor composition and mechanism of ABCG1-mediated cellular free cholesterol efflux. J. Lipid Res. 2009, 50, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Miyake, Y.; Kozutsumi, Y.; Nakamura, S.; Fujita, T.; Kawasaki, T. Serine palmitoyltransferase is the primary target of a sphingosine-like immunosuppressant, ISP-1/myriocin. Biochem. Biophys. Res. Commun. 1995, 211, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Cohen, B.E. Amphotericin B membrane action: Role for two types of ion channels in eliciting cell survival and lethal effects. J. Memb. Biol. 2010, 238, 1–20. [Google Scholar] [CrossRef]
- Yamaji, A.; Sekizawa, Y.; Emoto, K.; Sakuraba, H.; Inoue, K.; Kobayashi, H.; Umeda, M. Lysenin, a novel sphingomyelin-specific binding protein. J. Biol. Chem. 1998, 273, 5300–5306. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, R.J. Cholesterol-dependent cytolysins. Adv. Exp. Med. Biol. 2010, 677, 56–66. [Google Scholar] [PubMed]
- Czub, J.; Baginski, M. Modulation of amphotericin B membrane interaction by cholesterol and ergosterol—A molecular dynamics study. J. Phys. Chem. B 2006, 110, 16743–16753. [Google Scholar] [CrossRef] [PubMed]
- Romero, E.A.; Valdivieso, E.; Cohen, B.E. Formation of two different types of ion channels by amphotericin B in human erythrocyte membranes. J. Memb. Biol. 2009, 230, 69–81. [Google Scholar] [CrossRef]
- Alam, J.M.; Kobayashi, T.; Yamazaki, M. The single-giant unilamellar vesicle method reveals lysenin-induced pore formation in lipid membranes containing sphingomyelin. Biochemistry 2012, 51, 5160–5172. [Google Scholar] [CrossRef] [PubMed]
- Castro, B.M.; Silva, L.C.; Fedorov, A.; de Almeida, R.F.; Prieto, M. Cholesterol-rich fluid membranes solubilize ceramide domains: Implications for the structure and dynamics of mammalian intracellular and plasma membranes. J. Biol. Chem. 2009, 284, 22978–22987. [Google Scholar] [CrossRef] [PubMed]
- Megha; London, E. Ceramide selectively displaces cholesterol from ordered lipid domains (rafts): Implications for lipid raft structure and function. J. Biol. Chem. 2004, 279, 9997–10004. [Google Scholar]
- Pinto, S.N.; Laviad, E.L.; Stiban, J.; Kelly, S.L.; Merrill, A.H., Jr.; Prieto, M.; Futerman, A.H.; Silva, L.C. Changes in membrane biophysical properties induced by sphingomyelinase depend on the sphingolipid N-acyl chain. J. Lipid Res. 2014, 55, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Quazi, F.; Molday, R.S. Differential phospholipid substrates and directional transport by ATP-binding cassette proteins ABCA1, ABCA7, and ABCA4 and disease-causing mutants. J. Biol. Chem. 2013, 288, 34414–34426. [Google Scholar] [CrossRef] [PubMed]
- Contreras, F.X.; Sanchez-Magraner, L.; Alonso, A.; Goni, F.M. Transbilayer (flip-flop) lipid motion and lipid scrambling in membranes. FEBS Lett. 2010, 584, 1779–1786. [Google Scholar] [CrossRef] [PubMed]
- Bennett, W.F.; MacCallum, J.L.; Hinner, M.J.; Marrink, S.J.; Tieleman, D.P. Molecular view of cholesterol flip-flop and chemical potential in different membrane environments. J. Am. Chem. Soci. 2009, 131, 12714–12720. [Google Scholar] [CrossRef]
- Sturek, J.M.; Castle, J.D.; Trace, A.P.; Page, L.C.; Castle, A.M.; Evans-Molina, C.; Parks, J.S.; Mirmira, R.G.; Hedrick, C.C. An intracellular role for ABCG1-mediated cholesterol transport in the regulated secretory pathway of mouse pancreatic beta cells. J. Clin. Invest. 2010, 120, 2575–2589. [Google Scholar] [CrossRef] [PubMed]
- Tarling, E.J.; Edwards, P.A. ATP binding cassette transporter G1 (ABCG1) is an intracellular sterol transporter. PNAS 2011, 108, 19719–19724. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neufeld, E.B.; O'Brien, K.; Walts, A.D.; Stonik, J.A.; Malide, D.; Combs, C.A.; Remaley, A.T. The Human ABCG1 Transporter Mobilizes Plasma Membrane and Late Endosomal Non-Sphingomyelin-Associated-Cholesterol for Efflux and Esterification. Biology 2014, 3, 866-891. https://doi.org/10.3390/biology3040866
Neufeld EB, O'Brien K, Walts AD, Stonik JA, Malide D, Combs CA, Remaley AT. The Human ABCG1 Transporter Mobilizes Plasma Membrane and Late Endosomal Non-Sphingomyelin-Associated-Cholesterol for Efflux and Esterification. Biology. 2014; 3(4):866-891. https://doi.org/10.3390/biology3040866
Chicago/Turabian StyleNeufeld, Edward B., Katherine O'Brien, Avram D. Walts, John A. Stonik, Daniela Malide, Christian A. Combs, and Alan T. Remaley. 2014. "The Human ABCG1 Transporter Mobilizes Plasma Membrane and Late Endosomal Non-Sphingomyelin-Associated-Cholesterol for Efflux and Esterification" Biology 3, no. 4: 866-891. https://doi.org/10.3390/biology3040866