
NPPD: A Protein-Protein Docking Scoring Function Based on Dyadic Differences in Networks of Hydrophobic and Hydrophilic Amino Acid Residues

Abstract
:1. Introduction
2. Experimental Section

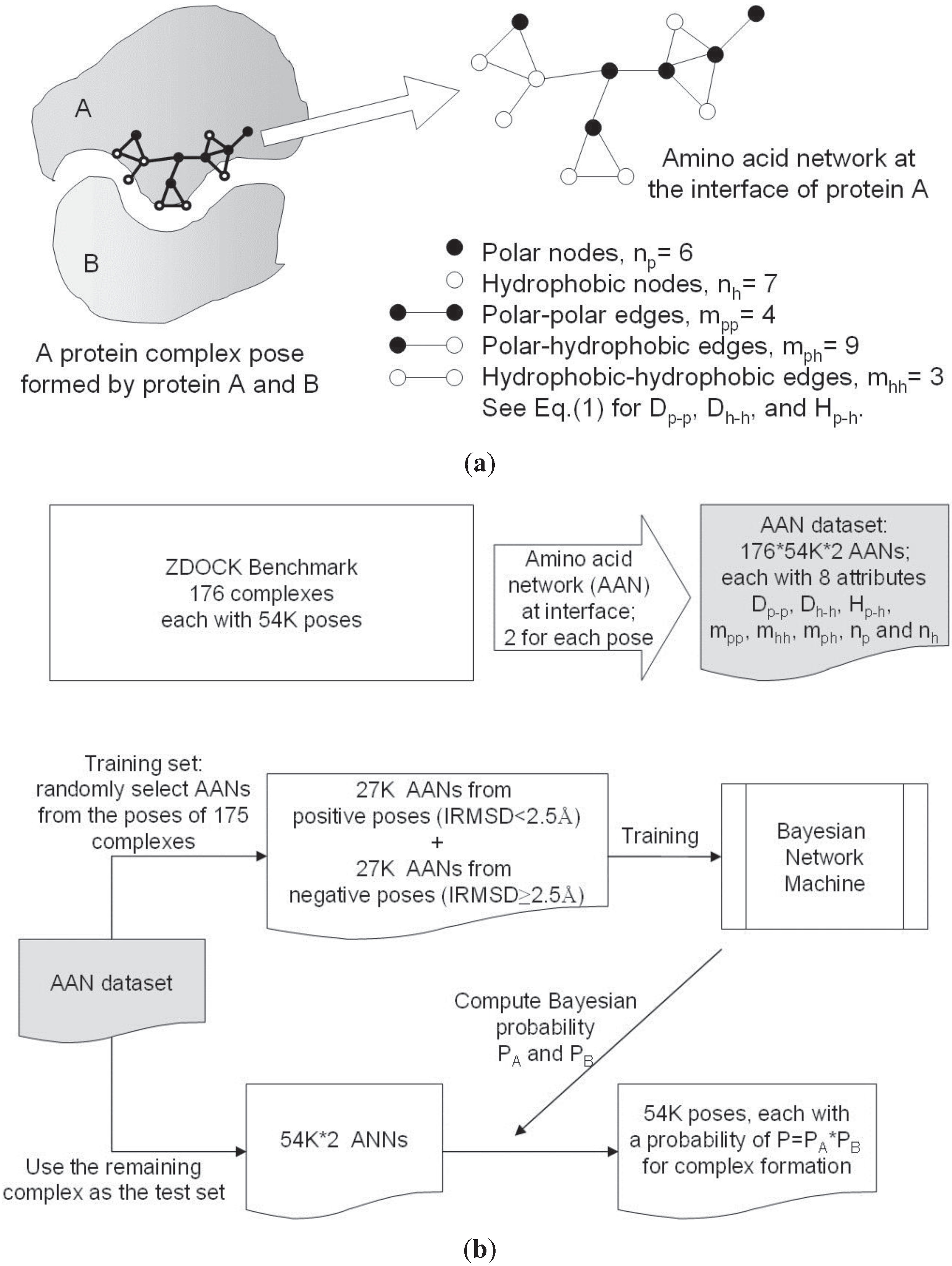
2.1. Docking Datasets, Poses, and Quality Measures
2.2. Amino Acid Networks and Network Parameters
2.3. Bayesian Network
3. Results and Discussion
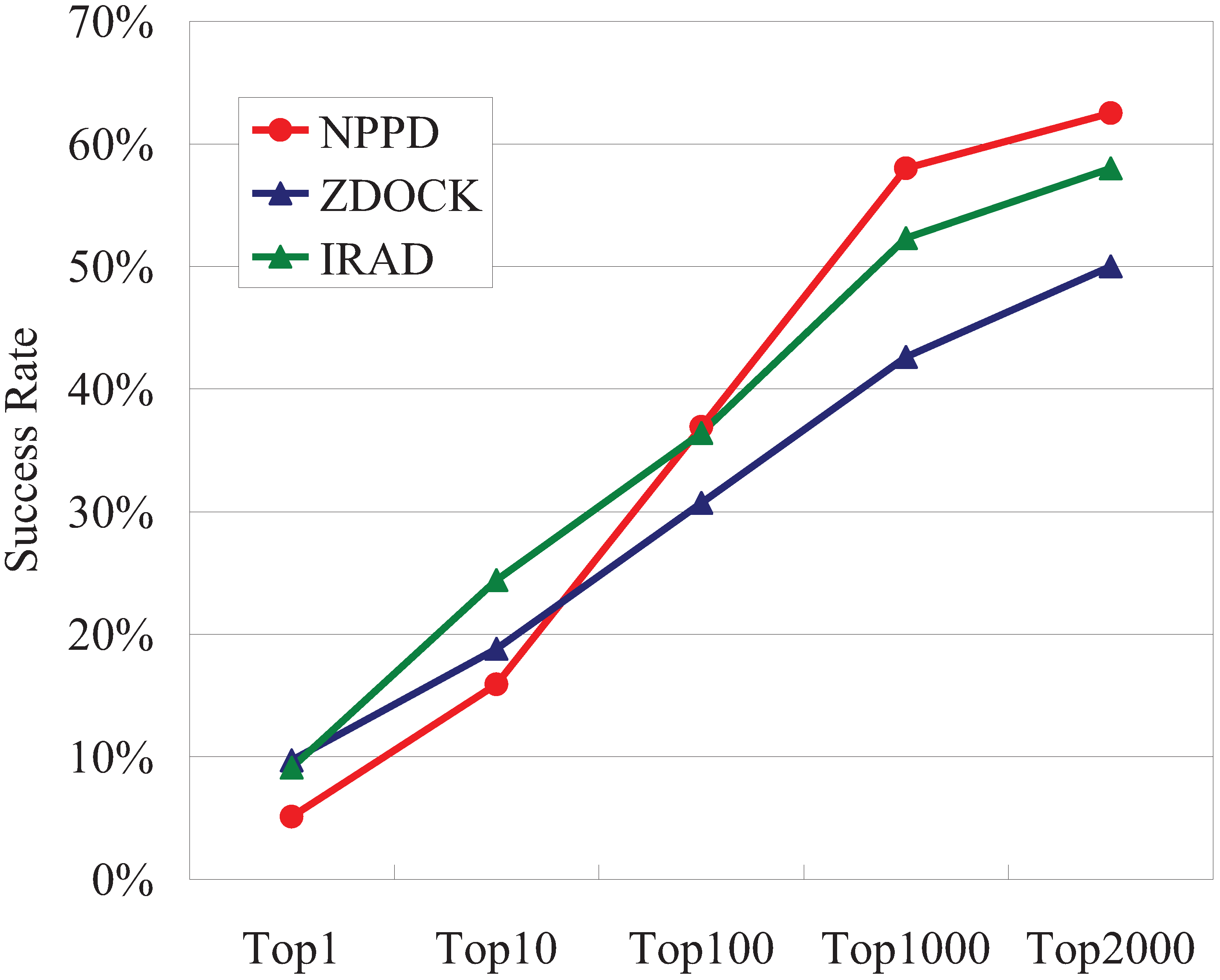
3.1. Performance of NPPD and IRAD


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Set | Top1 | Top10 | Top100 | Top1000 | Top2000 |
|---|---|---|---|---|---|
| NPPD (A) | 9 | 28 | 65 | 102 | 110 |
| IRAD (B) | 16 | 43 | 64 | 92 | 102 |
| Intersection (A∩B) | 3 | 15 | 44 | 80 | 95 |
| Union (A∪B) = a | 22 | 56 | 85 | 114 | 117 |
| Unique to NPPD or IRAD (A⊖B) = b | 19 | 41 | 41 | 34 | 22 |
| Complementarity = b/a | 86% | 73% | 48% | 30% | 19% |
3.2. Comparison with Other Network-Based Methods
| Conditions of docking poses | 176 Complexes | 43 Complexes | ||
| Pons et al. [37] | NPPD | Chang et al. [38] | NPPD | |
| Generation of docking poses | FTDock [16] | ZDOCK | RossettaDock 1.0 [75] | ZDOCK |
| Number of poses generated | 10,000 | 1000 | ||
| Criterion for a success hit | L-RMSD < 10 Å | L-RMSD < 5 Å | ||
| Top 1 success rate * | 5.0% (7.0%) | 8.0% | 2.3% (25.6%) | 11.6% |
| Top10 success rate * | 10.6% (29.8%) | 18.5% | 23.2% (53.4%) | 25.6% |
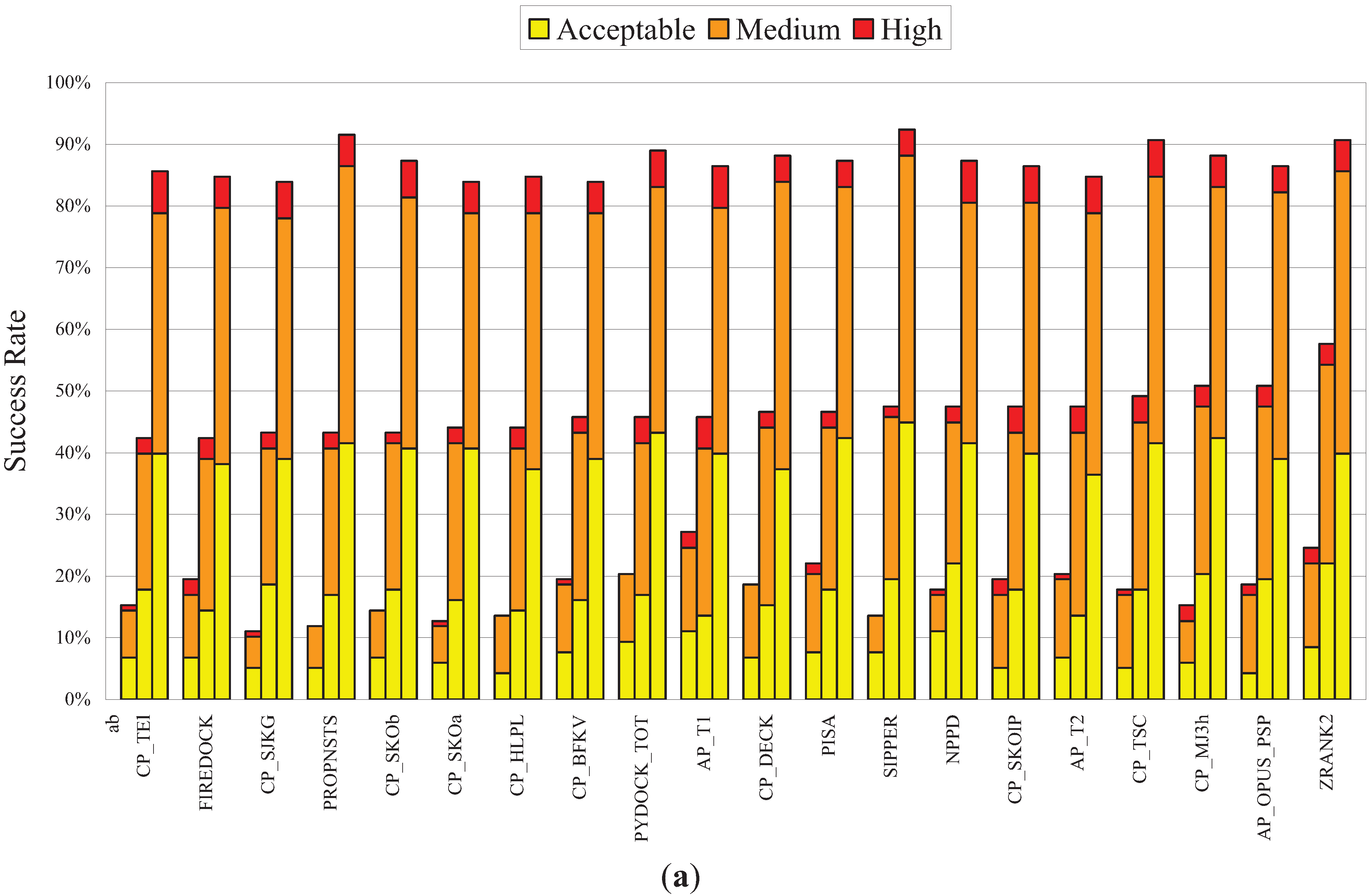
3.3. Performance of NPPD in a Comprehensive Evaluation of a Number of PPD Scoring Functions
3.4. Some Limitations and Prospects



4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mosca, R.; Pons, T.; Ceol, A.; Valencia, A.; Aloy, P. Towards a detailed atlas of protein-protein interactions. Curr. Opin. Struct. Biol. 2013, 23, 929–940. [Google Scholar] [CrossRef] [PubMed]
- Lensink, M.F.; Wodak, S.J. Docking, scoring, and affinity prediction in CAPRI. Proteins 2013, 81, 2082–2095. [Google Scholar] [CrossRef] [PubMed]
- Moal, I.H.; Moretti, R.; Baker, D.; Fernandez-Recio, J. Scoring functions for protein-protein interactions. Curr. Opin. Struct. Biol. 2013, 23, 862–867. [Google Scholar] [CrossRef] [PubMed]
- Shih, E.S.C.; Hwang, M.J. A critical assessment of information-guided protein-protein docking predictions. Mol. Cell Proteomics 2013, 12, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Shih, E.S.C.; Hwang, M.J. On the use of distance constraints in protein-protein docking computations. Proteins Struct. Funct. Bioinform. 2012, 80, 194–205. [Google Scholar] [CrossRef]
- Viswanath, S.; Ravikant, D.V.; Elber, R. Improving ranking of models for protein complexes with side chain modeling and atomic potentials. Proteins 2013, 81, 592–606. [Google Scholar] [CrossRef] [PubMed]
- Pallara, C.; Jimenez-Garcia, B.; Perez-Cano, L.; Romero-Durana, M.; Solernou, A.; Grosdidier, S.; Pons, C.; Moal, I.H.; Fernandez-Recio, J. Expanding the frontiers of protein-protein modeling: From docking and scoring to binding affinity predictions and other challenges. Proteins 2013, 81, 2192–2200. [Google Scholar] [CrossRef] [PubMed]
- Pons, C.; Talavera, D.; de la Cruz, X.; Orozco, M.; Fernandez-Recio, J. Scoring by intermolecular pairwise propensities of exposed residues (SIPPER): A new efficient potential for protein-protein docking. J. Chem. Inf. Model 2011, 51, 370–337. [Google Scholar] [CrossRef] [PubMed]
- Mitra, P.; Pal, D. Using correlated parameters for improved ranking of protein-protein docking decoys. J. Comput. Chem. 2011, 32, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Tobi, D. Designing coarse grained-and atom based-potentials for protein-protein docking. BMC Struct. Biol. 2010, 10, 40. [Google Scholar] [CrossRef] [PubMed]
- Demir-Kavuk, O.; Krull, F.; Chae, M.H.; Knapp, E.W. Predicting protein complex geometries with linear scoring functions. Genome Inform. 2010, 24, 21–30. [Google Scholar] [PubMed]
- Pierce, B.; Weng, Z. A combination of rescoring and refinement significantly improves protein docking performance. Proteins 2008, 72, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Andrusier, N.; Nussinov, R.; Wolfson, H.J. FireDock: Fast interaction refinement in molecular docking. Proteins 2007, 69, 139–159. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.M.; Blundell, T.L.; Fernandez-Recio, J. pyDock: Electrostatics and desolvation for effective scoring of rigid-body protein-protein docking. Proteins 2007, 68, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.; Gatchell, D.W.; Prasad, J.C.; Vajda, S. Combination of scoring functions improves discrimination in protein-protein docking. Proteins 2003, 53, 840–854. [Google Scholar] [CrossRef] [PubMed]
- Gabb, H.A.; Jackson, R.M.; Sternberg, M.J. Modelling protein docking using shape complementarity, electrostatics and biochemical information. J. Mol. Biol. 1997, 272, 106–120. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Vakser, I.A. DECK: Distance and environment-dependent, coarse-grained, knowledge-based potentials for protein-protein docking. BMC Bioinform. 2011, 12, 280. [Google Scholar] [CrossRef]
- Lu, H.; Lu, L.; Skolnick, J. Development of unified statistical potentials describing protein-protein interactions. Biophys. J. 2003, 84, 1895–1901. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, S.; Jernigan, R.L. Self-consistent estimation of inter-residue protein contact energies based on an equilibrium mixture approximation of residues. Proteins 1999, 34, 49–68. [Google Scholar] [CrossRef] [PubMed]
- Omori, S.; Kitao, A. CyClus: A fast, comprehensive cylindrical interface approximation clustering/reranking method for rigid-body protein-protein docking decoys. Proteins 2013, 81, 1005–1016. [Google Scholar] [CrossRef] [PubMed]
- Chuang, G.Y.; Kozakov, D.; Brenke, R.; Comeau, S.R.; Vajda, S. DARS (Decoys as the Reference State) potentials for protein-protein docking. Biophys. J. 2008, 95, 4217–4227. [Google Scholar] [CrossRef] [PubMed]
- Muller, W.; Sticht, H. A protein-specifically adapted scoring function for the reranking of docking solutions. Proteins 2007, 67, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Esmaielbeiki, R.; Nebel, J.C. Scoring docking conformations using predicted protein interfaces. BMC Bioinform. 2014, 15, 171. [Google Scholar] [CrossRef]
- Anishchenko, I.; Kundrotas, P.J.; Tuzikov, A.V.; Vakser, I.A. Protein models: The grand challenge of protein docking. Proteins 2014, 82, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Kundrotas, P.J.; Vakser, I.A. Global and local structural similarity in protein-protein complexes: Implications for template-based docking. Proteins 2013, 81, 2137–2142. [Google Scholar] [CrossRef] [PubMed]
- Torchala, M.; Moal, I.H.; Chaleil, R.A.; Agius, R.; Bates, P.A. A Markov-chain model description of binding funnels to enhance the ranking of docked solutions. Proteins 2013, 81, 2143–2149. [Google Scholar] [CrossRef] [PubMed]
- London, N.; Schueler-Furman, O. Funnel hunting in a rough terrain: Learning and discriminating native energy funnels. Structure 2008, 16, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Kozakov, D.; Schueler-Furman, O.; Vajda, S. Discrimination of near-native structures in protein-protein docking by testing the stability of local minima. Proteins 2008, 72, 993–1004. [Google Scholar] [CrossRef] [PubMed]
- Schneidman-Duhovny, D.; Rossi, A.; Avila-Sakar, A.; Kim, S.J.; Velazquez-Muriel, J.; Strop, P.; Liang, H.; Krukenberg, K.A.; Liao, M.; Kim, H.M.; et al. A method for integrative structure determination of protein-protein complexes. Bioinformatics 2012, 28, 3282–3289. [Google Scholar] [CrossRef] [PubMed]
- De Vries, S.J.; Bonvin, A.M. CPORT: A consensus interface predictor and its performance in prediction-driven docking with HADDOCK. PLOS ONE 2011, 6, e17695. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Koehl, P.; Hass, J.; Amenta, N. Surface-histogram: A new shape descriptor for protein-protein docking. Proteins 2012, 80, 221–238. [Google Scholar] [CrossRef] [PubMed]
- Shentu, Z.; al Hasan, M.; Bystroff, C.; Zaki, M.J. Context shapes: Efficient complementary shape matching for protein-protein docking. Proteins 2008, 70, 1056–1073. [Google Scholar] [CrossRef] [PubMed]
- Fink, F.; Hochrein, J.; Wolowski, V.; Merkl, R.; Gronwald, W. PROCOS: Computational analysis of protein-protein complexes. J. Comput. Chem. 2011, 32, 2575–2586. [Google Scholar] [CrossRef] [PubMed]
- Bourquard, T.; Bernauer, J.; Aze, J.; Poupon, A. A collaborative filtering approach for protein-protein docking scoring functions. PLOS ONE 2011, 6, e18541. [Google Scholar] [CrossRef] [PubMed]
- Chae, M.H.; Krull, F.; Lorenzen, S.; Knapp, E.W. Predicting protein complex geometries with a neural network. Proteins 2010, 78, 1026–1039. [Google Scholar] [CrossRef] [PubMed]
- Andreani, J.; Faure, G.; Guerois, R. InterEvScore: A novel coarse-grained interface scoring function using a multi-body statistical potential coupled to evolution. Bioinformatics 2013, 29, 1742–1749. [Google Scholar] [CrossRef] [PubMed]
- Pons, C.; Glaser, F.; Fernandez-Recio, J. Prediction of protein-binding areas by small-world residue networks and application to docking. BMC Bioinform. 2011, 12, 378. [Google Scholar] [CrossRef]
- Chang, S.; Jiao, X.; Li, C.H.; Gong, X.Q.; Chen, W.Z.; Wang, C.X. Amino acid network and its scoring application in protein-protein docking. Biophys. Chem. 2008, 134, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Perica, T.; Teichmann, S.A. Evolution of protein structures and interactions from the perspective of residue contact networks. Curr. Opin. Struct. Biol. 2013, 23, 954–963. [Google Scholar] [CrossRef] [PubMed]
- Di Paola, L.; de Ruvo, M.; Paci, P.; Santoni, D.; Giuliani, A. Protein contact networks: An emerging paradigm in chemistry. Chem. Rev. 2013, 113, 1598–1613. [Google Scholar] [CrossRef] [PubMed]
- Greene, L.H. Protein structure networks. Brief Funct. Genomics 2012, 11, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Giollo, M.; Martin, A.J.; Walsh, I.; Ferrari, C.; Tosatto, S.C. NeEMO: A method using residue interaction networks to improve prediction of protein stability upon mutation. BMC Genomics 2014, 15, S7. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, A.; Zbilut, J.P.; Tomita, M.; Giuliani, A. Proteins as networks: Usefulness of graph theory in protein science. Curr. Protein Pept. Sci. 2008, 9, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Zhou, J.; Sun, M.; Chen, J.; Hu, G.; Shen, B. The construction of an amino acid network for understanding protein structure and function. Amino Acids 2014, 46, 1419–1439. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Wang, J.; Chen, L.; Zhong, J.; Zhang, Z.; Pan, Y. Predicting Protein Functions by using unbalanced bi-random walk algorithm on protein-protein interaction network and functional interrelationship network. Curr. Protein Pept. Sci. 2014, 15, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Axe, J.M.; Yezdimer, E.M.; O’Rourke, K.F.; Kerstetter, N.E.; You, W.; Chang, C.E.; Boehr, D.D. Amino acid networks in a (beta/alpha)(8) barrel enzyme change during catalytic turnover. J. Am. Chem. Soc. 2014, 136, 6818–6821. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.C.; Park, K.; Kim, D. Analysis of the residue-residue coevolution network and the functionally important residues in proteins. Proteins 2008, 72, 863–872. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.; Hamer, R.; Reinert, G.; Deane, C.M. Local network patterns in protein-protein interfaces. PLOS ONE 2013, 8, e57031. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.E.; Hummer, G. Interface-resolved network of protein-protein interactions. PLOS Comput. Biol. 2013, 9, e1003065. [Google Scholar] [CrossRef] [PubMed]
- Goebels, F.; Frishman, D. Prediction of protein interaction types based on sequence and network features. BMC Syst. Biol. 2013, 7, S5. [Google Scholar] [CrossRef] [PubMed]
- Del Sol, A.; O’Meara, P. Small-world network approach to identify key residues in protein-protein interaction. Proteins 2005, 58, 672–682. [Google Scholar] [CrossRef] [PubMed]
- Maetschke, S.R.; Yuan, Z. Exploiting structural and topological information to improve prediction of RNA-protein binding sites. BMC Bioinform. 2009, 10, 341. [Google Scholar] [CrossRef]
- Sathyapriya, R.; Vijayabaskar, M.S.; Vishveshwara, S. Insights into protein-DNA interactions through structure network analysis. PLOS Comput. Biol. 2008, 4, e1000170. [Google Scholar] [CrossRef] [PubMed]
- Montiel Molina, H.M.; Millan-Pacheco, C.; Pastor, N.; del Rio, G. Computer-based screening of functional conformers of proteins. PLOS Comput. Biol. 2008, 4, e1000009. [Google Scholar] [CrossRef] [PubMed]
- Bode, C.; Kovacs, I.A.; Szalay, M.S.; Palotai, R.; Korcsmaros, T.; Csermely, P. Network analysis of protein dynamics. FEBS Lett. 2007, 581, 2776–2782. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, J.; Wang, W. Identifying folding nucleus based on residue contact networks of proteins. Proteins 2008, 71, 1899–1907. [Google Scholar] [CrossRef] [PubMed]
- Bagler, G.; Sinha, S. Assortative mixing in protein contact networks and protein folding kinetics. Bioinformatics 2007, 23, 1760–1707. [Google Scholar] [CrossRef] [PubMed]
- Vendruscolo, M.; Dokholyan, N.V.; Paci, E.; Karplus, M. Small-world view of the amino acids that play a key role in protein folding. Phys. Rev. E 2002, 65, 061910. [Google Scholar] [CrossRef]
- Bhattacharyya, M.; Bhat, C.R.; Vishveshwara, S. An automated approach to network features of protein structure ensembles. Protein Sci. 2013, 22, 1399–1416. [Google Scholar] [PubMed]
- Khor, S. Towards an integrated understanding of the structural characteristics of protein residue networks. Theory Biosci. 2012, 131, 61–75. [Google Scholar] [CrossRef] [PubMed]
- Estrada, E. Universality in protein residue networks. Biophys. J. 2010, 98, 890–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinda, K.V.; Vishveshwara, S. A network representation of protein structures: Implications for protein stability. Biophys. J. 2005, 89, 4159–4170. [Google Scholar] [CrossRef] [PubMed]
- Bagler, G.; Sinha, S. Network properties of protein structures. Phys. A 2005, 346, 27–33. [Google Scholar] [CrossRef]
- Shih, E.S.C.; Hwang, M.-J.; Institute of Biomedical Sciences, Academia Sinica, Taipei, Taiwan. Unpublished data. 2015.
- Hwang, H.; Vreven, T.; Janin, J.; Weng, Z. Protein-protein docking benchmark version 4.0. Proteins 2010, 78, 3111–3114. [Google Scholar] [CrossRef] [PubMed]
- Pierce, B.G.; Hourai, Y.; Weng, Z.P. Accelerating protein docking in ZDOCK using an advanced 3D convolution library. PLOS ONE 2011, 6, e24657. [Google Scholar] [CrossRef] [PubMed]
- Moal, I.H.; Torchala, M.; Bates, P.A.; Fernandez-Recio, J. The scoring of poses in protein-protein docking: Current capabilities and future directions. BMC Bioinform. 2013, 14, 286. [Google Scholar] [CrossRef]
- Torchala, M.; Bates, P.A. Predicting the structure of protein-protein complexes using the SwarmDock Web Server. Methods Mol. Biol. 2014, 1137, 181–197. [Google Scholar] [PubMed]
- Eisenberg, D.; Weiss, R.M.; Terwilliger, T.C.; Wilcox, W. Hydrophobic Moments and Protein-Structure. Faraday Symp. Chem. S 1982, 17, 109–120. [Google Scholar] [CrossRef]
- Park, J.; Barabasi, A.L. Distribution of node characteristics in complex networks. Proc. Natl. Acad. Sci. USA 2007, 104, 17916–17920. [Google Scholar] [CrossRef] [PubMed]
- Fienberg, S.E.; Meyer, M.M.; Wasserman, S.S. Statistical-Analysis of Multiple Sociometric Relations. J. Am. Stat. Assoc. 1985, 80, 51–67. [Google Scholar] [CrossRef]
- Hall, M.; Frank, E.; Holmes, G.; Pfahringer, B.; Reutemann, P.; Witten, I.H. The WEKA data mining software: An update. SIGKDD Explor. Newsl. 2009, 11, 10–18. [Google Scholar] [CrossRef]
- Needham, C.J.; Bradford, J.R.; Bulpitt, A.J.; Westhead, D.R. Inference in Bayesian networks. Nat. Biotechnol. 2006, 24, 51–53. [Google Scholar] [CrossRef] [PubMed]
- Vreven, T.; Hwang, H.; Weng, Z. Integrating atom-based and residue-based scoring functions for protein-protein docking. Protein Sci. 2011, 20, 1576–1586. [Google Scholar] [CrossRef] [PubMed]
- Gray, J.J.; Moughon, S.; Wang, C.; Schueler-Furman, O.; Kuhlman, B.; Rohl, C.A.; Baker, D. Protein-protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations. J. Mol. Biol. 2003, 331, 281–299. [Google Scholar] [CrossRef] [PubMed]
- Khashan, R.; Zheng, W.; Tropsha, A. Scoring protein interaction decoys using exposed residues (SPIDER): A novel multibody interaction scoring function based on frequent geometric patterns of interfacial residues. Proteins 2012, 80, 2207–2217. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, D.W.; Kozakov, D.; Vajda, S. Accelerating and focusing protein-protein docking correlations using multi-dimensional rotational FFT generating functions. Bioinformatics 2008, 24, 1865–1873. [Google Scholar] [CrossRef] [PubMed]
- Moreira, I.S.; Martins, J.M.; Coimbra, J.T.; Ramos, M.J.; Fernandes, P.A. A new scoring function for protein-protein docking that identifies native structures with unprecedented accuracy. Phys. Chem. Chem. Phys. 2015, 17, 2378–2387. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shih, E.S.C.; Hwang, M.-J. NPPD: A Protein-Protein Docking Scoring Function Based on Dyadic Differences in Networks of Hydrophobic and Hydrophilic Amino Acid Residues. Biology 2015, 4, 282-297. https://doi.org/10.3390/biology4020282
Shih ESC, Hwang M-J. NPPD: A Protein-Protein Docking Scoring Function Based on Dyadic Differences in Networks of Hydrophobic and Hydrophilic Amino Acid Residues. Biology. 2015; 4(2):282-297. https://doi.org/10.3390/biology4020282
Chicago/Turabian StyleShih, Edward S. C., and Ming-Jing Hwang. 2015. "NPPD: A Protein-Protein Docking Scoring Function Based on Dyadic Differences in Networks of Hydrophobic and Hydrophilic Amino Acid Residues" Biology 4, no. 2: 282-297. https://doi.org/10.3390/biology4020282