
Cardiac Channelopathies and Sudden Death: Recent Clinical and Genetic Advances

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
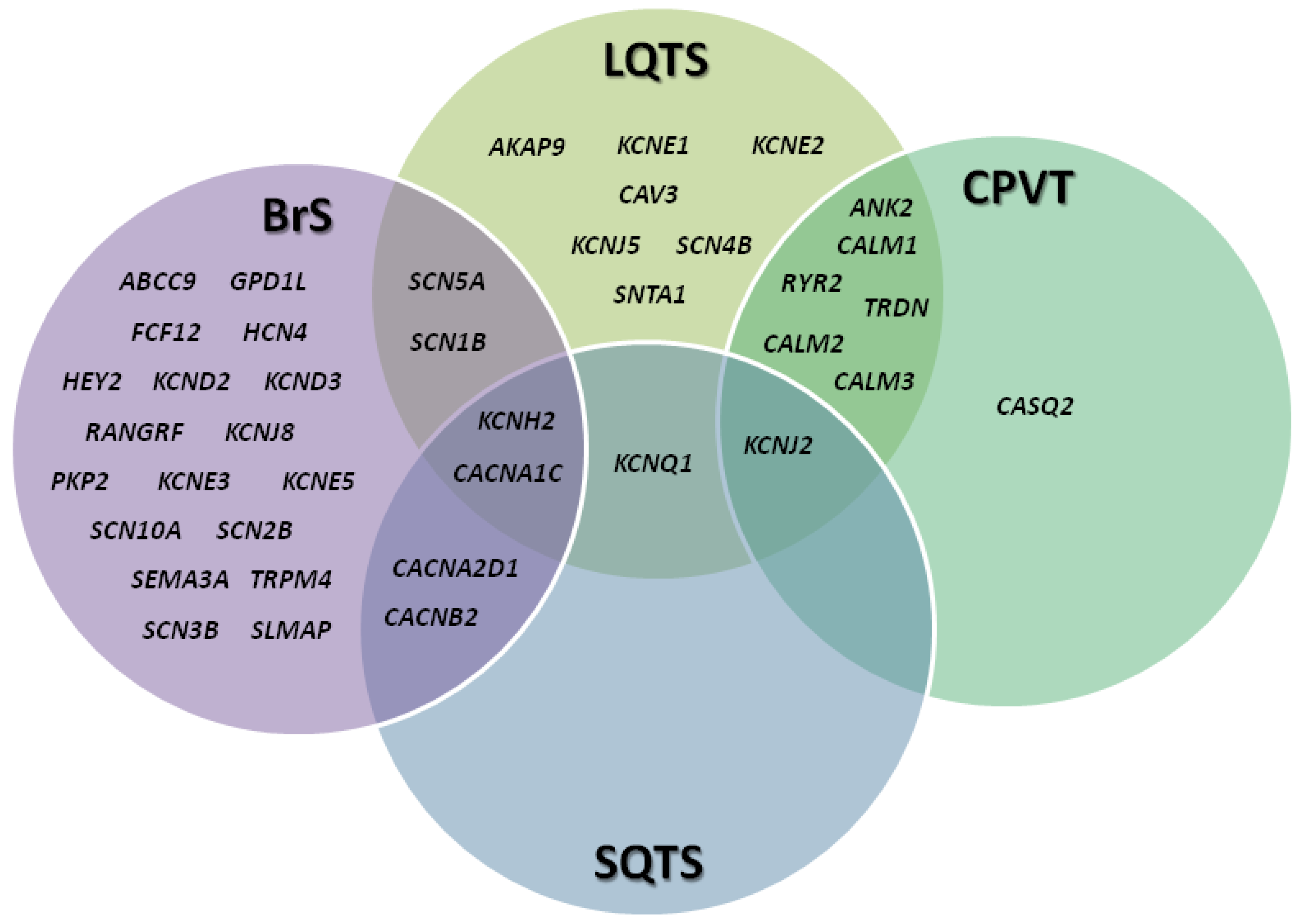
2. Channelopathies: An Overview
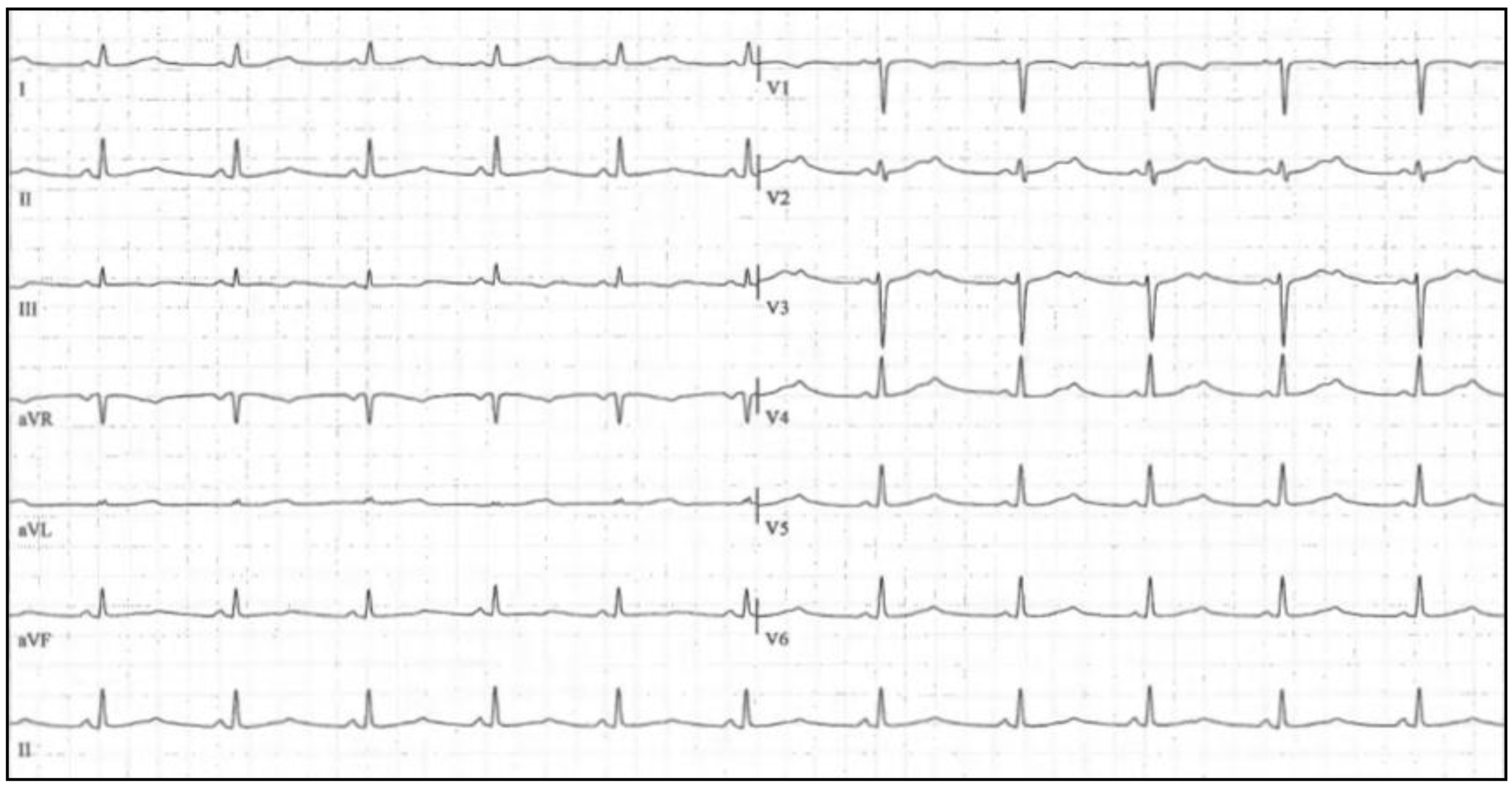
3. Brugada Syndrome
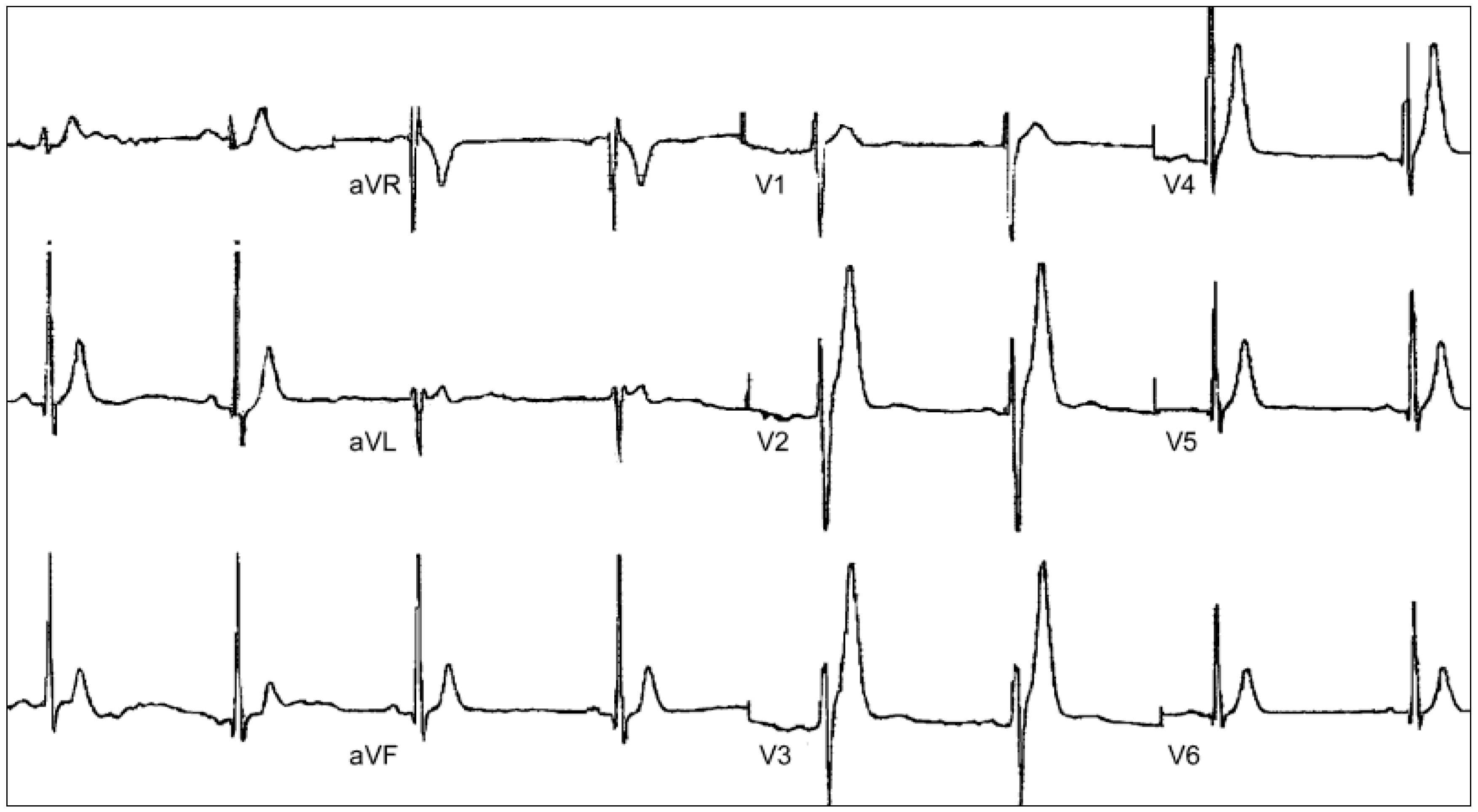
3.1. Clinical Presentation and Diagnosis
3.2. Genetics
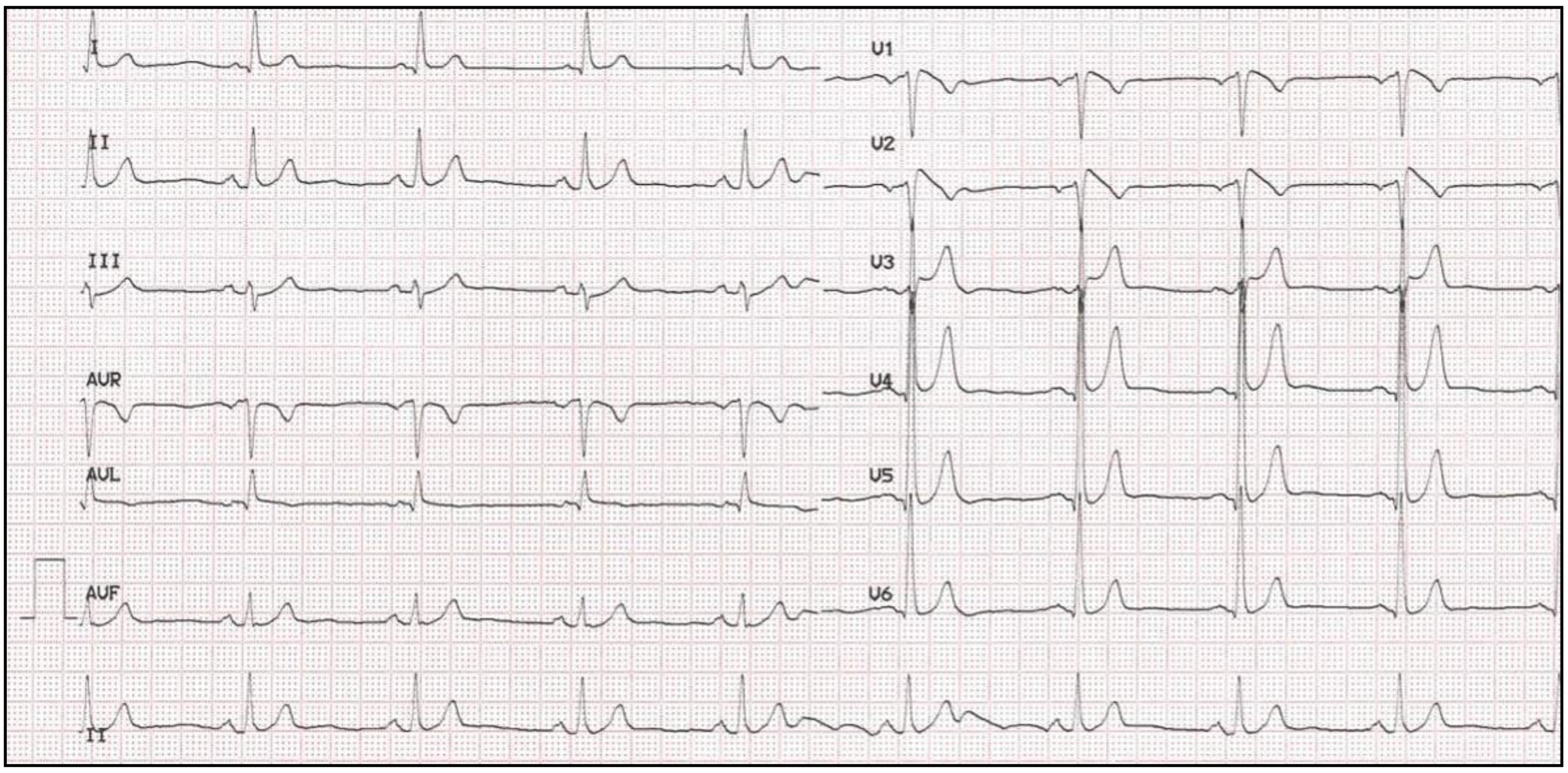
4. Long QT Syndrome
4.1. Clinical Presentation and Diagnosis
4.2. Genetics
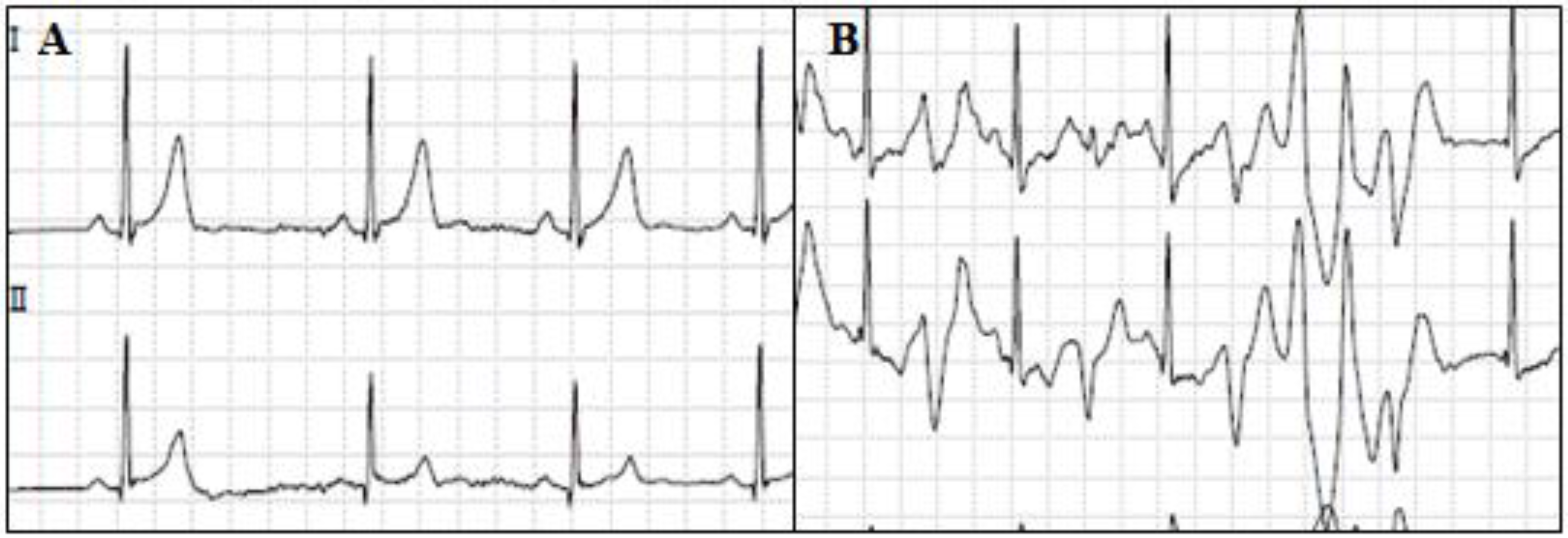
5. Short QT Syndrome
5.1. Clinical Presentation and Diagnosis
5.2. Genetics
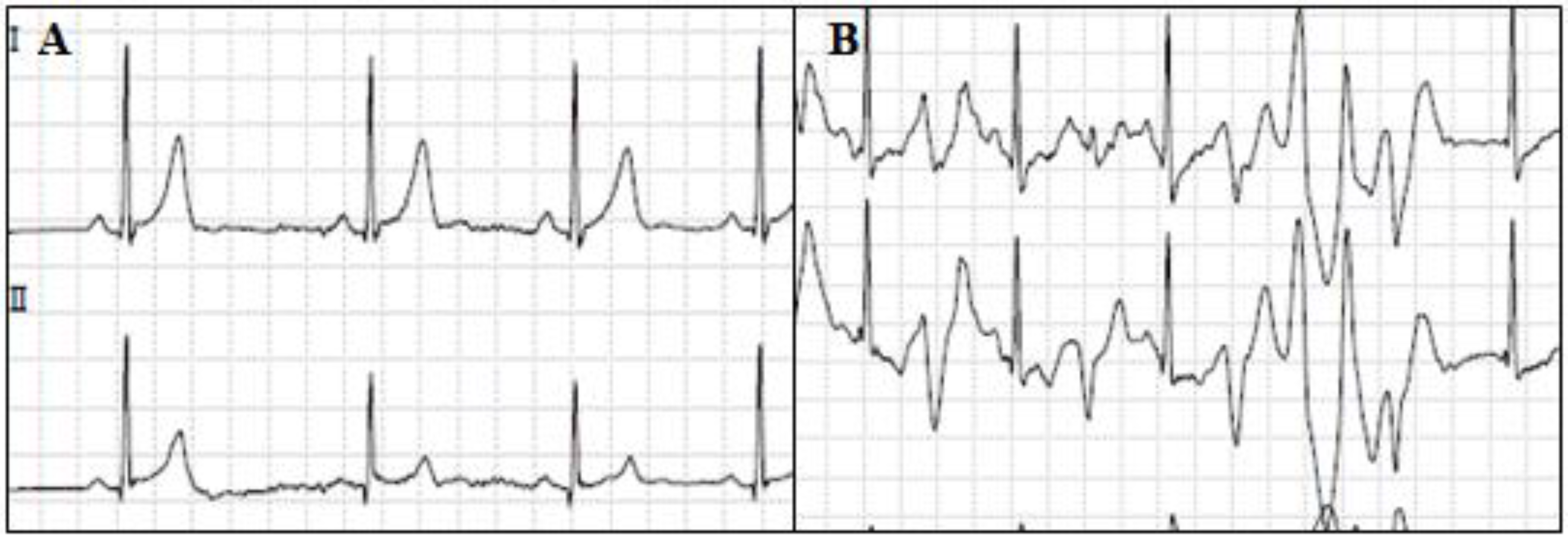
6. Catecholaminergic Polymorphic Ventricular Tachycardia
6.1. Clinical Presentation and Diagnosis
6.2. Genetics
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- De Luna, A.B.; Elosua, R. Sudden death. Rev. Esp. Cardiol. 2012, 65, 1039–1052. [Google Scholar]
- Pachon, M.; Almendral, J. Sudden death: Managing the patient who survives. Heart 2011, 97, 1619–1625. [Google Scholar] [CrossRef] [PubMed]
- Basso, C.; Carturan, E.; Pilichou, K.; Rizzo, S.; Corrado, D.; Thiene, G. Sudden cardiac death with normal heart: Molecular autopsy. Cardiovasc. Pathol. 2010, 19, 321–325. [Google Scholar] [CrossRef] [PubMed]
- Oliva, A.; Flores, J.; Merigioli, S.; LeDuc, L.; Benito, B.; Partemi, S.; Arzamendi, D.; Campuzano, O.; Leung, T.L.; Iglesias, A.; et al. Autopsy investigation and bayesian approach to coronary artery disease in victims of motor-vehicle accidents. Atherosclerosis 2011, 218, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Podrid, P.J.; Myerburg, R.J. Epidemiology and stratification of risk for sudden cardiac death. Clin. Cardiol. 2005, 28, I3–I11. [Google Scholar] [CrossRef] [PubMed]
- Nichol, G.; Rumsfeld, J.; Eigel, B.; Abella, B.S.; Labarthe, D.; Hong, Y.; O’Connor, R.E.; Mosesso, V.N.; Berg, R.A.; Leeper, B.B.; et al. Essential features of designating out-of-hospital cardiac arrest as a reportable event: A scientific statement from the American Heart Association Emergency Cardiovascular Care Committee; Council on Cardiopulmonary, Perioperative, And Critical Care; Council on Cardiovascular Nursing; Council on Clinical Cardiology; and Quality of Care and Outcomes Research Interdisciplinary Working Group. Circulation 2008, 117, 2299–2308. [Google Scholar] [PubMed]
- Chugh, S.S.; Senashova, O.; Watts, A.; Tran, P.T.; Zhou, Z.; Gong, Q.; Titus, J.L.; Hayflick, S.J. Postmortem molecular screening in unexplained sudden death. J. Am. Coll. Cardiol. 2004, 43, 1625–1629. [Google Scholar] [CrossRef] [PubMed]
- De Vreede-Swagemakers, J.J.; Gorgels, A.P.; Dubois-Arbouw, W.I.; van Ree, J.W.; Daemen, M.J.; Houben, L.G.; Wellens, H.J. Out-of-hospital cardiac arrest in the 1990’s: A population-based study in the maastricht area on incidence, characteristics and survival. J. Am. Coll. Cardiol. 1997, 30, 1500–1505. [Google Scholar] [CrossRef]
- Byrne, R.; Constant, O.; Smyth, Y.; Callagy, G.; Nash, P.; Daly, K.; Crowley, J. Multiple source surveillance incidence and aetiology of out-of-hospital sudden cardiac death in a rural population in the West of Ireland. Eur. Heart J. 2008, 29, 1418–1423. [Google Scholar] [CrossRef] [PubMed]
- Hua, W.; Zhang, L.F.; Wu, Y.F.; Liu, X.Q.; Guo, D.S.; Zhou, H.L.; Gou, Z.P.; Zhao, L.C.; Niu, H.X.; Chen, K.P.; et al. Incidence of sudden cardiac death in China: Analysis of 4 regional populations. J. Am. Coll. Cardiol. 2009, 54, 1110–1118. [Google Scholar] [CrossRef] [PubMed]
- Eckart, R.E.; Shry, E.A.; Burke, A.P.; McNear, J.A.; Appel, D.A.; Castillo-Rojas, L.M.; Avedissian, L.; Pearse, L.A.; Potter, R.N.; Tremaine, L.; et al. Sudden death in young adults: An autopsy-based series of a population undergoing active surveillance. J. Am. Coll. Cardiol. 2011, 58, 1254–1261. [Google Scholar] [CrossRef] [PubMed]
- Fishman, G.I.; Chugh, S.S.; Dimarco, J.P.; Albert, C.M.; Anderson, M.E.; Bonow, R.O.; Buxton, A.E.; Chen, P.S.; Estes, M.; Jouven, X.; et al. Sudden cardiac death prediction and prevention: Report from a National Heart, Lung, And Blood Institute And Heart Rhythm Society Workshop. Circulation 2010, 122, 2335–2348. [Google Scholar] [CrossRef] [PubMed]
- Myerburg, R.J.; Junttila, M.J. Sudden cardiac death caused by coronary heart disease. Circulation 2012, 125, 1043–1052. [Google Scholar] [CrossRef] [PubMed]
- Arzamendi, D.; Benito, B.; Tizon-Marcos, H.; Flores, J.; Tanguay, J.F.; Ly, H.; Doucet, S.; Leduc, L.; Leung, T.K.; Campuzano, O.; et al. Increase in sudden death from coronary artery disease in young adults. Am. Heart J. 2011, 161, 574–580. [Google Scholar] [CrossRef] [PubMed]
- Boczek, N.J.; Tester, D.J.; Ackerman, M.J. The molecular autopsy: An indispensable step following sudden cardiac death in the young? Herzschrittmacherther. Elektrophysiol. 2012, 23, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, O.; Sanchez-Molero, O.; Allegue, C.; Coll, M.; Mademont-Soler, I.; Selga, E.; Ferrer-Costa, C.; Mates, J.; Iglesias, A.; Sarquella-Brugada, G.; et al. Post-mortem genetic analysis in juvenile cases of sudden cardiac death. Forensic. Sci. Int. 2014, 245, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, O.; Allegue, C.; Partemi, S.; Iglesias, A.; Oliva, A.; Brugada, R. Negative autopsy and sudden cardiac death. Int. J. Leg. Med. 2014, 128, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Nerbonne, J.M.; Kass, R.S. Molecular physiology of cardiac repolarization. Physiol. Rev. 2005, 85, 1205–1253. [Google Scholar] [CrossRef] [PubMed]
- Roden, D.M.; Balser, J.R.; George, A.L., Jr.; Anderson, M.E. Cardiac ion channels. Annu. Rev. Physiol. 2002, 64, 431–475. [Google Scholar] [CrossRef] [PubMed]
- Amin, A.S.; Asghari-Roodsari, A.; Tan, H.L. Cardiac sodium channelopathies. Pflugers Arch. 2010, 460, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Antzelevitch, C.; Brugada, P.; Borggrefe, M.; Brugada, J.; Brugada, R.; Corrado, D.; Gussak, I.; LeMarec, H.; Nademanee, K.; Perez Riera, A.R.; et al. Brugada syndrome: Report of the second consensus conference. Heart Rhythm 2005, 2, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Berne, P.; Brugada, J. Brugada syndrome 2012. Circ. J. 2012, 76, 1563–1571. [Google Scholar] [CrossRef] [PubMed]
- Brugada, R.; Campuzano, O.; Sarquella-Brugada, G.; Brugada, J.; Brugada, P. Brugada syndrome. Methodist Debakey Cardiovasc J. 2014, 10, 25–28. [Google Scholar] [CrossRef] [PubMed]
- Coronel, R.; Casini, S.; Koopmann, T.T.; Wilms-Schopman, F.J.; Verkerk, A.O.; de Groot, J.R.; Bhuiyan, Z.; Bezzina, C.R.; Veldkamp, M.W.; Linnenbank, A.C.; et al. Right ventricular fibrosis and conduction delay in a patient with clinical signs of Brugada syndrome: A combined electrophysiological, genetic, histopathologic, and computational study. Circulation 2005, 112, 2769–2777. [Google Scholar] [CrossRef] [PubMed]
- Frustaci, A.; Russo, M.A.; Chimenti, C. Structural myocardial abnormalities in asymptomatic family members with Brugada syndrome and SCN5A gene mutation. Eur. Heart J. 2009. [Google Scholar] [CrossRef] [PubMed]
- Wilde, A.A.; Antzelevitch, C.; Borggrefe, M.; Brugada, J.; Brugada, R.; Brugada, P.; Corrado, D.; Hauer, R.N.; Kass, R.S.; Nademanee, K.; et al. Proposed diagnostic criteria for the brugada syndrome: Consensus report. Circulation 2002, 106, 2514–2519. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Wilde, A.A.; Horie, M.; Cho, Y.; Behr, E.R.; Berul, C.; Blom, N.; Brugada, J.; Chiang, C.E.; Huikuri, H.; et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: Document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm 2013, 10, 1932–1963. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Kirsch, G.E.; Zhang, D.; Brugada, R.; Brugada, J.; Brugada, P.; Potenza, D.; Moya, A.; Borggrefe, M.; Breithardt, G.; et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 1998, 392, 293–296. [Google Scholar] [PubMed]
- Brugada, P.; Brugada, J.; Roy, D. Brugada syndrome 1992-2012: 20 years of scientific excitement, and more. Eur. Heart J. 2013, 34, 3610–3615. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Koopmann, T.T.; Le Scouarnec, S.; Yang, T.; Ingram, C.R.; Schott, J.J.; Demolombe, S.; Probst, V.; Anselme, F.; Escande, D.; et al. Sodium channel beta1 subunit mutations associated with brugada syndrome and cardiac conduction disease in humans. J. Clin. Investig. 2008, 118, 2260–2268. [Google Scholar] [PubMed]
- Riuro, H.; Beltran-Alvarez, P.; Tarradas, A.; Selga, E.; Campuzano, O.; Verges, M.; Pagans, S.; Iglesias, A.; Brugada, J.; Brugada, P.; et al. A missense mutation in the sodium channel beta2 subunit reveals SCN2B as a new candidate gene for brugada syndrome. Hum. Mutat. 2013, 34, 961–966. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Barajas-Martinez, H.; Burashnikov, E.; Springer, M.; Wu, Y.; Varro, A.; Pfeiffer, R.; Koopmann, T.T.; Cordeiro, J.M.; Guerchicoff, A.; et al. A mutation in the beta 3 subunit of the cardiac sodium channel associated with Brugada ECG phenotype. Circ. Cardiovasc. Genet. 2009, 2, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Barajas-Martinez, H.; Pfeiffer, R.; Dezi, F.; Pfeiffer, J.; Buch, T.; Betzenhauser, M.J.; Belardinelli, L.; Kahlig, K.M.; Rajamani, S.; et al. Mutations in SCN10A are responsible for a large fraction of cases of Brugada syndrome. J. Am. Coll. Cardiol. 2014, 64, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Behr, E.R.; Savio-Galimberti, E.; Barc, J.; Holst, A.G.; Petropoulou, E.; Prins, B.P.; Jabbari, J.; Torchio, M.; Berthet, M.; Mizusawa, Y.; et al. Role of common and rare variants in SCN10A: Results from the Brugada syndrome qrs locus gene discovery collaborative study. Cardiovasc. Res. 2015, 106, 520–529. [Google Scholar] [CrossRef] [PubMed]
- Le Scouarnec, S.; Karakachoff, M.; Gourraud, J.B.; Lindenbaum, P.; Bonnaud, S.; Portero, V.; Duboscq-Bidot, L.; Daumy, X.; Simonet, F.; Teusan, R.; et al. Testing the burden of rare variation in arrhythmia-susceptibility genes provides new insights into molecular diagnosis for Brugada syndrome. Hum. Mol. Genet. 2015, 24, 2757–2763. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, M.; Ohno, S.; Makiyama, T.; Horie, M. Novel SCN10A variants associated with Brugada syndrome. Europace 2016, 18, 905–911. [Google Scholar] [CrossRef] [PubMed]
- Antzelevitch, C.; Pollevick, G.D.; Cordeiro, J.M.; Casis, O.; Sanguinetti, M.C.; Aizawa, Y.; Guerchicoff, A.; Pfeiffer, R.; Oliva, A.; Wollnik, B.; et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by st-segment elevation, short QT intervals, and sudden cardiac death. Circulation 2007, 115, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, J.M.; Marieb, M.; Pfeiffer, R.; Calloe, K.; Burashnikov, E.; Antzelevitch, C. Accelerated inactivation of the l-type calcium current due to a mutation in CACNB2B underlies Brugada syndrome. J. Mol. Cell Cardiol. 2009, 46, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Burashnikov, E.; Pfeiffer, R.; Barajas-Martinez, H.; Delpon, E.; Hu, D.; Desai, M.; Borggrefe, M.; Haissaguerre, M.; Kanter, R.; Pollevick, G.D.; et al. Mutations in the cardiac l-type calcium channel associated with inherited j-wave syndromes and sudden cardiac death. Heart Rhythm 2010, 7, 1872–1882. [Google Scholar] [CrossRef] [PubMed]
- Giudicessi, J.R.; Ye, D.; Tester, D.J.; Crotti, L.; Mugione, A.; Nesterenko, V.V.; Albertson, R.M.; Antzelevitch, C.; Schwartz, P.J.; Ackerman, M.J. Transient outward current (Ito) gain-of-function mutations in the KCND3-encoded Kv4.3 potassium channel and Brugada syndrome. Heart Rhythm 2011, 8, 1024–1032. [Google Scholar] [CrossRef] [PubMed]
- Perrin, M.J.; Adler, A.; Green, S.; Al-Zoughool, F.; Doroshenko, P.; Orr, N.; Uppal, S.; Healey, J.S.; Birnie, D.; Sanatani, S.; et al. Evaluation of genes encoding for the transient outward current (Ito) identifies the KCND2 gene as a cause of J-wave syndrome associated with sudden cardiac death. Circ. Cardiovasc. Genet. 2014, 7, 782–789. [Google Scholar] [CrossRef] [PubMed]
- Delpon, E.; Cordeiro, J.M.; Nunez, L.; Thomsen, P.E.; Guerchicoff, A.; Pollevick, G.D.; Wu, Y.; Kanters, J.K.; Larsen, C.T.; Hofman-Bang, J.; et al. Functional effects of KCNE3 mutation and its role in the development of Brugada syndrome. Circ. Arrhythm. Electrophysiol. 2008, 1, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Ohno, S.; Zankov, D.P.; Ding, W.G.; Itoh, H.; Makiyama, T.; Doi, T.; Shizuta, S.; Hattori, T.; Miyamoto, A.; Naiki, N.; et al. KCNE5 (KCNE1L) variants are novel modulators of Brugada syndrome and idiopathic ventricular fibrillation. Circ. Arrhythm. Electrophysiol. 2011, 4, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Barajas-Martinez, H.; Hu, D.; Ferrer, T.; Onetti, C.G.; Wu, Y.; Burashnikov, E.; Boyle, M.; Surman, T.; Urrutia, J.; Veltmann, C.; et al. Molecular genetic and functional association of Brugada and early repolarization syndromes with S422l missense mutation in KCNJ8. Heart Rhythm 2012, 9, 548–555. [Google Scholar] [CrossRef]
- Kawamura, M.; Ozawa, T.; Yao, T.; Ashihara, T.; Sugimoto, Y.; Yagi, T.; Itoh, H.; Ito, M.; Makiyama, T.; Horie, M. Dynamic change in ST-segment and spontaneous occurrence of ventricular fibrillation in Brugada syndrome with a novel nonsense mutation in the SCN5A gene during long-term follow-up. Circ. J. 2009, 73, 584–588. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Ohno, S.; Ding, W.G.; Fukuyama, M.; Miyamoto, A.; Itoh, H.; Makiyama, T.; Wu, J.; Bai, J.; Hasegawa, K.; et al. Gain-of-function KCNH2 mutations in patients with Brugada syndrome. J. Cardiovasc. Electrophysiol. 2014, 25, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Kattygnarath, D.; Maugenre, S.; Neyroud, N.; Balse, E.; Ichai, C.; Denjoy, I.; Dilanian, G.; Martins, R.P.; Fressart, V.; Berthet, M.; et al. Mog1: A new susceptibility gene for Brugada syndrome. Circ. Cardiovasc. Genet. 2011, 4, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Shy, D.; Gillet, L.; Abriel, H. Cardiac sodium channel NaV1.5 distribution in myocytes via interacting proteins: The multiple pool model. Biochim. Biophys. Acta 2013, 1833, 886–894. [Google Scholar] [CrossRef] [PubMed]
- London, B.; Michalec, M.; Mehdi, H.; Zhu, X.; Kerchner, L.; Sanyal, S.; Viswanathan, P.C.; Pfahnl, A.E.; Shang, L.L.; Madhusudanan, M.; et al. Mutation in glycerol-3-phosphate dehydrogenase 1 like gene (Gpd1-L) decreases cardiac Na+ current and causes inherited arrhythmias. Circulation 2007, 116, 2260–2268. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Sato, A.; Marcou, C.A.; Tester, D.J.; Ackerman, M.J.; Crotti, L.; Schwartz, P.J.; On, Y.K.; Park, J.E.; Nakamura, K.; et al. A novel disease gene for Brugada syndrome: Sarcolemmal membrane-associated protein gene mutations impair intracellular trafficking of hNav1.5. Circ. Arrhythm. Electrophysiol. 2012, 5, 1098–1107. [Google Scholar] [CrossRef] [PubMed]
- Cerrone, M.; Delmar, M. Desmosomes and the sodium channel complex: Implications for arrhythmogenic cardiomyopathy and Brugada syndrome. Trends Cardiovasc. Med. 2014, 24, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Cerrone, M.; Lin, X.; Zhang, M.; Agullo-Pascual, E.; Pfenniger, A.; Chkourko Gusky, H.; Novelli, V.; Kim, C.; Tirasawadichai, T.; Judge, D.P.; et al. Missense mutations in plakophilin-2 cause sodium current deficit and associate with a Brugada syndrome phenotype. Circulation 2014, 129, 1092–1103. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Chatel, S.; Simard, C.; Syam, N.; Salle, L.; Probst, V.; Morel, J.; Millat, G.; Lopez, M.; Abriel, H.; et al. Molecular genetics and functional anomalies in a series of 248 Brugada cases with 11 mutations in the trpm4 channel. PLoS ONE 2013, 8, e54131. [Google Scholar] [CrossRef] [PubMed]
- Hennessey, J.A.; Marcou, C.A.; Wang, C.; Wei, E.Q.; Tester, D.J.; Torchio, M.; Dagradi, F.; Crotti, L.; Schwartz, P.J.; Ackerman, M.J.; et al. FGF12 is a candidate Brugada syndrome locus. Heart Rhythm 2013, 10, 1886–1894. [Google Scholar] [CrossRef] [PubMed]
- Bezzina, C.R.; Barc, J.; Mizusawa, Y.; Remme, C.A.; Gourraud, J.B.; Simonet, F.; Verkerk, A.O.; Schwartz, P.J.; Crotti, L.; Dagradi, F.; et al. Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat. Genet. 2013, 45, 1044–1049. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Hirano, Y.; Higashiuesato, Y.; Aizawa, Y.; Hayashi, T.; Inagaki, N.; Tana, T.; Ohya, Y.; Takishita, S.; Muratani, H.; et al. Role of HCN4 channel in preventing ventricular arrhythmia. J. Hum. Genet. 2009, 54, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Boczek, N.J.; Ye, D.; Johnson, E.K.; Wang, W.; Crotti, L.; Tester, D.J.; Dagradi, F.; Mizusawa, Y.; Torchio, M.; Alders, M.; et al. Characterization of SEMA3A-encoded semaphorin as a naturally occurring kv4.3 protein inhibitor and its contribution to Brugada syndrome. Circ. Res. 2014, 115, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Eastaugh, L.J.; James, P.A.; Phelan, D.G.; Davis, A.M. Brugada syndrome caused by a large deletion in SCN5A only detected by multiplex ligation-dependent probe amplification. J. Cardiovasc. Electrophysiol. 2011, 22, 1073–1076. [Google Scholar] [CrossRef] [PubMed]
- Mademont-Soler, I.; Pinsach-Abuin, M.L.; Riuro, H.; Mates, J.; Perez-Serra, A.; Coll, M.; Porres, J.M.; Del Olmo, B.; Iglesias, A.; Selga, E.; et al. Large genomic imbalances in Brugada syndrome. PLoS ONE 2016, 11, e0163514. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Molina, E.; Lacunza, J.; Ruiz-Espejo, F.; Sabater, M.; Garcia-Alberola, A.; Gimeno, J.R.; Canizares, F.; Garcia, A.; Martinez, P.; Valdes, M.; et al. A study of the SCN5A gene in a cohort of 76 patients with Brugada syndrome. Clin. Genet. 2013, 83, 530–538. [Google Scholar] [CrossRef] [PubMed]
- Koopmann, T.T.; Beekman, L.; Alders, M.; Meregalli, P.G.; Mannens, M.M.; Moorman, A.F.; Wilde, A.A.; Bezzina, C.R. Exclusion of multiple candidate genes and large genomic rearrangements in SCN5A in a dutch Brugada syndrome cohort. Heart Rhythm 2007, 4, 752–755. [Google Scholar] [CrossRef] [PubMed]
- Selga, E.; Campuzano, O.; Pinsach-Abuin, M.L.; Perez-Serra, A.; Mademont-Soler, I.; Riuro, H.; Pico, F.; Coll, M.; Iglesias, A.; Pagans, S.; et al. Comprehensive genetic characterization of a spanish Brugada syndrome cohort. PLoS ONE 2015, 10, e0132888. [Google Scholar] [CrossRef] [PubMed]
- Kapplinger, J.D.; Tester, D.J.; Alders, M.; Benito, B.; Berthet, M.; Brugada, J.; Brugada, P.; Fressart, V.; Guerchicoff, A.; Harris-Kerr, C.; et al. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm 2010, 7, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Wilde, A.A.; Postema, P.G.; Di Diego, J.M.; Viskin, S.; Morita, H.; Fish, J.M.; Antzelevitch, C. The pathophysiological mechanism underlying Brugada syndrome: Depolarization versus repolarization. J. Mol. Cell Cardiol. 2010, 49, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Blomstrom-Lundqvist, C.; Mazzanti, A.; Blom, N.; Borggrefe, M.; Camm, J.; Elliott, P.M.; Fitzsimons, D.; Hatala, R.; Hindricks, G.; et al. 2015 ESC guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Rev. Esp. Cardiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Jervell, A.; Lange-Nielsen, F. Congenital deaf-mutism, functional heart disease with prolongation of the QT interval and sudden death. Am. Heart J. 1957, 54, 59–68. [Google Scholar] [CrossRef]
- Levine, S.A.; Woodworth, C.R. Congenital deaf-mutism, prolonged qt interval, syncopal attacks and sudden death. N. Engl. J. Med. 1958, 259, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Romano, C.; Gemme, G.; Pongiglione, R. Rare cardiac arrythmias of the pediatric age. II. Syncopal attacks due to paroxysmal ventricular fibrillation. (presentation of 1st case in italian pediatric literature). Clin. Pediatr. 1963, 45, 656–683. [Google Scholar]
- Ward, O.C. A new familial cardiac syndrome in children. J. Ir. Med. Assoc. 1964, 54, 103–106. [Google Scholar] [PubMed]
- Schwartz, P.J.; Periti, M.; Malliani, A. The long QT syndrome. Am. Heart J. 1975, 89, 378–390. [Google Scholar] [CrossRef]
- Kaufman, E.S. Arrhythmic risk in congenital long QT syndrome. J. Electrocardiol. 2011, 44, 645–649. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, P.J.; Stramba-Badiale, M.; Crotti, L.; Pedrazzini, M.; Besana, A.; Bosi, G.; Gabbarini, F.; Goulene, K.; Insolia, R.; Mannarino, S.; et al. Prevalence of the congenital long-QT syndrome. Circulation 2009, 120, 1761–1767. [Google Scholar] [CrossRef] [PubMed]
- Rautaharju, P.M.; Zhou, S.H.; Wong, S.; Calhoun, H.P.; Berenson, G.S.; Prineas, R.; Davignon, A. Sex differences in the evolution of the electrocardiographic qt interval with age. Can. J. Cardiol. 1992, 8, 690–695. [Google Scholar] [PubMed]
- Hashiba, K. Sex differences in phenotypic manifestation and gene transmission in the romano-ward syndrome. Ann. N. Y. Acad. Sci. 1992, 644, 142–156. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, M.H.; Timothy, K.W.; Frankovich, D.; Fromm, B.S.; Keating, M.; Locati, E.H.; Taggart, R.T.; Towbin, J.A.; Moss, A.J.; Schwartz, P.J.; et al. Age-gender influence on the rate-corrected qt interval and the qt-heart rate relation in families with genotypically characterized long QT syndrome. J. Am. Coll. Cardiol. 1997, 29, 93–99. [Google Scholar] [CrossRef]
- Rodriguez, I.; Kilborn, M.J.; Liu, X.K.; Pezzullo, J.C.; Woosley, R.L. Drug-induced qt prolongation in women during the menstrual cycle. JAMA 2001, 285, 1322–1326. [Google Scholar] [CrossRef] [PubMed]
- Locati, E.H.; Zareba, W.; Moss, A.J.; Schwartz, P.J.; Vincent, G.M.; Lehmann, M.H.; Towbin, J.A.; Priori, S.G.; Napolitano, C.; Robinson, J.L.; et al. Age- and sex-related differences in clinical manifestations in patients with congenital long-QT syndrome: Findings from the international lqts registry. Circulation 1998, 97, 2237–2244. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.; Moss, A.J.; McNitt, S.; Zareba, W.; Andrews, M.L.; Qi, M.; Robinson, J.L.; Goldenberg, I.; Ackerman, M.J.; Benhorin, J.; et al. Long QT syndrome and pregnancy. J. Am. Coll Cardiol. 2007, 49, 1092–1098. [Google Scholar] [CrossRef] [PubMed]
- Makkar, R.R.; Fromm, B.S.; Steinman, R.T.; Meissner, M.D.; Lehmann, M.H. Female gender as a risk factor for torsades de pointes associated with cardiovascular drugs. JAMA 1993, 270, 2590–2597. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, M.H.; Hardy, S.; Archibald, D.; quart, B.; MacNeil, D.J. Sex difference in risk of torsade de pointes with d,l-sotalol. Circulation 1996, 94, 2535–2541. [Google Scholar] [CrossRef] [PubMed]
- Pratt, C.M.; Waldo, A.L.; Camm, A.J. Can antiarrhythmic drugs survive survival trials? Am. J. Cardiol. 1998, 81, 24D–34D. [Google Scholar] [CrossRef]
- Medeiros-Domingo, A.; Iturralde-Torres, P.; Ackerman, M.J. Clinical and genetic characteristics of long QT syndrome. Rev. Esp. Cardiol. 2007, 60, 739–752. [Google Scholar] [CrossRef] [PubMed]
- Roden, D.M. Cellular basis of drug-induced torsades de pointes. Br. J. Pharmacol. 2008, 154, 1502–1507. [Google Scholar] [CrossRef] [PubMed]
- Roden, D.M. Clinical practice. Long-QT syndrome. N. Engl. J. Med. 2008, 358, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Morita, H.; Wu, J.; Zipes, D.P. The QT syndromes: Long and short. Lancet 2008, 372, 750–763. [Google Scholar] [CrossRef]
- Goldenberg, I.; Zareba, W.; Moss, A.J. Long QT syndrome. Curr. Probl. Cardiol. 2008, 33, 629–694. [Google Scholar] [CrossRef] [PubMed]
- Vohra, J. The long QT syndrome. Heart Lung Circ. 2007, 16, S5–S12. [Google Scholar] [CrossRef] [PubMed]
- Brink, P.A.; Crotti, L.; Corfield, V.; Goosen, A.; Durrheim, G.; Hedley, P.; Heradien, M.; Geldenhuys, G.; Vanoli, E.; Bacchini, S.; et al. Phenotypic variability and unusual clinical severity of congenital long-QT syndrome in a founder population. Circulation 2005, 112, 2602–2610. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Napolitano, C.; Vicentini, A. Inherited arrhythmia syndromes: Applying the molecular biology and genetic to the clinical management. J. Interv. Card. Electrophysiol. 2003, 9, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Moss, A.J.; Schwartz, P.J.; Crampton, R.S.; Tzivoni, D.; Locati, E.H.; MacCluer, J.; Hall, W.J.; Weitkamp, L.; Vincent, G.M.; Garson, A., Jr.; et al. The long qt syndrome. Prospective longitudinal study of 328 families. Circulation 1991, 84, 1136–1144. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, P.J. Idiopathic long QT syndrome: Progress and questions. Am. Heart. J. 1985, 109, 399–411. [Google Scholar] [CrossRef]
- Schwartz, P.J.; Moss, A.J.; Vincent, G.M.; Crampton, R.S. Diagnostic criteria for the long QT syndrome. An update. Circulation 1993, 88, 782–784. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, P.J. The congenital long QT syndromes from genotype to phenotype: Clinical implications. J. Intern. Med. 2006, 259, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, P.J. Practical issues in the management of the long qt syndrome: Focus on diagnosis and therapy. Swiss Med. Wkly. 2013. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Timothy, K.W.; Vincent, G.M.; Lehmann, M.H.; Fox, J.; Giuli, L.C.; Shen, J.; Splawski, I.; Priori, S.G.; Compton, S.J.; et al. Spectrum of ST-T-wave patterns and repolarization parameters in congenital long-QT syndrome: Ecg findings identify genotypes. Circulation 2000, 102, 2849–2855. [Google Scholar] [CrossRef] [PubMed]
- Moss, A.J.; Zareba, W.; Benhorin, J.; Locati, E.H.; Hall, W.J.; Robinson, J.L.; Schwartz, P.J.; Towbin, J.A.; Vincent, G.M.; Lehmann, M.H. ECG T-wave patterns in genetically distinct forms of the hereditary long QT syndrome. Circulation 1995, 92, 2929–2934. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Benson, D.W.; Tristani-Firouzi, M.; Ptacek, L.J.; Tawil, R.; Schwartz, P.J.; George, A.L.; Horie, M.; Andelfinger, G.; Snow, G.L.; et al. Electrocardiographic features in andersen-tawil syndrome patients with kcnj2 mutations: Characteristic T-U-wave patterns predict the KCNJ2 genotype. Circulation 2005, 111, 2720–2726. [Google Scholar] [CrossRef] [PubMed]
- Lupoglazoff, J.M.; Denjoy, I.; Villain, E.; Fressart, V.; Simon, F.; Bozio, A.; Berthet, M.; Benammar, N.; Hainque, B.; Guicheney, P. Long QT syndrome in neonates: Conduction disorders associated with herg mutations and sinus bradycardia with KCNQ1 mutations. J. Am. Coll. Cardiol. 2004, 43, 826–830. [Google Scholar] [CrossRef] [PubMed]
- Donger, C.; Denjoy, I.; Berthet, M.; Neyroud, N.; Cruaud, C.; Bennaceur, M.; Chivoret, G.; Schwartz, K.; Coumel, P.; Guicheney, P. KVLQT1 c-terminal missense mutation causes a forme fruste long-QT syndrome. Circulation 1997, 96, 2778–2781. [Google Scholar] [CrossRef] [PubMed]
- Paulussen, A.D.; Gilissen, R.A.; Armstrong, M.; Doevendans, P.A.; Verhasselt, P.; Smeets, H.J.; Schulze-Bahr, E.; Haverkamp, W.; Breithardt, G.; Cohen, N.; et al. Genetic variations of KCNQ1, KCNH2, SCN5A, KCNE1, and KCNE2 in drug-induced long qt syndrome patients. J. Mol. Med. 2004, 82, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, P.J.; Malliani, A. Electrical alternation of the T-wave: Clinical and experimental evidence of its relationship with the sympathetic nervous system and with the long Q-T syndrome. Am. Heart J. 1975, 89, 45–50. [Google Scholar] [CrossRef]
- Keating, M.; Atkinson, D.; Dunn, C.; Timothy, K.; Vincent, G.M.; Leppert, M. Linkage of a cardiac arrhythmia, the long QT syndrome, and the harvey RAS-1 gene. Science 1991, 252, 704–706. [Google Scholar] [CrossRef] [PubMed]
- Towbin, J.A. Molecular genetic aspects of the romano-ward long QT syndrome. Tex. Heart Inst. J. 1994, 21, 42–47. [Google Scholar] [PubMed]
- Towbin, J.A.; Li, H.; Taggart, R.T.; Lehmann, M.H.; Schwartz, P.J.; Satler, C.A.; Ayyagari, R.; Robinson, J.L.; Moss, A.; Hejtmancik, J.F. Evidence of genetic heterogeneity in romano-ward long QT syndrome. Analysis of 23 families. Circulation 1994, 90, 2635–2644. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Atkinson, D.; Towbin, J.A.; Splawski, I.; Lehmann, M.H.; Li, H.; Timothy, K.; Taggart, R.T.; Schwartz, P.J.; Vincent, G.M.; et al. Two long QT syndrome loci map to chromosomes 3 and 7 with evidence for further heterogeneity. Nat. Genet. 1994, 8, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Kapplinger, J.D.; Tester, D.J.; Salisbury, B.A.; Carr, J.L.; Harris-Kerr, C.; Pollevick, G.D.; Wilde, A.A.; Ackerman, M.J. Spectrum and prevalence of mutations from the first 2,500 consecutive unrelated patients referred for the familion long qt syndrome genetic test. Heart Rhythm 2009, 6, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Barhanin, J.; Lesage, F.; Guillemare, E.; Fink, M.; Lazdunski, M.; Romey, G. K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature 1996, 384, 78–80. [Google Scholar] [CrossRef] [PubMed]
- Bellocq, C.; van Ginneken, A.C.; Bezzina, C.R.; Alders, M.; Escande, D.; Mannens, M.M.; Baro, I.; Wilde, A.A. Mutation in the KCNQ1 gene leading to the short QT-interval syndrome. Circulation 2004, 109, 2394–2397. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Curran, M.E.; Splawski, I.; Burn, T.C.; Millholland, J.M.; VanRaay, T.J.; Shen, J.; Timothy, K.W.; Vincent, G.M.; de Jager, T.; et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat. Genet. 1996, 12, 17–23. [Google Scholar] [CrossRef]
- Curran, M.E.; Splawski, I.; Timothy, K.W.; Vincent, G.M.; Green, E.D.; Keating, M.T. A molecular basis for cardiac arrhythmia: Herg mutations cause long QT syndrome. Cell 1995, 80, 795–803. [Google Scholar] [CrossRef]
- Wang, Q.; Shen, J.; Splawski, I.; Atkinson, D.; Li, Z.; Robinson, J.L.; Moss, A.J.; Towbin, J.A.; Keating, M.T. SCN5A mutations associated with an inherited cardiac arrhythmia, long qt syndrome. Cell 1995, 80, 805–811. [Google Scholar] [CrossRef]
- Mohler, P.J.; Schott, J.J.; Gramolini, A.O.; Dilly, K.W.; Guatimosim, S.; duBell, W.H.; Song, L.S.; Haurogne, K.; Kyndt, F.; Ali, M.E.; et al. Ankyrin-b mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature 2003, 421, 634–639. [Google Scholar] [CrossRef] [PubMed]
- Splawski, I.; Tristani-Firouzi, M.; Lehmann, M.H.; Sanguinetti, M.C.; Keating, M.T. Mutations in the hmink gene cause long QT syndrome and suppress iks function. Nat. Genet. 1997, 17, 338–340. [Google Scholar] [CrossRef] [PubMed]
- Abbott, G.W.; Sesti, F.; Splawski, I.; Buck, M.E.; Lehmann, M.H.; Timothy, K.W.; Keating, M.T.; Goldstein, S.A. MIRP1 forms IKR potassium channels with HERG and is associated with cardiac arrhythmia. Cell 1999, 97, 175–187. [Google Scholar] [CrossRef]
- Andersen, E.D.; Krasilnikoff, P.A.; Overvad, H. Intermittent muscular weakness, extrasystoles, and multiple developmental anomalies: A new syndrome? Acta Paediatr. Scand. 1971, 60, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Tawil, R.; Ptacek, L.J.; Pavlakis, S.G.; DeVivo, D.C.; Penn, A.S.; Ozdemir, C.; Griggs, R.C. Andersen’s syndrome: Potassium-sensitive periodic paralysis, ventricular ectopy, and dysmorphic features. Ann. Neurol. 1994, 35, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Boczek, N.J.; Best, J.M.; Tester, D.J.; Giudicessi, J.R.; Middha, S.; Evans, J.M.; Kamp, T.J.; Ackerman, M.J. Exome sequencing and systems biology converge to identify novel mutations in the l-type calcium channel, CACNA1C, linked to autosomal dominant long QT syndrome. Circ. Cardiovasc. Genet. 2013, 6, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, M.; Ohno, S.; Wang, Q.; Shirayama, T.; Itoh, H.; Horie, M. Nonsense-mediated mrna decay due to a CACNA1C splicing mutation in a patient with Brugada syndrome. Heart Rhythm 2014, 11, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Splawski, I.; Timothy, K.W.; Sharpe, L.M.; Decher, N.; Kumar, P.; Bloise, R.; Napolitano, C.; Schwartz, P.J.; Joseph, R.M.; Condouris, K.; et al. Ca(v)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 2004, 119, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Splawski, I.; Timothy, K.W.; Decher, N.; Kumar, P.; Sachse, F.B.; Beggs, A.H.; Sanguinetti, M.C.; Keating, M.T. Severe arrhythmia disorder caused by cardiac L-type calcium channel mutations. Proc. Natl. Acad. Sci. USA 2005, 102, 8089–8096. [Google Scholar] [CrossRef] [PubMed]
- Boczek, N.J.; Miller, E.M.; Ye, D.; Nesterenko, V.V.; Tester, D.J.; Antzelevitch, C.; Czosek, R.J.; Ackerman, M.J.; Ware, S.M. Novel timothy syndrome mutation leading to increase in CACNA1C window current. Heart Rhythm 2015, 12, 211–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vatta, M.; Ackerman, M.J.; Ye, B.; Makielski, J.C.; Ughanze, E.E.; Taylor, E.W.; Tester, D.J.; Balijepalli, R.C.; Foell, J.D.; Li, Z.; et al. Mutant caveolin-3 induces persistent late sodium current and is associated with long-QT syndrome. Circulation 2006, 114, 2104–2112. [Google Scholar] [CrossRef] [PubMed]
- Medeiros-Domingo, A.; Kaku, T.; Tester, D.J.; Iturralde-Torres, P.; Itty, A.; Ye, B.; Valdivia, C.; Ueda, K.; Canizales-Quinteros, S.; Tusie-Luna, M.T.; et al. SCN4B-encoded sodium channel beta4 subunit in congenital long-QT syndrome. Circulation 2007, 116, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Valdivia, C.; Medeiros-Domingo, A.; Tester, D.J.; Vatta, M.; Farrugia, G.; Ackerman, M.J.; Makielski, J.C. Syntrophin mutation associated with long QT syndrome through activation of the nnos-SCN5A macromolecular complex. Proc. Natl. Acad. Sci. USA 2008, 105, 9355–9360. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Marquardt, M.L.; Tester, D.J.; Sampson, K.J.; Ackerman, M.J.; Kass, R.S. Mutation of an a-kinase-anchoring protein causes long-QT syndrome. Proc. Natl. Acad. Sci. USA 2007, 104, 20990–20995. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.X.; Sandow, S.L.; Doceul, V.; Chen, Y.H.; Harper, H.; Hamilton, B.; Meadows, H.J.; Trezise, D.J.; Clare, J.J. Improved functional expression of recombinant human ether-a-go-go (hERG) k+ channels by cultivation at reduced temperature. BMC Biotechnol. 2007. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liang, B.; Liu, J.; Li, J.; Grunnet, M.; Olesen, S.P.; Rasmussen, H.B.; Ellinor, P.T.; Gao, L.; Lin, X.; et al. Identification of a KIR3.4 mutation in congenital long QT syndrome. Am. J. Hum. Genet. 2010, 86, 872–880. [Google Scholar] [CrossRef] [PubMed]
- Crotti, L.; Johnson, C.N.; Graf, E.; De Ferrari, G.M.; Cuneo, B.F.; Ovadia, M.; Papagiannis, J.; Feldkamp, M.D.; Rathi, S.G.; Kunic, J.D.; et al. Calmodulin mutations associated with recurrent cardiac arrest in infants. Circulation 2013, 127, 1009–1017. [Google Scholar] [CrossRef] [PubMed]
- Makita, N.; Yagihara, N.; Crotti, L.; Johnson, C.N.; Beckmann, B.M.; Roh, M.S.; Shigemizu, D.; Lichtner, P.; Ishikawa, T.; Aiba, T.; et al. Novel calmodulin mutations associated with congenital arrhythmia susceptibility. Circ. Cardiovasc. Genet. 2014, 7, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Reed, G.J.; Boczek, N.J.; Etheridge, S.P.; Ackerman, M.J. Calm3 mutation associated with long QT syndrome. Heart Rhythm 2015, 12, 419–422. [Google Scholar] [CrossRef] [PubMed]
- Riuro, H.; Campuzano, O.; Arbelo, E.; Iglesias, A.; Batlle, M.; Perez-Villa, F.; Brugada, J.; Perez, G.J.; Scornik, F.S.; Brugada, R. A missense mutation in the sodium channel beta1b subunit reveals SCN1B as a susceptibility gene underlying long qt syndrome. Heart Rhythm 2014, 11, 1202–1209. [Google Scholar] [CrossRef] [PubMed]
- Kauferstein, S.; Kiehne, N.; Erkapic, D.; Schmidt, J.; Hamm, C.W.; Bratzke, H.; Pitschner, H.F.; Kuniss, M.; Neumann, T. A novel mutation in the cardiac ryanodine receptor gene (RYR2) in a patient with an unequivocal lqts. Int. J. Cardiol. 2011, 146, 249–250. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, P.J.; Spazzolini, C.; Crotti, L.; Bathen, J.; Amlie, J.P.; Timothy, K.; Shkolnikova, M.; Berul, C.I.; Bitner-Glindzicz, M.; Toivonen, L.; et al. The jervell and lange-nielsen syndrome: Natural history, molecular basis, and clinical outcome. Circulation 2006, 113, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Gussak, I.; Brugada, P.; Brugada, J.; Wright, R.S.; Kopecky, S.L.; Chaitman, B.R.; Bjerregaard, P. Idiopathic short QT interval: A new clinical syndrome? Cardiology 2000, 94, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Gaita, F.; Giustetto, C.; Bianchi, F.; Wolpert, C.; Schimpf, R.; Riccardi, R.; Grossi, S.; Richiardi, E.; Borggrefe, M. Short QT syndrome: A familial cause of sudden death. Circulation 2003, 108, 965–970. [Google Scholar] [CrossRef] [PubMed]
- Mazzanti, A.; O’Rourke, S.; Ng, K.; Miceli, C.; Borio, G.; Curcio, A.; Esposito, F.; Napolitano, C.; Priori, S.G. The usual suspects in sudden cardiac death of the young: A focus on inherited arrhythmogenic diseases. Expert Rev. Cardiovasc. Ther. 2014, 12, 499–519. [Google Scholar] [CrossRef] [PubMed]
- Kobza, R.; Roos, M.; Niggli, B.; Abacherli, R.; Lupi, G.A.; Frey, F.; Schmid, J.J.; Erne, P. Prevalence of long and short QT in a young population of 41,767 predominantly male swiss conscripts. Heart Rhythm 2009, 6, 652–657. [Google Scholar] [CrossRef] [PubMed]
- Funada, A.; Hayashi, K.; Ino, H.; Fujino, N.; Uchiyama, K.; Sakata, K.; Masuta, E.; Sakamoto, Y.; Tsubokawa, T.; Yamagishi, M. Assessment of qt intervals and prevalence of short QT syndrome in japan. Clin. Cardiol. 2008, 31, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Anttonen, O.; Junttila, M.J.; Rissanen, H.; Reunanen, A.; Viitasalo, M.; Huikuri, H.V. Prevalence and prognostic significance of short qt interval in a middle-aged finnish population. Circulation 2007, 116, 714–720. [Google Scholar] [CrossRef] [PubMed]
- Rudic, B.; Schimpf, R.; Borggrefe, M. Short qt syndrome—Review of diagnosis and treatment. Arrhythm. Electrophysiol. Rev. 2014, 3, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, A.; Hayashi, H.; Yoshino, T.; Kawaguchi, T.; Taniguchi, A.; Itoh, H.; Sugimoto, Y.; Itoh, M.; Makiyama, T.; Xue, J.Q.; et al. Clinical and electrocardiographic characteristics of patients with short qt interval in a large hospital-based population. Heart Rhythm 2012, 9, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Giustetto, C.; Di Monte, F.; Wolpert, C.; Borggrefe, M.; Schimpf, R.; Sbragia, P.; Leone, G.; Maury, P.; Anttonen, O.; Haissaguerre, M.; et al. Short qt syndrome: Clinical findings and diagnostic-therapeutic implications. Eur. Heart J. 2006, 27, 2440–2447. [Google Scholar] [CrossRef] [PubMed]
- Nierenberg, D.W. Spironolactone and metabolic acidosis. Ann. Intern. Med. 1979, 91, 321–322. [Google Scholar] [CrossRef] [PubMed]
- Holbrook, M.; Malik, M.; Shah, R.R.; Valentin, J.P. Drug induced shortening of the QT/QTC interval: An emerging safety issue warranting further modelling and evaluation in drug research and development? J. Pharmacol. Toxicol. Methods 2009, 59, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Viskin, S.; Justo, D.; Zeltser, D. Drug-induced prolongation of the QT interval. N. Engl. J. Med. 2004, 350, 2618–2621. [Google Scholar] [PubMed]
- Viskin, S.; Zeltser, D.; Ish-Shalom, M.; Katz, A.; Glikson, M.; Justo, D.; Tekes-Manova, D.; Belhassen, B. Is idiopathic ventricular fibrillation a short QT syndrome? Comparison of qt intervals of patients with idiopathic ventricular fibrillation and healthy controls. Heart Rhythm 2004, 1, 587–591. [Google Scholar] [CrossRef] [PubMed]
- Schimpf, R.; Wolpert, C.; Gaita, F.; Giustetto, C.; Borggrefe, M. Short QT syndrome. Cardiovasc. Res. 2005, 67, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Schimpf, R.; Borggrefe, M.; Wolpert, C. Clinical and molecular genetics of the short QT syndrome. Curr. Opin. Cardiol. 2008, 23, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Tulumen, E.; Giustetto, C.; Wolpert, C.; Maury, P.; Anttonen, O.; Probst, V.; Blanc, J.J.; Sbragia, P.; Scrocco, C.; Rudic, B.; et al. PQ segment depression in patients with short QT syndrome: A novel marker for diagnosing short qt syndrome? Heart Rhythm 2014, 11, 1024–1030. [Google Scholar] [CrossRef] [PubMed]
- Borggrefe, M.; Wolpert, C.; Antzelevitch, C.; Veltmann, C.; Giustetto, C.; Gaita, F.; Schimpf, R. Short QT syndrome. Genotype-phenotype correlations. J. Electrocardiol. 2005, 38, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Giustetto, C.; Schimpf, R.; Mazzanti, A.; Scrocco, C.; Maury, P.; Anttonen, O.; Probst, V.; Blanc, J.J.; Sbragia, P.; Dalmasso, P.; et al. Long-term follow-up of patients with short QT syndrome. J. Am. Coll. Cardiol. 2011, 58, 587–595. [Google Scholar] [CrossRef]
- Gollob, M.H.; Redpath, C.J.; Roberts, J.D. The short QT syndrome: Proposed diagnostic criteria. J. Am. Coll. Cardiol. 2011, 57, 802–812. [Google Scholar] [CrossRef] [PubMed]
- Brugada, R.; Hong, K.; Dumaine, R.; Cordeiro, J.; Gaita, F.; Borggrefe, M.; Menendez, T.M.; Brugada, J.; Pollevick, G.D.; Wolpert, C.; et al. Sudden death associated with short-QT syndrome linked to mutations in herg. Circulation 2004, 109, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Pandit, S.V.; Rivolta, I.; Berenfeld, O.; Ronchetti, E.; Dhamoon, A.; Napolitano, C.; Anumonwo, J.; di Barletta, M.R.; Gudapakkam, S.; et al. A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ. Res. 2005, 96, 800–807. [Google Scholar] [CrossRef] [PubMed]
- Templin, C.; Ghadri, J.R.; Rougier, J.S.; Baumer, A.; Kaplan, V.; Albesa, M.; Sticht, H.; Rauch, A.; Puleo, C.; Hu, D.; et al. Identification of a novel loss-of-function calcium channel gene mutation in short qt syndrome (SQTS6). Eur. Heart J. 2011, 32, 1077–1088. [Google Scholar] [CrossRef] [PubMed]
- Berg, K.J. Multifocal ventricular extrasytoles with adams-stokes syndrome in siblings. Am. Heart J. 1960, 60, 965–970. [Google Scholar] [CrossRef]
- Reid, D.S.; Tynan, M.; Braidwood, L.; Fitzgerald, G.R. Bidirectional tachycardia in a child. A study using his bundle electrography. Br. Heart J. 1975, 37, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Leenhardt, A.; Lucet, V.; Denjoy, I.; Grau, F.; Ngoc, D.D.; Coumel, P. Catecholaminergic polymorphic ventricular tachycardia in children. A 7-year follow-up of 21 patients. Circulation 1995, 91, 1512–1519. [Google Scholar] [CrossRef] [PubMed]
- Lieve, K.V.; van der Werf, C.; Wilde, A.A. Catecholaminergic polymorphic ventricular tachycardia. Circ. J. 2016, 80, 1285–1291. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, C.; Priori, S.G. Diagnosis and treatment of catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 2007, 4, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Napolitano, C.; Memmi, M.; Colombi, B.; Drago, F.; Gasparini, M.; DeSimone, L.; Coltorti, F.; Bloise, R.; Keegan, R.; et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation 2002, 106, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Lehnart, S.E.; Wehrens, X.H.; Laitinen, P.J.; Reiken, S.R.; Deng, S.X.; Cheng, Z.; Landry, D.W.; Kontula, K.; Swan, H.; Marks, A.R. Sudden death in familial polymorphic ventricular tachycardia associated with calcium release channel (ryanodine receptor) leak. Circulation 2004, 109, 3208–3214. [Google Scholar] [CrossRef] [PubMed]
- Refaat, M.M.; Hassanieh, S.; Scheinman, M. Catecholaminergic polymorphic ventricular tachycardia. Card. Electrophysiol. Clin. 2016, 8, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Terentyev, D.; Nori, A.; Santoro, M.; Viatchenko-Karpinski, S.; Kubalova, Z.; Gyorke, I.; Terentyeva, R.; Vedamoorthyrao, S.; Blom, N.A.; Valle, G.; et al. Abnormal interactions of calsequestrin with the ryanodine receptor calcium release channel complex linked to exercise-induced sudden cardiac death. Circ. Res. 2006, 98, 1151–1158. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Napolitano, C. Cardiac and skeletal muscle disorders caused by mutations in the intracellular Ca2+ release channels. J. Clin. Investig. 2005, 115, 2033–2038. [Google Scholar] [CrossRef] [PubMed]
- Postma, A.V.; Denjoy, I.; Kamblock, J.; Alders, M.; Lupoglazoff, J.M.; Vaksmann, G.; Dubosq-Bidot, L.; Sebillon, P.; Mannens, M.M.; Guicheney, P.; et al. Catecholaminergic polymorphic ventricular tachycardia: Ryr2 mutations, bradycardia, and follow up of the patients. J. Med. Genet. 2005, 42, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Aizawa, Y.; Komura, S.; Okada, S.; Chinushi, M.; Morita, H.; Ohe, T. Distinct U wave changes in patients with catecholaminergic polymorphic ventricular tachycardia (CPVT). Int. Heart J. 2006, 47, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Refaat, M.M.; Hotait, M.; Tseng, Z.H. Utility of the exercise electrocardiogram testing in sudden cardiac death risk stratification. Ann. Noninvasive Electrocardiol. 2014, 19, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, C.; Priori, S.G.; Bloise, R. Catecholaminergic polymorphic ventricular tachycardia. In GeneReviews®; Pagon, R.A., Adam, M.P., Ardinger, H.H., Wallace, S.E., Amemiya, A., Bean, L.J.H., Bird, T.D., Ledbetter, N., Mefford, H.C., Smith, R.J.H., et al., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Liu, N.; Ruan, Y.; Priori, S.G. Catecholaminergic polymorphic ventricular tachycardia. Prog. Cardiovasc. Dis. 2008, 51, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Napolitano, C.; Tiso, N.; Memmi, M.; Vignati, G.; Bloise, R.; Sorrentino, V.; Danieli, G.A. Mutations in the cardiac ryanodine receptor gene (HRYR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation 2001, 103, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Lahat, H.; Pras, E.; Olender, T.; Avidan, N.; Ben-Asher, E.; Man, O.; Levy-Nissenbaum, E.; Khoury, A.; Lorber, A.; Goldman, B.; et al. A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in bedouin families from israel. Am. J. Hum. Genet. 2001, 69, 1378–1384. [Google Scholar] [CrossRef] [PubMed]
- Postma, A.V.; Denjoy, I.; Hoorntje, T.M.; Lupoglazoff, J.M.; Da Costa, A.; Sebillon, P.; Mannens, M.M.; Wilde, A.A.; Guicheney, P. Absence of calsequestrin 2 causes severe forms of catecholaminergic polymorphic ventricular tachycardia. Circ. Res. 2002, 91, e21–e26. [Google Scholar] [CrossRef] [PubMed]
- Sumitomo, N. Current topics in catecholaminergic polymorphic ventricular tachycardia. J. Arrhythm. 2016, 32, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Nyegaard, M.; Overgaard, M.T.; Sondergaard, M.T.; Vranas, M.; Behr, E.R.; Hildebrandt, L.L.; Lund, J.; Hedley, P.L.; Camm, A.J.; Wettrell, G.; et al. Mutations in calmodulin cause ventricular tachycardia and sudden cardiac death. Am. J. Hum. Genet. 2012, 91, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Hurtado, N.; Boczek, N.J.; Kryshtal, D.O.; Johnson, C.N.; Sun, J.; Nitu, F.R.; Cornea, R.L.; Chazin, W.J.; Calvert, M.L.; Tester, D.J.; et al. Novel CPVT-associated calmodulin mutation in CALM3 (CALM3-A103V) activates arrhythmogenic ca waves and sparks. Circ. Arrhythm. Electrophysiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Roux-Buisson, N.; Cacheux, M.; Fourest-Lieuvin, A.; Fauconnier, J.; Brocard, J.; Denjoy, I.; Durand, P.; Guicheney, P.; Kyndt, F.; Leenhardt, A.; et al. Absence of triadin, a protein of the calcium release complex, is responsible for cardiac arrhythmia with sudden death in human. Hum. Mol. Genet. 2012, 21, 2759–2767. [Google Scholar] [PubMed]
- Vega, A.L.; Tester, D.J.; Ackerman, M.J.; Makielski, J.C. Protein kinase a-dependent biophysical phenotype for V227F-KCNJ2 mutation in catecholaminergic polymorphic ventricular tachycardia. Circ. Arrhythm. Electrophysiol. 2009, 2, 540–547. [Google Scholar] [CrossRef] [PubMed]
- Mohler, P.J.; Splawski, I.; Napolitano, C.; Bottelli, G.; Sharpe, L.; Timothy, K.; Priori, S.G.; Keating, M.T.; Bennett, V. A cardiac arrhythmia syndrome caused by loss of ankyrin-b function. Proc. Natl. Acad. Sci. USA 2004, 101, 9137–9142. [Google Scholar] [CrossRef] [PubMed]
- Bhuiyan, Z.A.; Hamdan, M.A.; Shamsi, E.T.; Postma, A.V.; Mannens, M.M.; Wilde, A.A.; Al-Gazali, L. A novel early onset lethal form of catecholaminergic polymorphic ventricular tachycardia maps to chromosome 7p14-p22. J. Cardiovasc. Electrophysiol. 2007, 18, 1060–1066. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernández-Falgueras, A.; Sarquella-Brugada, G.; Brugada, J.; Brugada, R.; Campuzano, O. Cardiac Channelopathies and Sudden Death: Recent Clinical and Genetic Advances. Biology 2017, 6, 7. https://doi.org/10.3390/biology6010007
Fernández-Falgueras A, Sarquella-Brugada G, Brugada J, Brugada R, Campuzano O. Cardiac Channelopathies and Sudden Death: Recent Clinical and Genetic Advances. Biology. 2017; 6(1):7. https://doi.org/10.3390/biology6010007
Chicago/Turabian StyleFernández-Falgueras, Anna, Georgia Sarquella-Brugada, Josep Brugada, Ramon Brugada, and Oscar Campuzano. 2017. "Cardiac Channelopathies and Sudden Death: Recent Clinical and Genetic Advances" Biology 6, no. 1: 7. https://doi.org/10.3390/biology6010007