Incomplete Penetrance and Variable Expressivity: Hallmarks in Channelopathies Associated with Sudden Cardiac Death

Abstract
:1. Introduction
2. Channelopathies
2.1. Long QT Syndrome
2.2. Brugada Syndrome
2.3. Catecholaminergic Polymorphic Ventricular Tachycardia
2.4. Short QT Syndrome
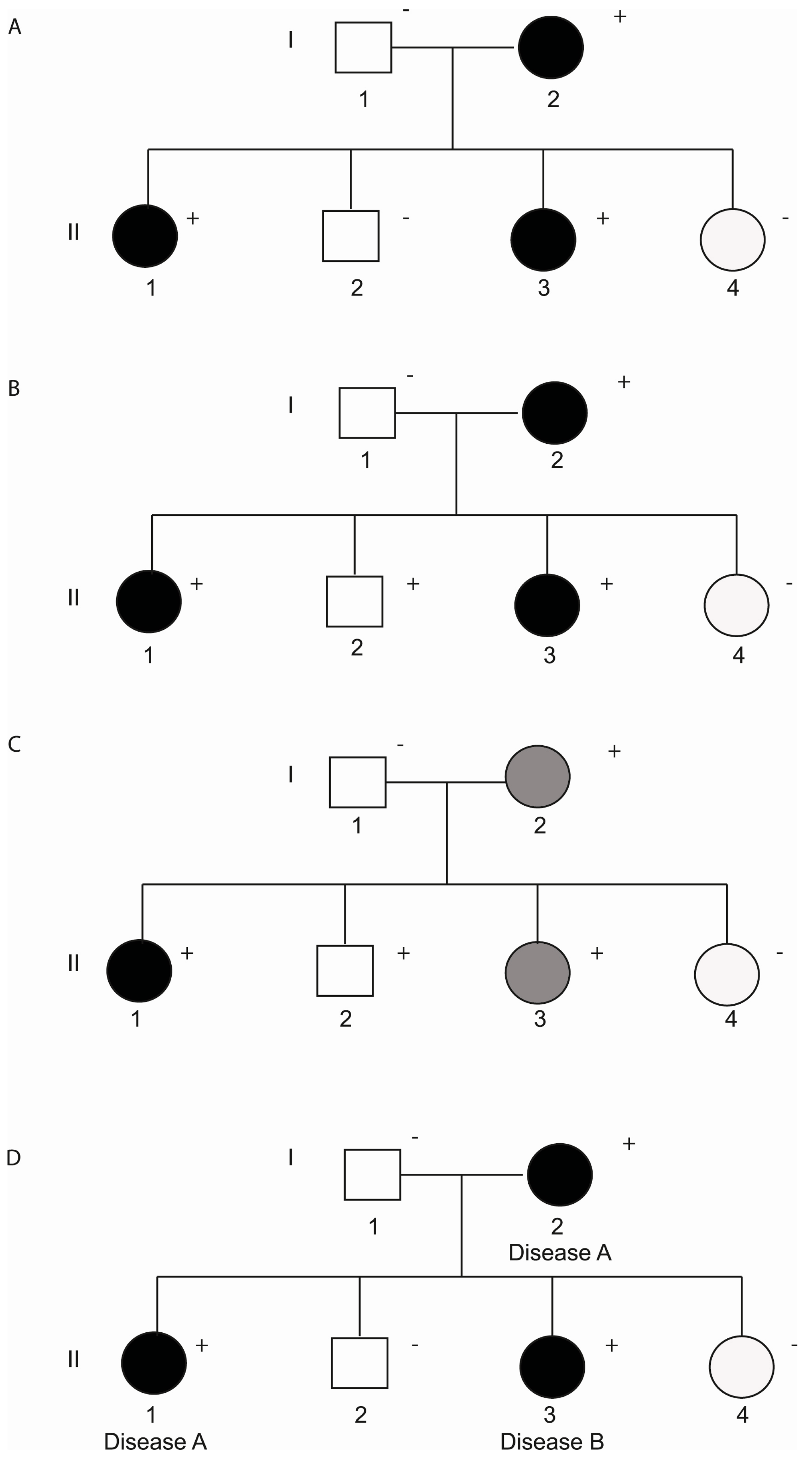
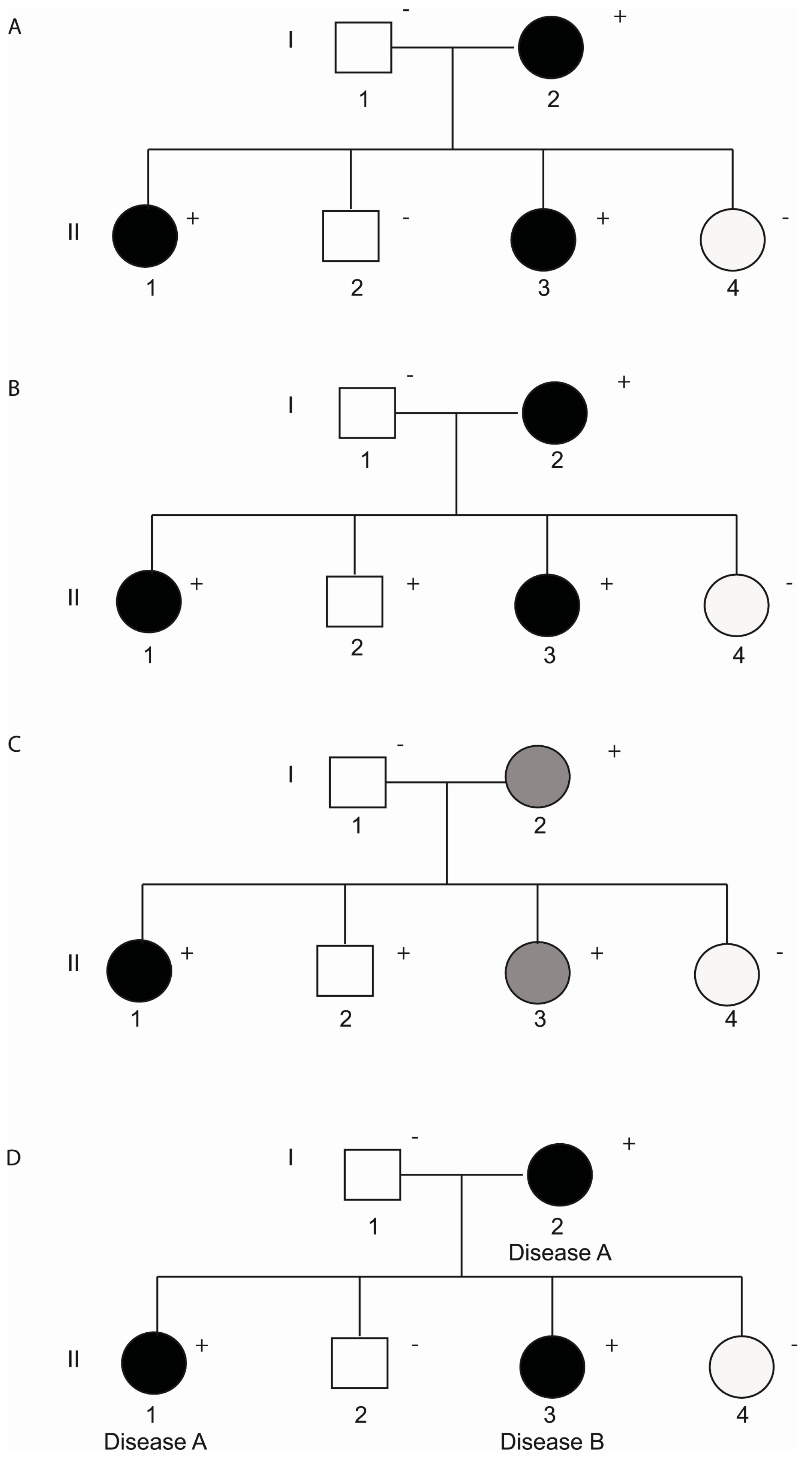
3. Incomplete Penetrance and Variable Expressivity
3.1. Non-Genetic Modifiers
3.2. Genetic Modifiers
3.2.1. Coding Variants
3.2.2. Non-Coding Variants
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bayes de Luna, A.; Elosua, R. Sudden death. Rev. Esp. Cardiol. 2012, 65, 1039–1052. [Google Scholar] [CrossRef] [PubMed]
- Marsman, R.F.; Tan, H.L.; Bezzina, C.R. Genetics of sudden cardiac death caused by ventricular arrhythmias. Nat. Rev. Cardiol. 2014, 11, 96–111. [Google Scholar] [CrossRef] [PubMed]
- Martens, E.; Sinner, M.F.; Siebermair, J.; Raufhake, C.; Beckmann, B.M.; Veith, S.; Duvel, D.; Steinbeck, G.; Kaab, S. Incidence of sudden cardiac death in Germany: Results from an emergency medical service registry in Lower Saxony. Europace 2014, 16, 1752–1758. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.J.; Blaha, M.J.; Chiuve, S.E.; Cushman, M.; Das, S.R.; Deo, R.; de Ferranti, S.D.; Floyd, J.; Fornage, M.; Gillespie, C.; et al. Heart Disease and Stroke Statistics-2017 Update: A Report From the American Heart Association. Circulation 2017, 135, e146–e603. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, O.; Sarquella-Brugada, G.; Brugada, R.; Brugada, J. Genetics of channelopathies associated with sudden cardiac death. Glob. Cardiol. Sci. Pract. 2015, 2015, 39. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Blomstrom-Lundqvist, C.; Mazzanti, A.; Blom, N.; Borggrefe, M.; Camm, J.; Elliott, P.M.; Fitzsimons, D.; Hatala, R.; Hindricks, G.; et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur. Heart J. 2015, 36, 2793–2867. [Google Scholar] [CrossRef] [PubMed]
- Lieve, K.V.; Wilde, A.A. Inherited ion channel diseases: A brief review. Europace 2015. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, P.J.; Stramba-Badiale, M.; Crotti, L.; Pedrazzini, M.; Besana, A.; Bosi, G.; Gabbarini, F.; Goulene, K.; Insolia, R.; Mannarino, S.; et al. Prevalence of the congenital long-QT syndrome. Circulation 2009, 120, 1761–1767. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, M.J.; Priori, S.G.; Willems, S.; Berul, C.; Brugada, R.; Calkins, H.; Camm, A.J.; Ellinor, P.T.; Gollob, M.; Hamilton, R.; et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm 2011, 8, 1308–1339. [Google Scholar] [CrossRef] [PubMed]
- Mizusawa, Y. Recent advances in genetic testing and counseling for inherited arrhythmias. J. Arrhythm. 2016, 32, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Falgueras, A.; Sarquella-Brugada, G.; Brugada, J.; Brugada, R.; Campuzano, O. Cardiac Channelopathies and Sudden Death: Recent Clinical and Genetic Advances. Biology 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, M.J.; Marcou, C.A.; Tester, D.J. Personalized medicine: Genetic diagnosis for inherited cardiomyopathies/channelopathies. Rev. Esp. Cardiol. 2013, 66, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Holst, A.G.; Jensen, H.K.; Eschen, O.; Henriksen, F.L.; Kanters, J.; Bundgaard, H.; Svendsen, J.H.; Haunso, S.; Tfelt-Hansen, J. Low disease prevalence and inappropriate implantable cardioverter defibrillator shock rate in Brugada syndrome: A nationwide study. Europace 2012, 14, 1025–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nademanee, K. Sudden unexplained death syndrome in Southeast Asia. Am. J. Cardiol. 1997, 79, 10–11. [Google Scholar] [CrossRef]
- Wanguemert, F.; Bosch Calero, C.; Perez, C.; Campuzano, O.; Beltran-Alvarez, P.; Scornik, F.S.; Iglesias, A.; Berne, P.; Allegue, C.; Ruiz Hernandez, P.M.; et al. Clinical and molecular characterization of a cardiac ryanodine receptor founder mutation causing catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 2015, 12, 1636–1643. [Google Scholar] [CrossRef] [PubMed]
- Lieve, K.V.; van der Werf, C.; Wilde, A.A. Catecholaminergic Polymorphic Ventricular Tachycardia. Circ. J. 2016, 80, 1285–1291. [Google Scholar] [CrossRef] [PubMed]
- Roux-Buisson, N.; Cacheux, M.; Fourest-Lieuvin, A.; Fauconnier, J.; Brocard, J.; Denjoy, I.; Durand, P.; Guicheney, P.; Kyndt, F.; Leenhardt, A.; et al. Absence of triadin, a protein of the calcium release complex, is responsible for cardiac arrhythmia with sudden death in human. Hum. Mol. Genet. 2012, 21, 2759–2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gussak, I.; Brugada, P.; Brugada, J.; Wright, R.S.; Kopecky, S.L.; Chaitman, B.R.; Bjerregaard, P. Idiopathic short QT interval: A new clinical syndrome? Cardiology 2000, 94, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Mazzanti, A.; O’Rourke, S.; Ng, K.; Miceli, C.; Borio, G.; Curcio, A.; Esposito, F.; Napolitano, C.; Priori, S.G. The usual suspects in sudden cardiac death of the young: A focus on inherited arrhythmogenic diseases. Expert Rev. Cardiovasc. Ther. 2014, 12, 499–519. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Wilde, A.A.; Horie, M.; Cho, Y.; Behr, E.R.; Berul, C.; Blom, N.; Brugada, J.; Chiang, C.E.; Huikuri, H.; et al. Executive summary: HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes. Europace 2013, 15, 1389–1406. [Google Scholar] [CrossRef] [PubMed]
- Veltmann, C.; Barajas-Martinez, H.; Wolpert, C.; Borggrefe, M.; Schimpf, R.; Pfeiffer, R.; Caceres, G.; Burashnikov, E.; Antzelevitch, C.; Hu, D. Further Insights in the Most Common SCN5A Mutation Causing Overlapping Phenotype of Long QT Syndrome, Brugada Syndrome, and Conduction Defect. J. Am. Heart Assoc. 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Giudicessi, J.R.; Ackerman, M.J. Determinants of incomplete penetrance and variable expressivity in heritable cardiac arrhythmia syndromes. Transl. Res. 2013, 161, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Hoogendijk, M.G. Diagnostic dilemmas: Overlapping features of brugada syndrome and arrhythmogenic right ventricular cardiomyopathy. Front. Physiol. 2012, 3, 144. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Napolitano, C.; Schwartz, P.J.; Bloise, R.; Crotti, L.; Ronchetti, E. The elusive link between LQT3 and Brugada syndrome: The role of flecainide challenge. Circulation 2000, 102, 945–947. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Napolitano, C.; Gasparini, M.; Pappone, C.; Della Bella, P.; Brignole, M.; Giordano, U.; Giovannini, T.; Menozzi, C.; Bloise, R.; et al. Clinical and genetic heterogeneity of right bundle branch block and ST-segment elevation syndrome: A prospective evaluation of 52 families. Circulation 2000, 102, 2509–2515. [Google Scholar] [CrossRef] [PubMed]
- Postma, A.V.; Denjoy, I.; Kamblock, J.; Alders, M.; Lupoglazoff, J.M.; Vaksmann, G.; Dubosq-Bidot, L.; Sebillon, P.; Mannens, M.M.; Guicheney, P.; et al. Catecholaminergic polymorphic ventricular tachycardia: RYR2 mutations, bradycardia, and follow up of the patients. J. Med. Genet. 2005, 42, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Van der Werf, C.; Nederend, I.; Hofman, N.; van Geloven, N.; Ebink, C.; Frohn-Mulder, I.M.; Alings, A.M.; Bosker, H.A.; Bracke, F.A.; van den Heuvel, F.; et al. Familial evaluation in catecholaminergic polymorphic ventricular tachycardia: Disease penetrance and expression in cardiac ryanodine receptor mutation-carrying relatives. Circ. Arrhythm. Electrophysiol. 2012, 5, 748–756. [Google Scholar] [CrossRef] [PubMed]
- Benito, B.; Sarkozy, A.; Mont, L.; Henkens, S.; Berruezo, A.; Tamborero, D.; Arzamendi, D.; Berne, P.; Brugada, R.; Brugada, P.; et al. Gender differences in clinical manifestations of Brugada syndrome. J. Am. Coll. Cardiol. 2008, 52, 1567–1573. [Google Scholar] [CrossRef] [PubMed]
- Mazzanti, A.; Kanthan, A.; Monteforte, N.; Memmi, M.; Bloise, R.; Novelli, V.; Miceli, C.; O’Rourke, S.; Borio, G.; Zienciuk-Krajka, A.; et al. Novel insight into the natural history of short QT syndrome. J. Am. Coll. Cardiol. 2014, 63, 1300–1308. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Napolitano, C.; Memmi, M.; Colombi, B.; Drago, F.; Gasparini, M.; DeSimone, L.; Coltorti, F.; Bloise, R.; Keegan, R.; et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation 2002, 106, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Probst, V.; Denjoy, I.; Meregalli, P.G.; Amirault, J.C.; Sacher, F.; Mansourati, J.; Babuty, D.; Villain, E.; Victor, J.; Schott, J.J.; et al. Clinical aspects and prognosis of Brugada syndrome in children. Circulation 2007, 115, 2042–2048. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Du, Y.; Yang, P.; Lin, S.; Xi, Y.; Yang, Z.; Ma, A. Age-dependent alterations of voltage-gated Na(+) channel isoforms in rat sinoatrial node. Mech. Ageing Dev. 2015, 152, 80–90. [Google Scholar] [CrossRef] [PubMed]
- McMillan, M.R.; Day, T.G.; Bartsota, M.; Mead-Regan, S.; Bryant, R.; Mangat, J.; Abrams, D.; Lowe, M.; Kaski, J.P. Feasibility and outcomes of ajmaline provocation testing for Brugada syndrome in children in a specialist paediatric inherited cardiovascular diseases centre. Open Heart 2014, 1, e000023. [Google Scholar] [CrossRef] [PubMed]
- Conte, G.; de Asmundis, C.; Ciconte, G.; Julia, J.; Sieira, J.; Chierchia, G.B.; Brugada, P. Follow-up from childhood to adulthood of individuals with family history of Brugada syndrome and normal electrocardiograms. JAMA 2014, 312, 2039–2041. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, C.; Novelli, V.; Francis, M.D.; Priori, S.G. Genetic modulators of the phenotype in the long QT syndrome: State of the art and clinical impact. Curr. Opin. Genet. Dev. 2015, 33, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Bezzina, C.R.; Rook, M.B.; Groenewegen, W.A.; Herfst, L.J.; van der Wal, A.C.; Lam, J.; Jongsma, H.J.; Wilde, A.A.; Mannens, M.M. Compound heterozygosity for mutations (W156X and R225W) in SCN5A associated with severe cardiac conduction disturbances and degenerative changes in the conduction system. Circ. Res. 2003, 92, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Risgaard, B.; Jabbari, R.; Refsgaard, L.; Holst, A.G.; Haunso, S.; Sadjadieh, A.; Winkel, B.G.; Olesen, M.S.; Tfelt-Hansen, J. High prevalence of genetic variants previously associated with Brugada syndrome in new exome data. Clin. Genet. 2013, 84, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Refsgaard, L.; Holst, A.G.; Sadjadieh, G.; Haunso, S.; Nielsen, J.B.; Olesen, M.S. High prevalence of genetic variants previously associated with LQT syndrome in new exome data. Eur. J. Hum. Genet. 2012, 20, 905–908. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, O.; Sarquella-Brugada, G.; Mademont-Soler, I.; Allegue, C.; Cesar, S.; Ferrer-Costa, C.; Coll, M.; Mates, J.; Iglesias, A.; Brugada, J.; et al. Identification of Genetic Alterations, as Causative Genetic Defects in Long QT Syndrome, Using Next Generation Sequencing Technology. PLoS ONE 2014, 9, e114894. [Google Scholar] [CrossRef] [PubMed]
- Mademont-Soler, I.; Pinsach-Abuin, M.L.; Riuro, H.; Mates, J.; Perez-Serra, A.; Coll, M.; Porres, J.M.; Del Olmo, B.; Iglesias, A.; Selga, E.; et al. Large Genomic Imbalances in Brugada Syndrome. PLoS ONE 2016, 11, e0163514. [Google Scholar] [CrossRef] [PubMed]
- Nishio, Y.; Makiyama, T.; Itoh, H.; Sakaguchi, T.; Ohno, S.; Gong, Y.Z.; Yamamoto, S.; Ozawa, T.; Ding, W.G.; Toyoda, F.; et al. D85N, a KCNE1 polymorphism, is a disease-causing gene variant in long QT syndrome. J. Am. Coll. Cardiol. 2009, 54, 812–819. [Google Scholar] [CrossRef] [PubMed]
- Bezzina, C.R.; Barc, J.; Mizusawa, Y.; Remme, C.A.; Gourraud, J.B.; Simonet, F.; Verkerk, A.O.; Schwartz, P.J.; Crotti, L.; Dagradi, F.; et al. Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat. Genet. 2013. [Google Scholar] [CrossRef]
- Amin, A.S.; Giudicessi, J.R.; Tijsen, A.J.; Spanjaart, A.M.; Reckman, Y.J.; Klemens, C.A.; Tanck, M.W.; Kapplinger, J.D.; Hofman, N.; Sinner, M.F.; et al. Variants in the 3’ untranslated region of the KCNQ1-encoded Kv7.1 potassium channel modify disease severity in patients with type 1 long QT syndrome in an allele-specific manner. Eur. Heart J. 2012, 33, 714–723. [Google Scholar] [CrossRef] [PubMed]
- Crotti, L.; Lahtinen, A.M.; Spazzolini, C.; Mastantuono, E.; Monti, M.C.; Morassutto, C.; Parati, G.; Heradien, M.; Goosen, A.; Lichtner, P.; et al. Genetic Modifiers for the Long-QT Syndrome: How Important Is the Role of Variants in the 3’ Untranslated Region of KCNQ1? Circ. Cardiovasc. Genet. 2016, 9, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Bronze-da-Rocha, E. MicroRNAs expression profiles in cardiovascular diseases. BioMed Res. Int. 2014, 2014, 985408. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Non-Genetic Modifiers | |
|---|---|
| Gender | Worse prognosis in males |
| Age | Severe phenotypes in early manifestations, age-related penetrance to the ajmaline provocation test |
| Exogenous factors | Fever, excessive alcohol and large meals |
| Genetic Modifiers | |
| Coding variants—Rare Variants (MAF < 1%) | Additive effect of multiple independent mutations |
| CNVs | |
| New genes involved with the disease | |
| Coding variants—Common Variants (MAF > 1%) | Second hits (i.e., p.H558R_SCN5A; p.K897T_KCNH2; p.D85N_KCNE1 |
| Non-coding variants | 5′UTR and 3′UTR variants, microRNAs |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coll, M.; Pérez-Serra, A.; Mates, J.; Del Olmo, B.; Puigmulé, M.; Fernandez-Falgueras, A.; Iglesias, A.; Picó, F.; Lopez, L.; Brugada, R.; et al. Incomplete Penetrance and Variable Expressivity: Hallmarks in Channelopathies Associated with Sudden Cardiac Death. Biology 2018, 7, 3. https://doi.org/10.3390/biology7010003
Coll M, Pérez-Serra A, Mates J, Del Olmo B, Puigmulé M, Fernandez-Falgueras A, Iglesias A, Picó F, Lopez L, Brugada R, et al. Incomplete Penetrance and Variable Expressivity: Hallmarks in Channelopathies Associated with Sudden Cardiac Death. Biology. 2018; 7(1):3. https://doi.org/10.3390/biology7010003
Chicago/Turabian StyleColl, Monica, Alexandra Pérez-Serra, Jesus Mates, Bernat Del Olmo, Marta Puigmulé, Anna Fernandez-Falgueras, Anna Iglesias, Ferran Picó, Laura Lopez, Ramon Brugada, and et al. 2018. "Incomplete Penetrance and Variable Expressivity: Hallmarks in Channelopathies Associated with Sudden Cardiac Death" Biology 7, no. 1: 3. https://doi.org/10.3390/biology7010003