NF-κB Activation Exacerbates, but Is not Required for Murine Bmpr2-Related Pulmonary Hypertension

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
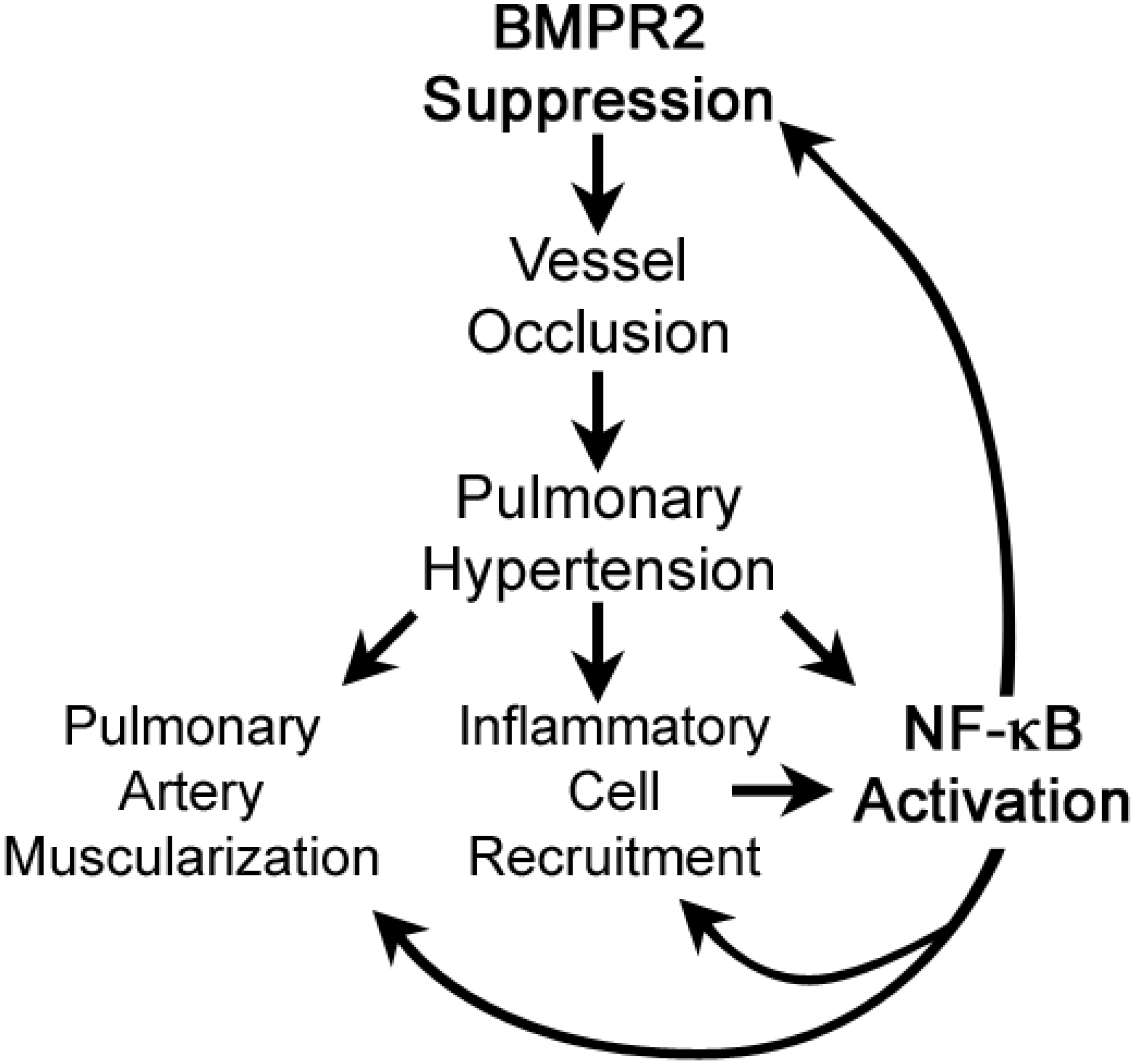
Abstract
:1. Introduction
2. Experimental Section
2.1. Electrophoretic Mobility Shift Assay (EMSA)
2.2. IP Lipopolysaccharide (LPS)
2.3. Western Blotting
2.4. Quantitative RT-PCR
2.5. Luciferase Assays
2.6. Echocardiography and Hemodynamic Phenotyping
2.7. dnIkB Experiments
2.8. RelA Virus Administration
2.9. IKTA Mice
2.10. Morphometry
2.11. Statistical Methods
3. Results and Discussions
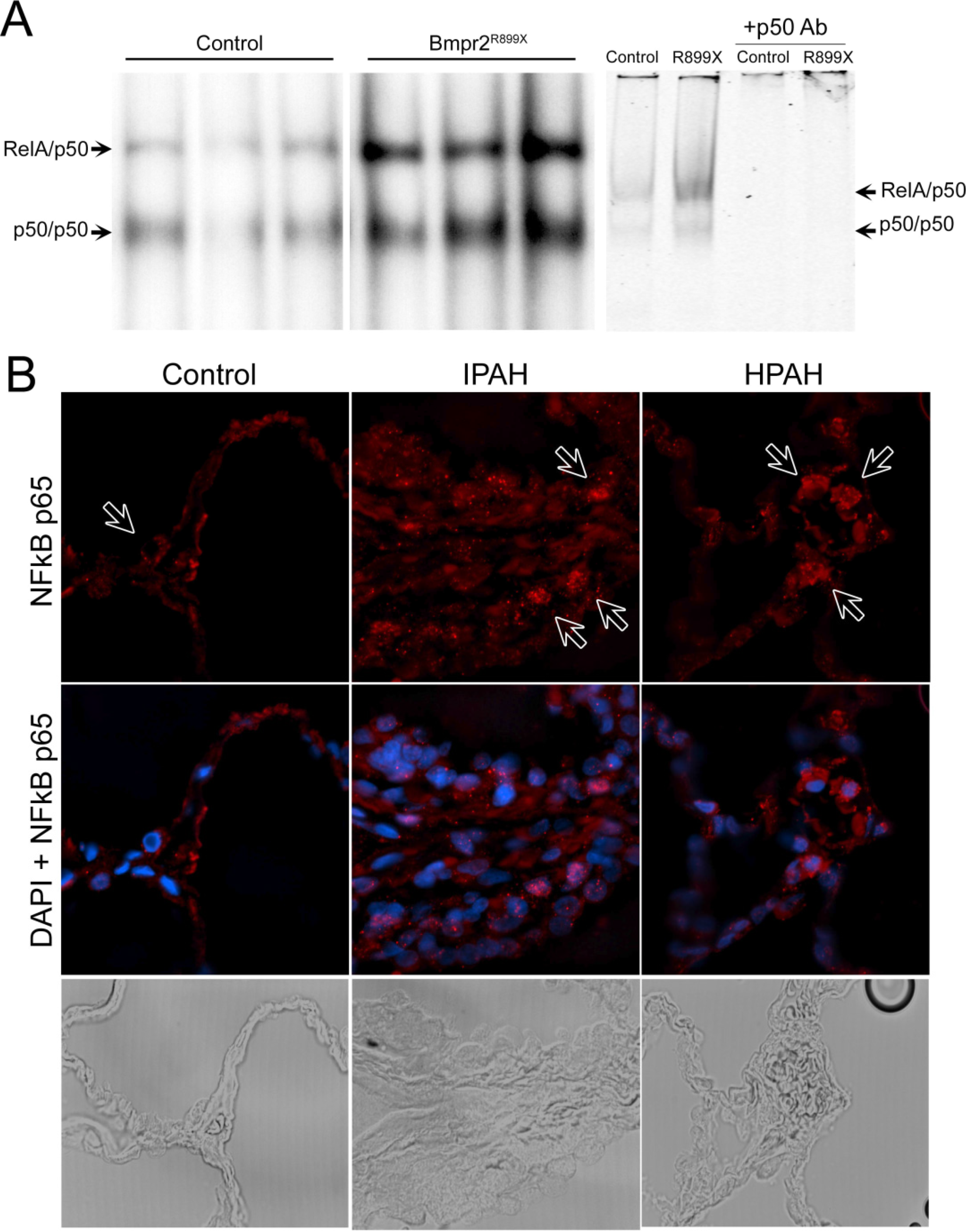
3.1. NF-κB Activation is Present in Human and Murine Pulmonary Hypertension
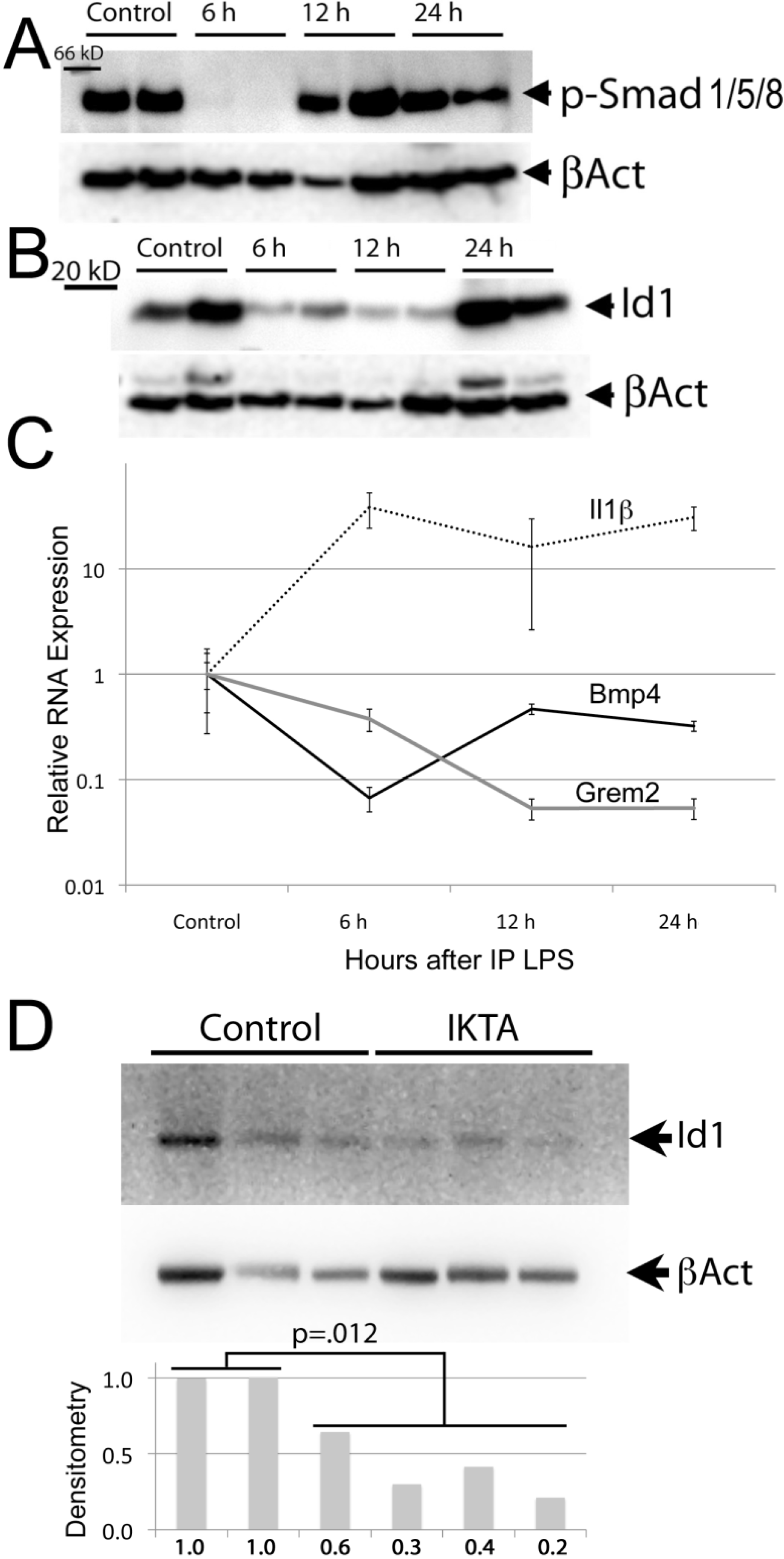
3.2. The BMP Pathway is Suppressed during Acute Inflammation


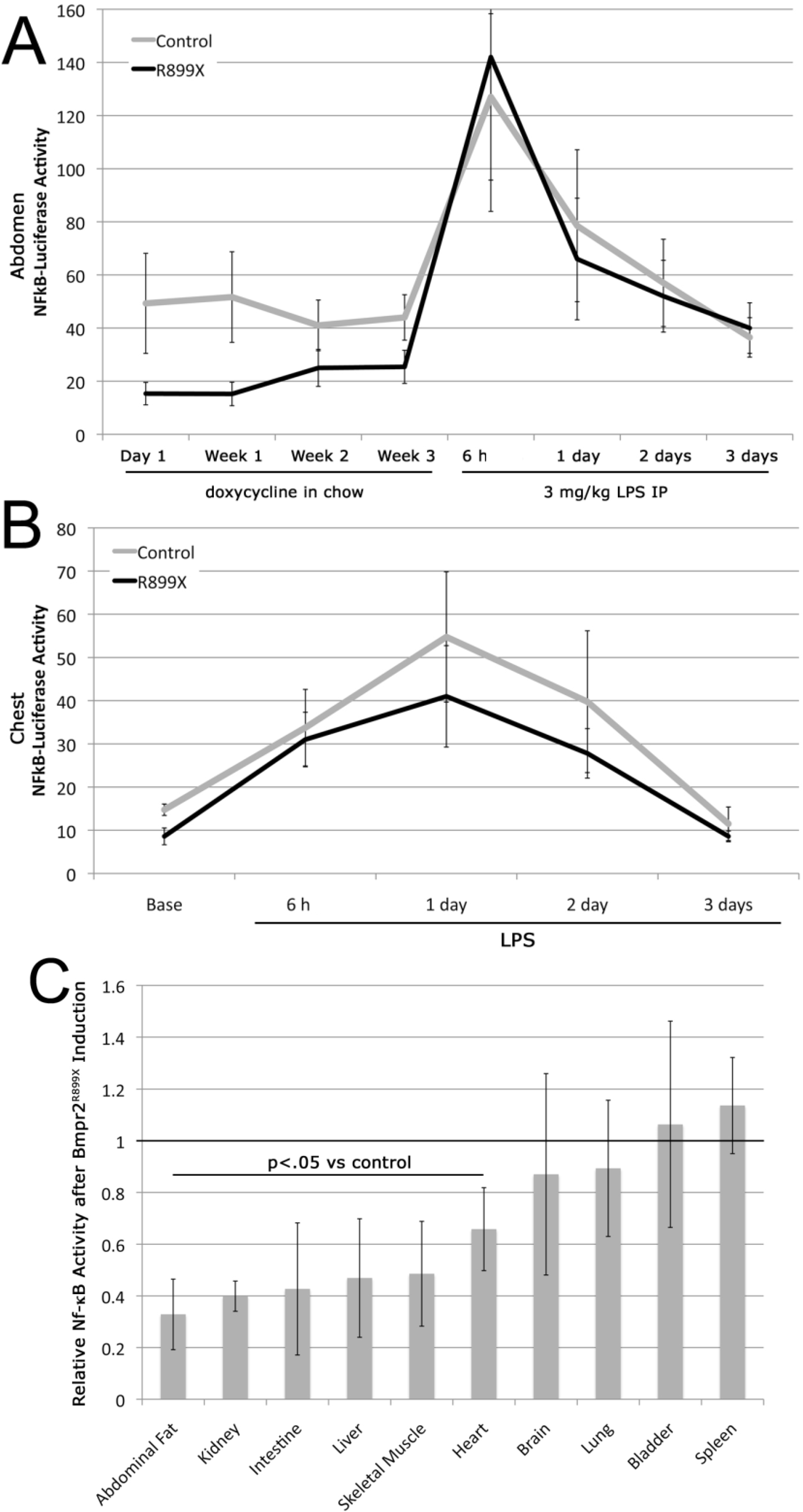
3.3. Expression of the Bmpr2R899X Transgene Suppresses Basal NF-κB Activation
3.4. NF-κB Activation Is not Required for Murine Bmpr2-Related PH


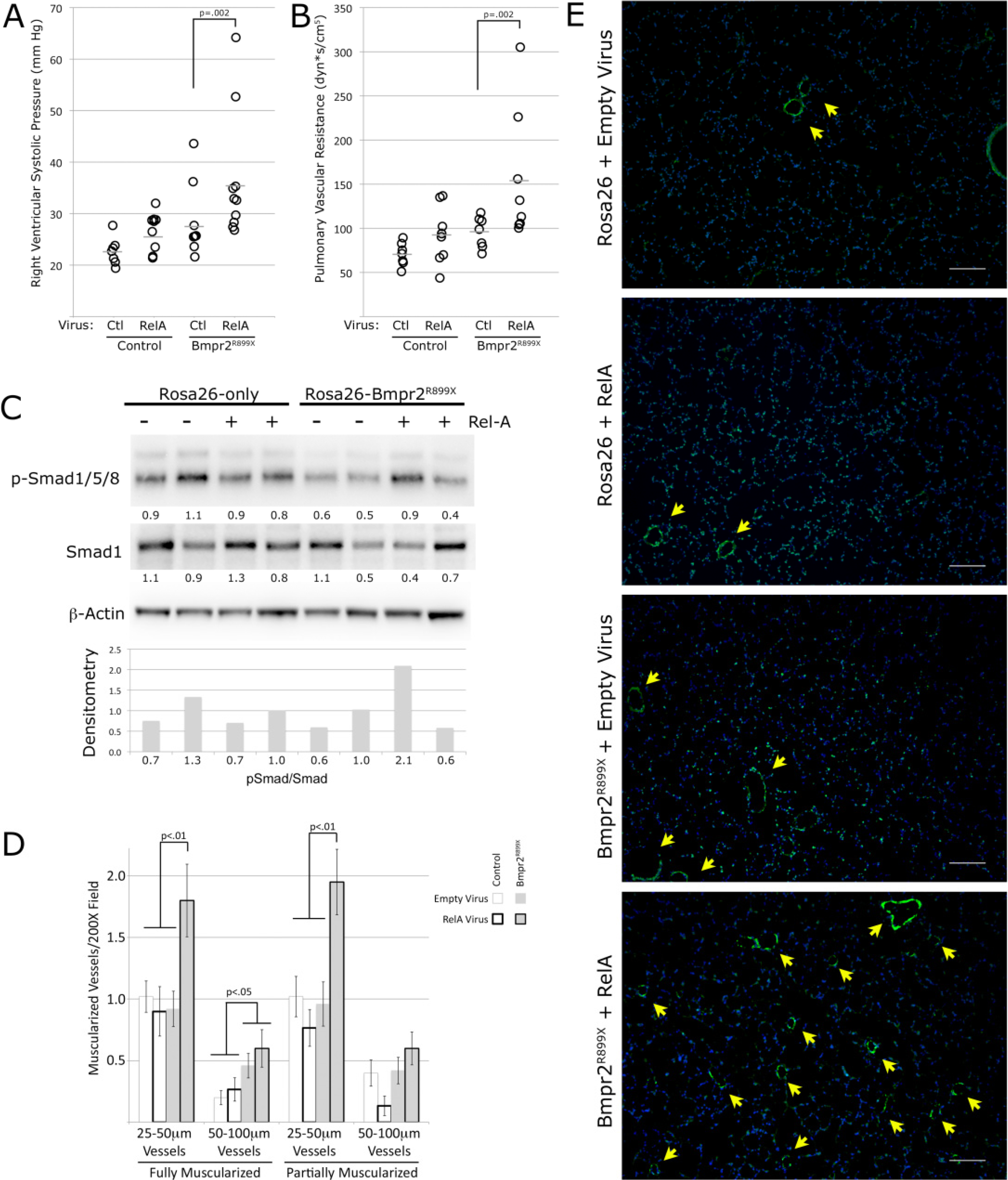
3.5. NF-κB Activation Exacerbates Murine Bmpr2-Related PH

3.5. Discussion

4. Conclusions
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Stenmark, K.R.; Meyrick, B.; Galie, N.; Mooi, W.J.; McMurtry, I.F. Animal models of pulmonary arterial hypertension: The hope for etiological discovery and pharmacological cure. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L1013–L1032. [Google Scholar] [CrossRef]
- Moosmang, S.; Schulla, V.; Welling, A.; Feil, R.; Feil, S.; Wegener, J.W.; Hofmann, F.; Klugbauer, N. Dominant role of smooth muscle L-type calcium channel Cav1.2 for blood pressure regulation. EMBO J. 2003, 22, 6027–6034. [Google Scholar] [CrossRef]
- Bonnet, S.; Michelakis, E.D.; Porter, C.J.; Andrade-Navarro, M.A.; Thebaud, B.; Haromy, A.; Harry, G.; Moudgil, R.; McMurtry, M.S.; Weir, E.K.; et al. An abnormal mitochondrial-hypoxia inducible factor-1alpha-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: Similarities to human pulmonary arterial hypertension. Circulation 2006, 113, 2630–2641. [Google Scholar] [CrossRef]
- Maurer, B.; Reich, N.; Juengel, A.; Kriegsmann, J.; Gay, R.E.; Schett, G.; Michel, B.A.; Gay, S.; Distler, J.H.; Distler, O. Fra-2 transgenic mice as a novel model of pulmonary hypertension associated with systemic sclerosis. Ann. Rheum. Dis. 2012, 71, 1382–1387. [Google Scholar] [CrossRef] [Green Version]
- Crosby, A.; Jones, FM.; Southwood, M.; Stewart, S.; Schermuly, R.; Butrous, G.; Dunne, D.W.; Morrell, N.W. Pulmonary vascular remodeling correlates with lung eggs and cytokines in murine schistosomiasis. Am. J. Respir. Crit. Care Med. 2010, 81, 279–288. [Google Scholar]
- Cogan, J.D.; Vnencak-Jones, C.L.; Phillips, J.A., 3rd.; Lane, K.B.; Wheeler, L.A.; Robbins, I.M.; Garrison, G.; Hedges, L.K.; Loyd, J.E. Gross BMPR2 gene rearrangements constitute a new cause for primary pulmonary hypertension. Genet. Med. 2005, 7, 169–174. [Google Scholar] [CrossRef]
- Lane, K.B.; Machado, R.D.; Pauciulo, M.W.; Thomson, J.R.; Phillips, J.A., 3rd.; Loyd, J.E.; Nichols, W.C.; Trembath, R.C. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat. Genet. 2000, 26, 81–84. [Google Scholar] [CrossRef]
- Austin, E.D.; Menon, S.; Hemnes, A.R.; Robinson, L.R.; Talati, M.; Fox, K.L.; Cogan, J.D.; Hamid, R.; Hedges, L.K.; Robbins, I.; et al. Idiopathic and heritable PAH perturb common molecular pathways, correlated with increased MSX1 inexpression. Pulm. Circ. 2011, 1, 389–398. [Google Scholar] [CrossRef]
- Tada, Y.; Majka, S.; Carr, M.; Harral, J.; Crona, D.; Kuriyama, T.; West, J. Molecular effects of loss of BMPR2 signaling in smooth muscle in a transgenic mouse model of PAH. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L1556–L1563. [Google Scholar] [CrossRef]
- West, J.; Harral, J.; Lane, K.; Deng, Y.; Ickes, B.; Crona, D.; Albu, S.; Stewart, D.; Fagan, K. Mice expressing BMPR2R899X transgene in smooth muscle develop pulmonary vascular lesions. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 295, L744–L755. [Google Scholar] [CrossRef]
- Majka, S.; Hagen, M.; Blackwell, T.; Harral, J.; Johnson, J.A.; Gendron, R.; Paradis, H.; Crona, D.; Loyd, J.E.; Nozik-Grayck, E.; et al. Physiologic and molecular consequences of endothelial Bmpr2 mutation. Respir. Res. 2011, 12, 84. [Google Scholar] [CrossRef]
- Talati, M.; West, J.; Blackwell, T.R.; Loyd, J.E.; Meyrick, B. BMPR2 mutation alters the lung macrophage endothelin-1 cascade in a mouse model and patients with heritable pulmonary artery hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 299, L363–L373. [Google Scholar] [CrossRef]
- Hagen, M.; Fagan, K.; Steudel, W.; Carr, M.; Lane, K.; Rodman, D.M.; West, J. Interaction of interleukin-6 and the BMP pathway in pulmonary smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L1473–L1479. [Google Scholar] [CrossRef]
- Tuder, R.M.; Groves, B.; Badesch, D.B.; Voelkel, N.F. Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hypertension. Am. J. Pathol. 1994, 144, 275–285. [Google Scholar]
- Perros, F.; Dorfmuller, P.; Montani, D.; Hammad, H.; Waelput, W.; Girerd, B.; Raymond, N.; Mercier, O.; Mussot, S.; Cohen-Kaminsky, S.; et al. Pulmonary lymphoid neogenesis in idiopathic pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2012, 185, 311–321. [Google Scholar] [CrossRef]
- Tamosiuniene, R.; Tian, W.; Dhillon, G.; Wang, L.; Sung, Y.K.; Gera, L.; Patterson, A.J.; Agrawal, R.; Rabinovitch, M.; Ambler, K.; et al. Regulatory T cells limit vascular endothelial injury and prevent pulmonary hypertension. Circ. Res. 2011, 109, 867–879. [Google Scholar] [CrossRef]
- Yu, Y.; Keller, S.H.; Remillard, C.V.; Safrina, O.; Nicholson, A.; Zhang, S.L.; Jiang, W.; Vangala, N.; Landsberg, J.W.; Wang, J.Y.; et al. A functional single-nucleotide polymorphism in the TRPC6 gene promoter associated with idiopathic pulmonary arterial hypertension. Circulation 2009, 119, 2313–2322. [Google Scholar] [CrossRef]
- Sawada, H.; Saito, T.; Nickel, N.P.; Alastalo, T.P.; Glotzbach, J.P.; Chan, R.; Haghighat, L.; Fuchs, G.; Januszyk, M.; Cao, A.; et al. Reduced BMPR2 expression induces GM-CSF translation and macrophage recruitment in humans and mice to exacerbate pulmonary hypertension. J. Exp. Med. 2014, 211, 263–280. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. NF-kappaB in immunobiology. Cell Res. 2011, 21, 223–244. [Google Scholar] [CrossRef]
- Johnson, J.A.; Hemnes, A.R.; Perrien, D.S.; Schuster, M.; Robinson, L.J.; Gladson, S.; Loibner, H.; Bai, S.; Blackwell, T.R.; Tada, Y.; et al. Cytoskeletal defects in Bmpr2-associated pulmonary arterial hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L474–L484. [Google Scholar] [CrossRef]
- West, J. Cross talk between Smad, MAPK, and actin in the etiology of pulmonary arterial hypertension. Adv. Exp. Med. Biol. 2010, 661, 265–278. [Google Scholar] [CrossRef]
- Rudarakanchana, N.; Flanagan, J.A.; Chen, H.; Upton, P.D.; Machado, R.; Patel, D.; Trembath, R.C.; Morrell, N.W. Functional analysis of bone morphogenetic protein type II receptor mutations underlying primary pulmonary hypertension. Hum. Mol. Genet. 2002, 11, 1517–1525. [Google Scholar] [CrossRef]
- Blackwell, T.S.; Blackwell, T.R.; Christman, J.W. Impaired activation of nuclear factor-kappaB in endotoxin-tolerant rats is associated with down-regulation of chemokine gene expression and inhibition of neutrophilic lung inflammation. J. Immunol. 1997, 158, 5934–5940. [Google Scholar]
- Sadikot, R.T.; Jansen, E.D.; Blackwell, T.R.; Zoia, O.; Yull, F.; Christman, J.W.; Blackwell, T.S. High-dose dexamethasone accentuates nuclear factor-kappa b activation in endotoxin-treated mice. Am. J. Respir. Crit. Care Med. 2001, 164, 873–878. [Google Scholar] [CrossRef]
- Sadikot, R.T.; Han, W.; Everhart, M.B.; Zoia, O.; Peebles, R.S.; Jansen, E.D.; Yull, F.E.; Christman, J.W.; Blackwell, T.S. Selective I kappa B kinase expression in airway epithelium generates neutrophilic lung inflammation. J. Immunol. 2003, 170, 1091–1098. [Google Scholar] [CrossRef]
- Spurney, C.F.; Knoblach, S.; Pistilli, E.E.; Nagaraju, K.; Martin, G.R.; Hoffman, E.P. Dystrophin-deficient cardiomyopathy in mouse: Expression of Nox4 and Lox are associated with fibrosis and altered functional parameters in the heart. Neuromuscul. Disord. 2008, 18, 371–381. [Google Scholar] [CrossRef]
- Tanaka, N.; Dalton, N.; Mao, L.; Rockman, H.A.; Peterson, K.L.; Gottshall, K.R.; Hunter, J.J.; Chien, K.R.; Ross, J., Jr. Transthoracic echocardiography in models of cardiac disease in the mouse. Circulation 1996, 94, 1109–1117. [Google Scholar] [CrossRef]
- Cheng, D.S.; Han, W.; Chen, S.M.; Sherrill, T.P.; Chont, M.; Park, G.Y.; Sheller, J.R.; Polosukhin, V.V.; Christman, J.W.; Yull, F.E.; et al. Airway epithelium controls lung inflammation and injury through the NF-kappa B pathway. J. Immunol. 2007, 178, 6504–6513. [Google Scholar] [CrossRef]
- Zaynagetdinov, R.; Stathopoulos, G.T.; Sherrill, T.P.; Cheng, D.S.; McLoed, A.G.; Ausborn, J.A.; Polosukhin, V.V.; Connelly, L.; Zhou, W.; Fingleton, B.; et al. Epithelial nuclear factor-kappaB signaling promotes lung carcinogenesis via recruitment of regulatory T lymphocytes. Oncogene 2012, 31, 3164–3176. [Google Scholar] [CrossRef]
- Sawada, H.; Mitani, Y.; Maruyama, J.; Jiang, B.H.; Ikeyama, Y.; Dida, F.A.; Yamamoto, H.; Imanaka-Yoshida, K.; Shimpo, H.; Mizoguchi, A.; et al. A nuclear factor-kappaB inhibitor pyrrolidine dithiocarbamate ameliorates pulmonary hypertension in rats. Chest 2007, 132, 1265–1274. [Google Scholar] [CrossRef]
- Price, L.C.; Caramori, G.; Perros, F.; Meng, C.; Gambaryan, N.; Dorfmuller, P.; Montani, D.; Casolari, P.; Zhu, J.; Dimopoulos, K.; et al. Nuclear factor kappa-B is activated in the pulmonary vessels of patients with end-stage idiopathic pulmonary arterial hypertension. PLoS One 2013, 8, e75415. [Google Scholar] [CrossRef]
- Bosmann, M.; Russkamp, N.F.; Ward, P.A. Fingerprinting of the TLR4-induced acute inflammatory response. Exp. Mol. Pathol. 2012, 93, 319–323. [Google Scholar] [CrossRef]
- West, J.; Gladson, S.; Johnson, J.; Blackwell, T.; Hemnes, A. BMPR2 Mutation Causes Defects in Glucocorticoid Receptor Shuttling. Am. J. Respir. Crit. Care Med. 2011, 183, A4955. [Google Scholar]
- Sorriento, D.; Santulli, G.; Fusco, A.; Anastasio, A.; Trimarco, B.; Iaccarino, G. Intracardiac injection of AdGRK5-NT reduces left ventricular hypertrophy by inhibiting NF-kappaB-dependent hypertrophic gene expression. Hypertension 2010, 56, 696–704. [Google Scholar] [CrossRef]
- Muraoka, R.S.; Bushdid, P.B.; Brantley, D.M.; Yull, F.E.; Kerr, L.D. Mesenchymal expression of nuclear factor-kappaB inhibits epithelial growth and branching in the embryonic chick lung. Dev. Biol. 2000, 225, 322–338. [Google Scholar] [CrossRef]
- Sadikot, R.T.; Zeng, H.; Joo, M.; Everhart, M.B.; Sherrill, T.P.; Li, B.; Cheng, D.S.; Yull, F.E.; Christman, J.W.; Blackwell, T.S. Targeted immunomodulation of the NF-kappaB pathway in airway epithelium impacts host defense against Pseudomonas aeruginosa. J. Immunol. 2006, 176, 4923–4930. [Google Scholar] [CrossRef]
- Ortiz, L.A.; Champion, H.C.; Lasky, J.A.; Gambelli, F.; Gozal, E.; Hoyle, G.W.; Beasley, M.B.; Hyman, A.L.; Friedman, M.; Kadowitz, P.J. Enalapril protects mice from pulmonary hypertension by inhibiting TNF-mediated activation of NF-kappaB and AP-1. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 282, L1209–L1221. [Google Scholar]
- Stacher, E.; Graham, B.B.; Hunt, J.M.; Gandjeva, A.; Groshong, S.D.; McLaughlin, V.V.; Jessup, M.; Grizzle, W.E.; Aldred, M.A.; Cool, C.D.; et al. Modern age pathology of pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2012, 186, 261–272. [Google Scholar] [CrossRef]
- Geraci, M.W.; Moore, M.; Gesell, T.; Yeager, M.E.; Alger, L.; Golpon, H.; Gao, B.; Loyd, J.E.; Tuder, R.M.; Voelkel, N.F. Gene expression patterns in the lungs of patients with primary pulmonary hypertension: A gene microarray analysis. Circ. Res. 2001, 88, 555–562. [Google Scholar] [CrossRef]
- Hamid, R.; Cogan, J.D.; Hedges, L.K.; Austin, E.; Phillips, J.A., 3rd.; Newman, J.H.; Loyd, J.E. Penetrance of pulmonary arterial hypertension is modulated by the expression of normal BMPR2 allele. Hum. Mutat. 2009, 30, 649–654. [Google Scholar] [CrossRef]
- Cogan, J.; Austin, E.; Hedges, L.; Womack, B.; West, J.; Loyd, J.; Hamid, R. Role of BMPR2 alternative splicing in heritable pulmonary arterial hypertension penetrance. Circulation 2012, 126, 1907–1916. [Google Scholar] [CrossRef]
- Talati, M.; West, J.; Zaynagetdinov, R.; Hong, C.C.; Han, W.; Blackwell, T.; Robinson, L.; Blackwell, T.S.; Lane, K. BMP Pathway Regulation of and by Macrophages. PLoS One 2014, 9, e94119. [Google Scholar] [CrossRef]
- Cool, C.D.; Voelkel, N.F.; Bull, T. Viral infection and pulmonary hypertension: Is there an association? Exp. Rev. Respir. Med. 2011, 5, 207–216. [Google Scholar] [CrossRef]
- Lu, M.; Lin, S.C.; Huang, Y.; Kang, Y.J.; Rich, R.; Lo, Y.C.; Myszka, D.; Han, J.; Wu, H. XIAP induces NF-kappaB activation via the BIR1/TAB1 interaction and BIR1 dimerization. Mol. Cell Biol. 2007, 26, 689–702. [Google Scholar]
- Matluk, N.; Rochira, J.A.; Karaczyn, A.; Adams, T.; Verdi, J.M. A role for NRAGE in NF-kappaB activation through the non-canonical BMP pathway. BMC Biol. 2010, 8, 7. [Google Scholar] [CrossRef]
- Humbert, M.; Monti, G.; Brenot, F.; Sitbon, O.; Portier, A.; Grangeot-Keros, L.; Duroux, P.; Galanaud, P.; Simonneau, G.; Emilie, D. Increased interleukin-1 and interleukin-6 serum concentrations in severe primary pulmonary hypertension. Am. J. Respir. Crit. Care Med. 1995, 151, 1628–1631. [Google Scholar] [CrossRef]
- Pinto, R.F.; Higuchi Mde, L.; Aiello, V.D. Decreased numbers of T-lymphocytes and predominance of recently recruited macrophages in the walls of peripheral pulmonary arteries from 26 patients with pulmonary hypertension secondary to congenital cardiac shunts. Cardiovasc. Pathol. 2004, 13, 268–275. [Google Scholar] [CrossRef]
- Itoh, T.; Nagaya, N.; Ishibashi-Ueda, H.; Kyotani, S.; Oya, H.; Sakamaki, F.; Kimura, H.; Nakanishi, N. Increased plasma monocyte chemoattractant protein-1 level in idiopathic pulmonary arterial hypertension. Respirology 2006, 11, 158–163. [Google Scholar] [CrossRef]
- Sanchez, O.; Marcos, E.; Perros, F.; Fadel, E.; Tu, L.; Humbert, M.; Dartevelle, P.; Simonneau, G.; Adnot, S.; Eddahibi, S. Role of endothelium-derived CC chemokine ligand 2 in idiopathic pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2007, 176, 1041–1047. [Google Scholar] [CrossRef]
- Steiner, M.K.; Syrkina, O.L.; Kolliputi, N.; Mark, E.J.; Hales, C.A.; Waxman, A.B. Interleukin-6 overexpression induces pulmonary hypertension. Circ. Res. 2009, 104, 236–244. [Google Scholar] [CrossRef]
- Courboulin, A.; Barrier, M.; Perreault, T.; Bonnet, P.; Tremblay, V.L.; Paulin, R.; Tremblay, E.; Lambert, C.; Jacob, M.H.; Bonnet, S.N.; et al. Plumbagin reverses proliferation and resistance to apoptosis in experimental PAH. Eur. Respir. J. 2012, 40, 618–629. [Google Scholar] [CrossRef]
- Lee, J.E.; Yang, Y.M.; Liang, F.X.; Gough, D.J.; Levy, D.E.; Sehgal, P.B. Nongenomic STAT5-dependent effects on Golgi apparatus and endoplasmic reticulum structure and function. Am. J. Physiol. Cell Physiol. 2012, 302, C804–C820. [Google Scholar] [CrossRef]
- Brown, R.D.; Ambler, S.K.; Li, M.; Sullivan, T.M.; Henry, L.N.; Crossno, J.T., Jr.; Long, C.S.; Garrington, T.P.; Stenmark, K.R. MAP kinase kinase kinase-2 (MEKK2) regulates hypertrophic remodeling of the right ventricle in hypoxia-induced pulmonary hypertension. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H269–H281. [Google Scholar] [CrossRef]
- Nasim, M.T.; Ogo, T.; Chowdhury, H.M.; Zhao, L.; Chen, C.N.; Rhodes, C.; Trembath, R.C. BMPR-II deficiency elicits pro-proliferative and anti-apoptotic responses through the activation of TGFbeta-TAK1-MAPK pathways in PAH. Hum. Mol. Genet. 2012, 21, 2548–2558. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Talati, M.; Mutlak, H.; Lane, K.B.; Han, W.; Hemnes, A.; Mutlak, O.; Blackwell, T.; Zaynagetdinov, R.; Blackwell, T.S.; West, J. NF-κB Activation Exacerbates, but Is not Required for Murine Bmpr2-Related Pulmonary Hypertension. Diseases 2014, 2, 148-167. https://doi.org/10.3390/diseases2020148
Talati M, Mutlak H, Lane KB, Han W, Hemnes A, Mutlak O, Blackwell T, Zaynagetdinov R, Blackwell TS, West J. NF-κB Activation Exacerbates, but Is not Required for Murine Bmpr2-Related Pulmonary Hypertension. Diseases. 2014; 2(2):148-167. https://doi.org/10.3390/diseases2020148
Chicago/Turabian StyleTalati, Megha, Haitham Mutlak, Kirk B. Lane, Wei Han, Anna Hemnes, Outi Mutlak, Tom Blackwell, Rinat Zaynagetdinov, Timothy S. Blackwell, and James West. 2014. "NF-κB Activation Exacerbates, but Is not Required for Murine Bmpr2-Related Pulmonary Hypertension" Diseases 2, no. 2: 148-167. https://doi.org/10.3390/diseases2020148