SUMOylation Regulates Transcription by the Progesterone Receptor A Isoform in a Target Gene Selective Manner

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasmids
2.2. Transcription Assays
2.3. Immunoblotting
2.4. Stable Cell Lines
2.5. Microarray Analysis
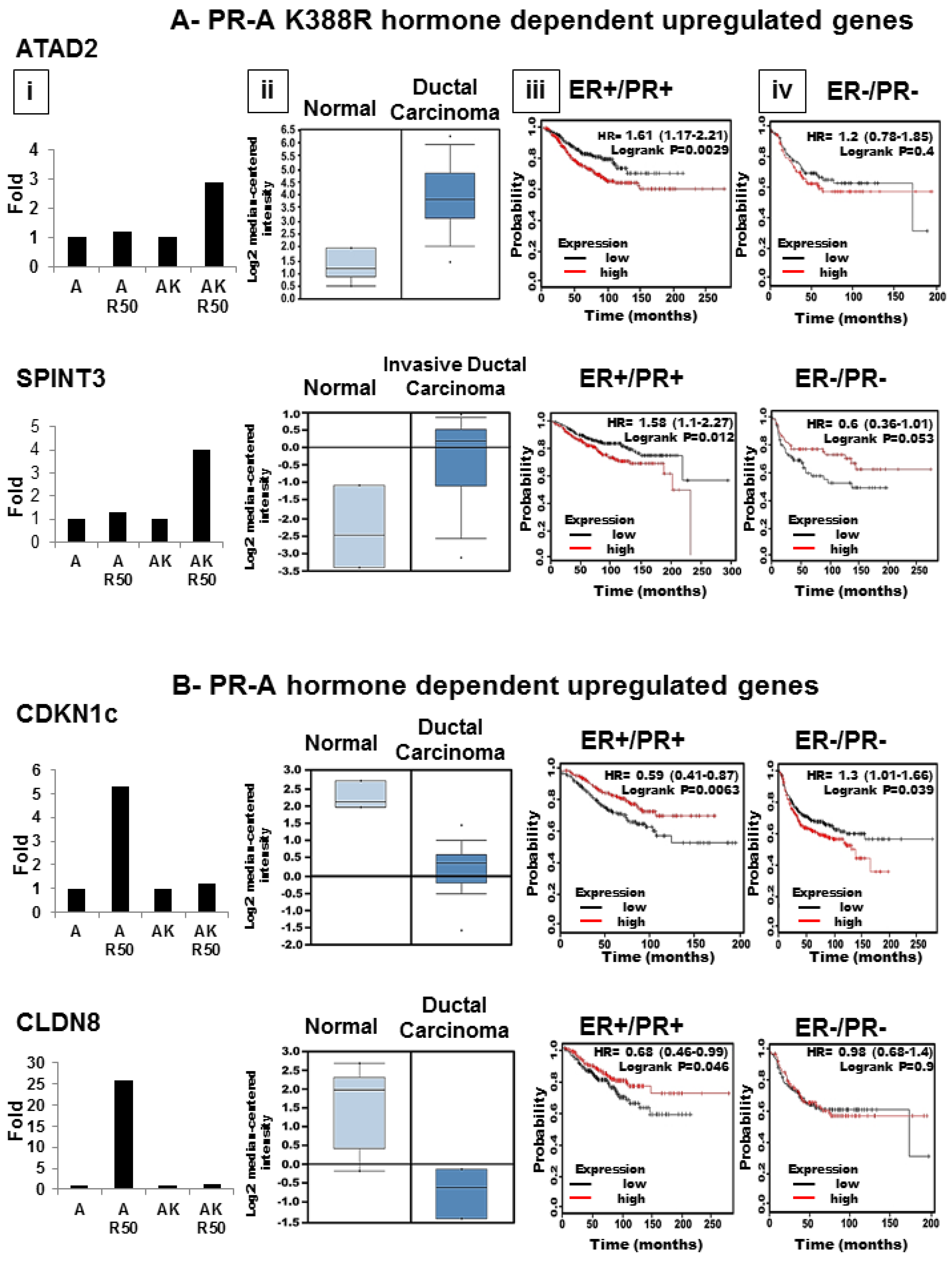
2.6. Gene Expression in Human Tumor Samples
3. Results
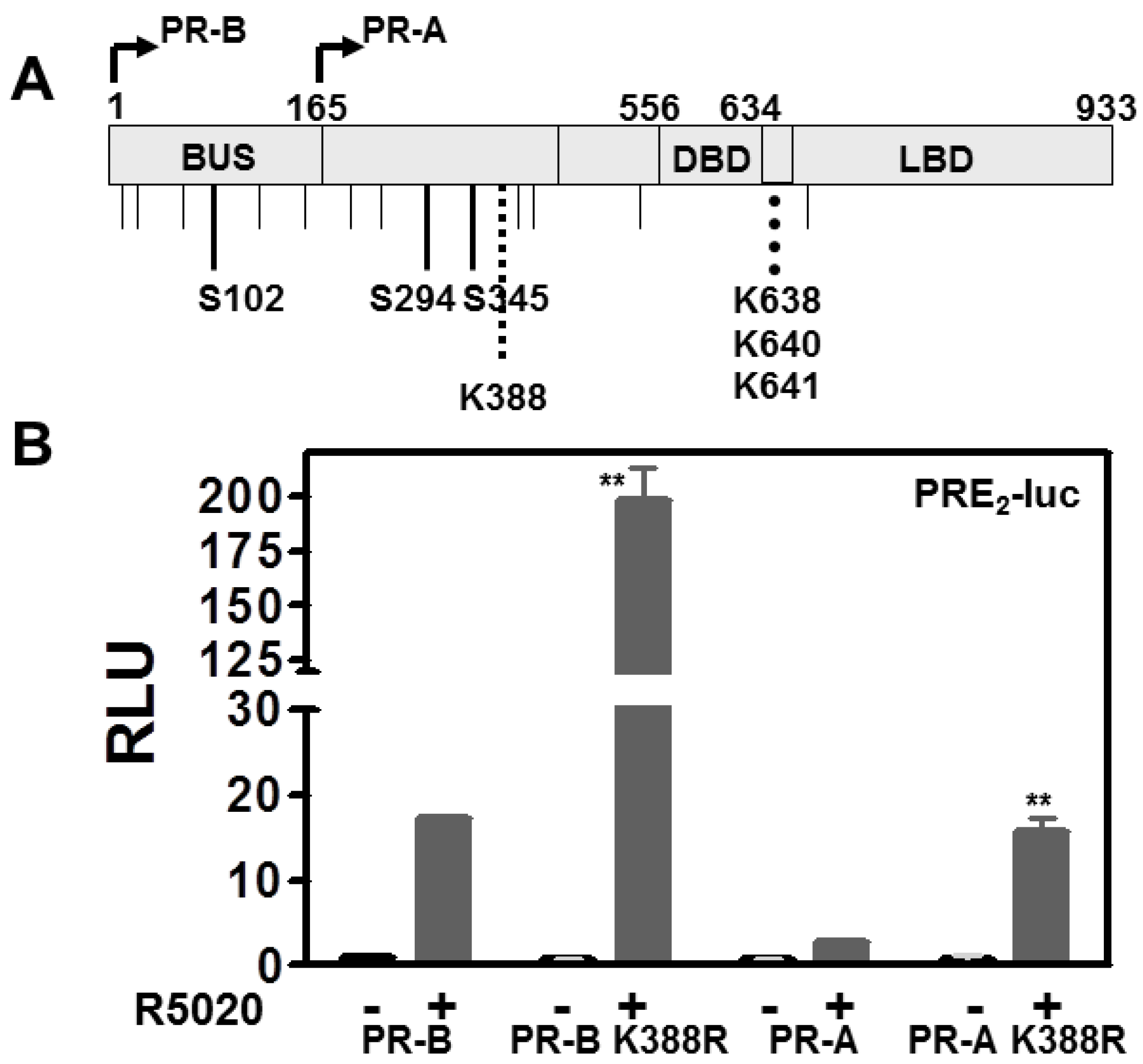
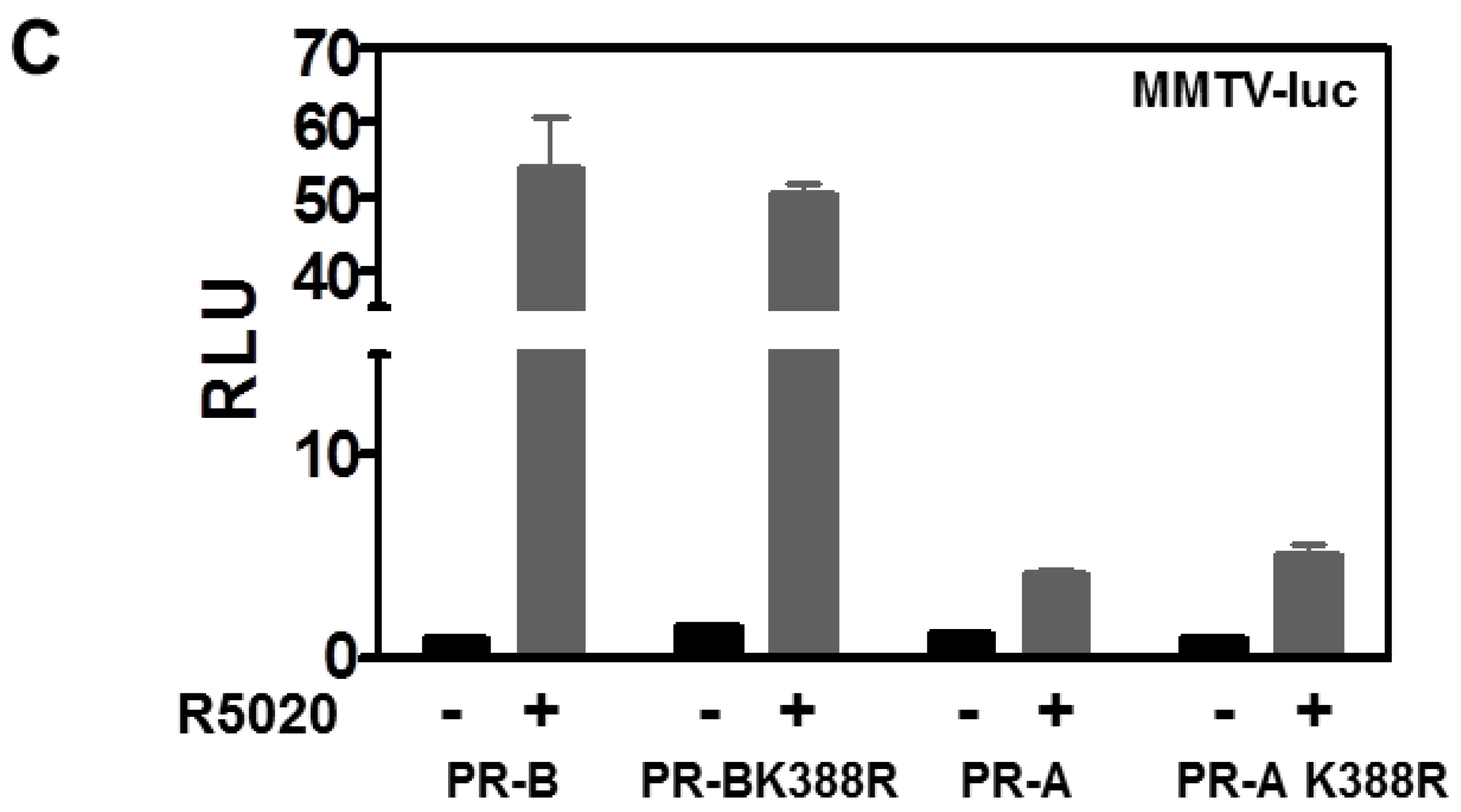
3.1. SUMOylation Suppresses PR Transcriptional Activity in a Promoter Dependent Manner
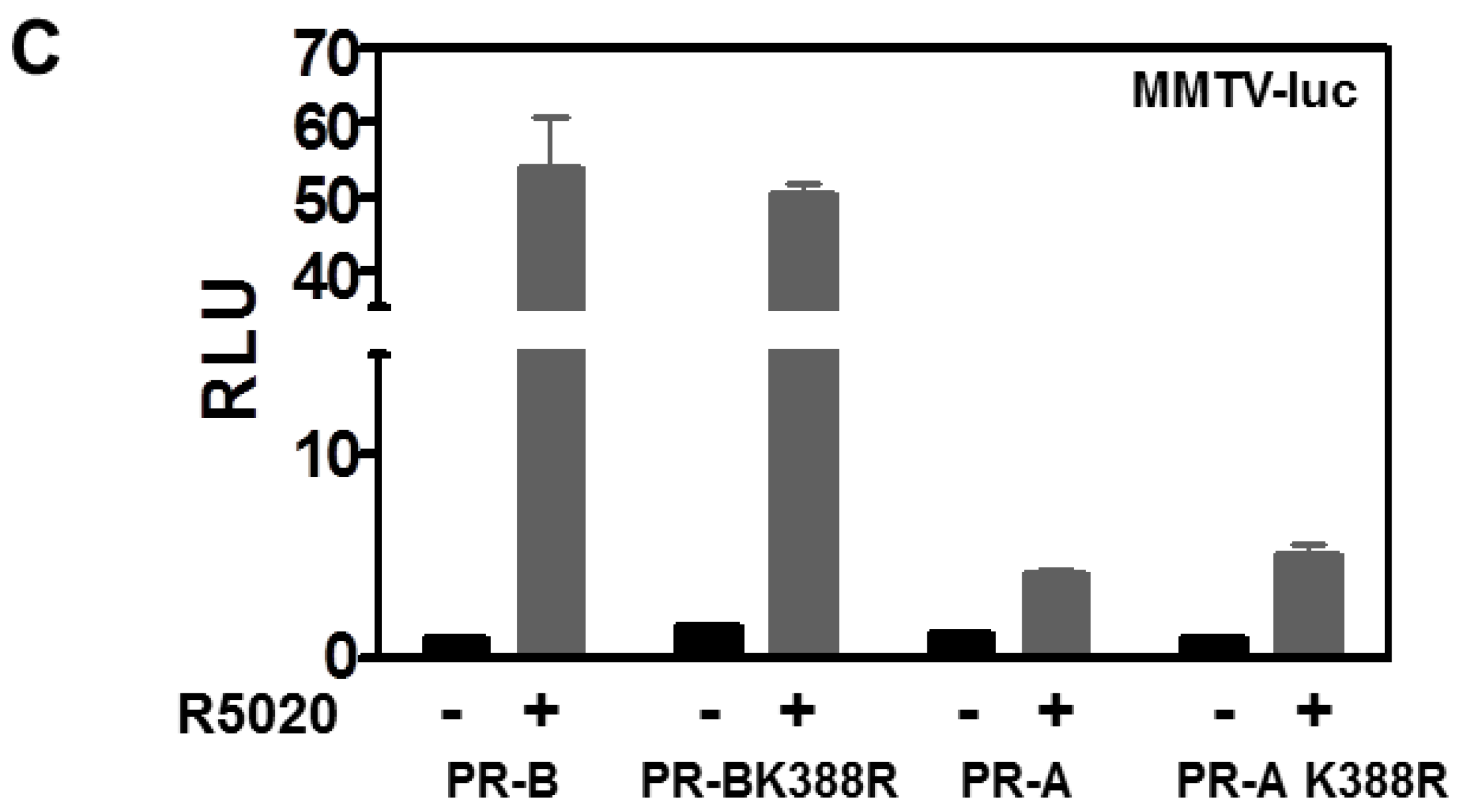
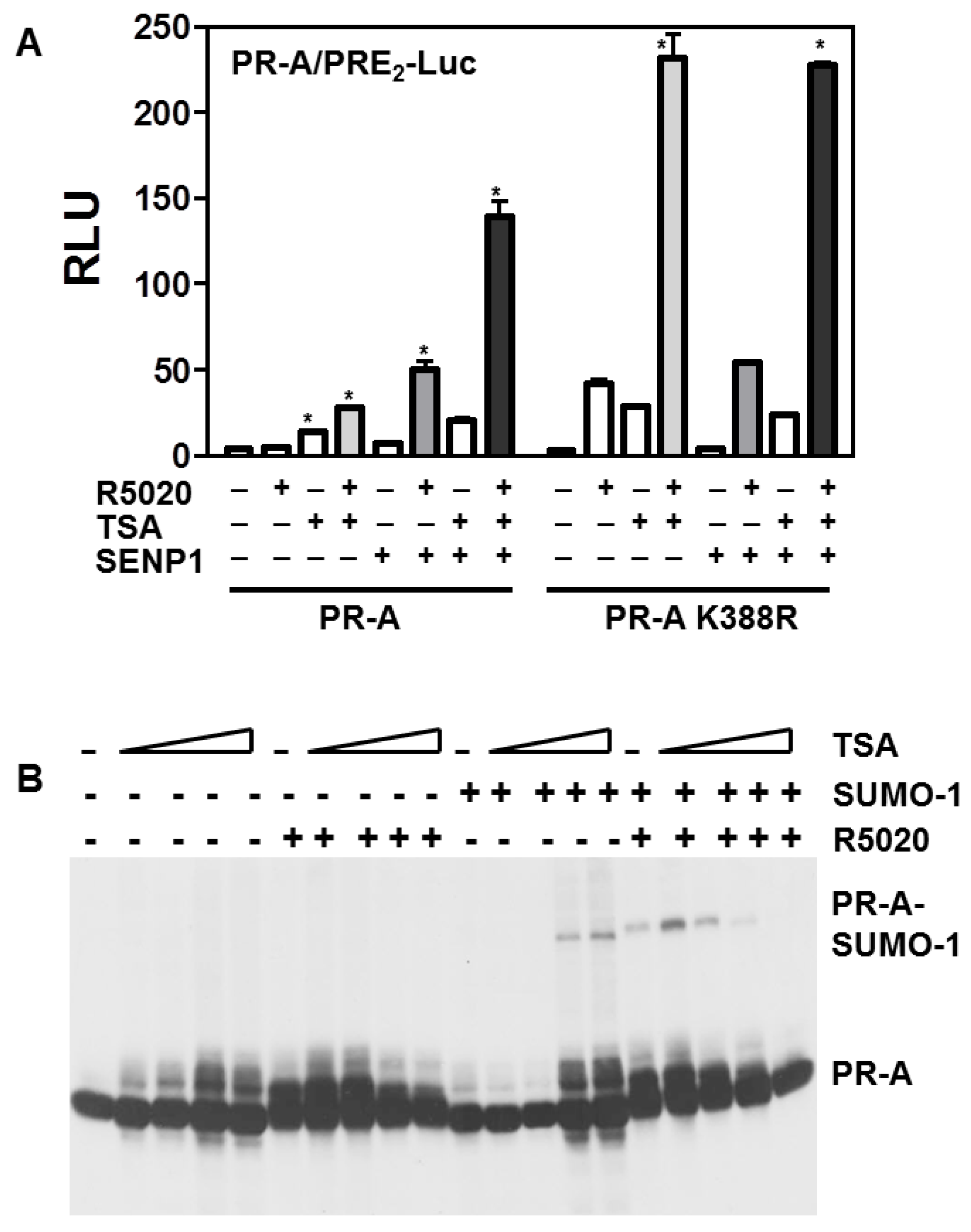
3.2. SENP1 deSUMOylates PR-A, Which Enhances Its Transcriptional Activity
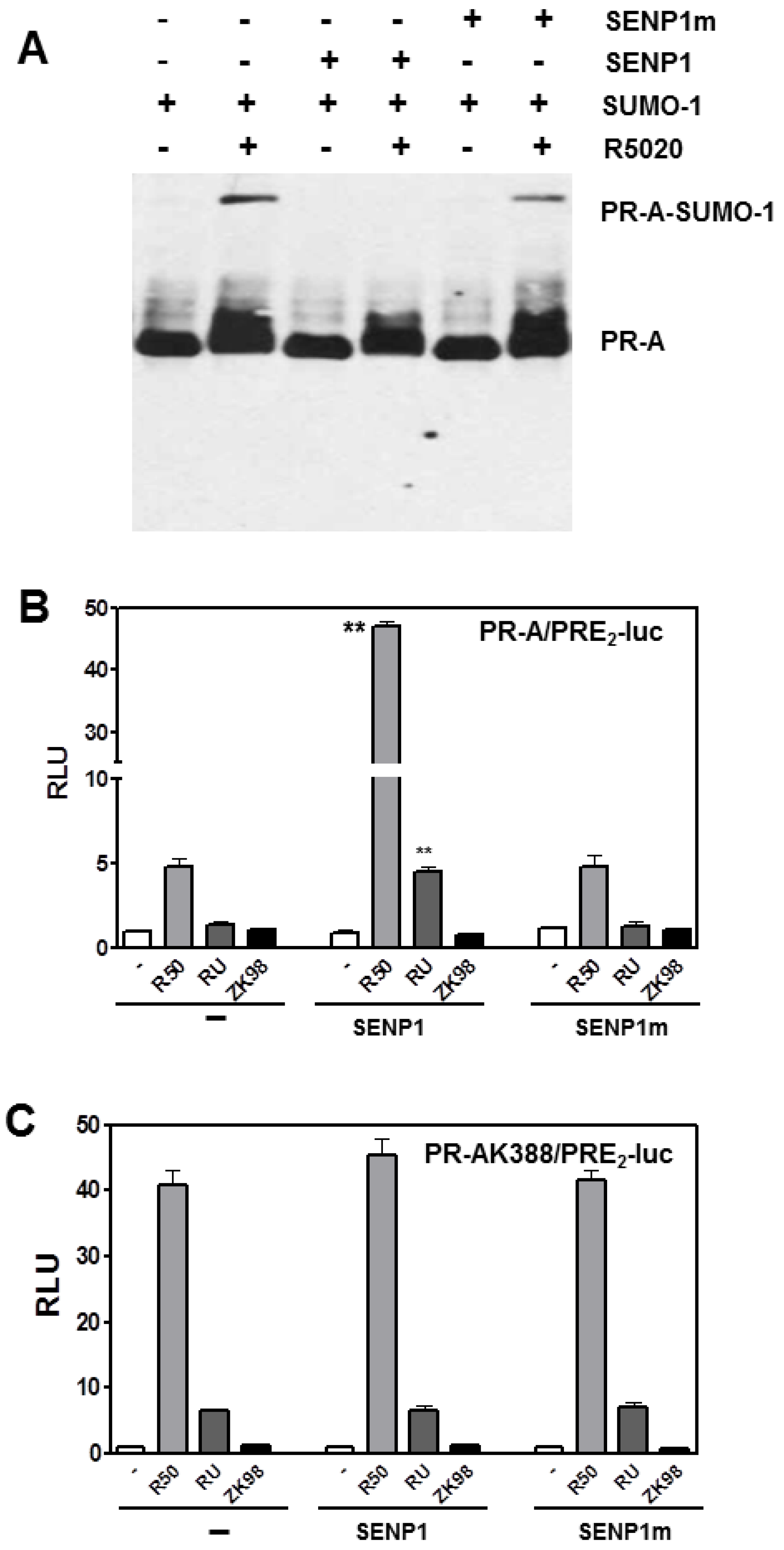
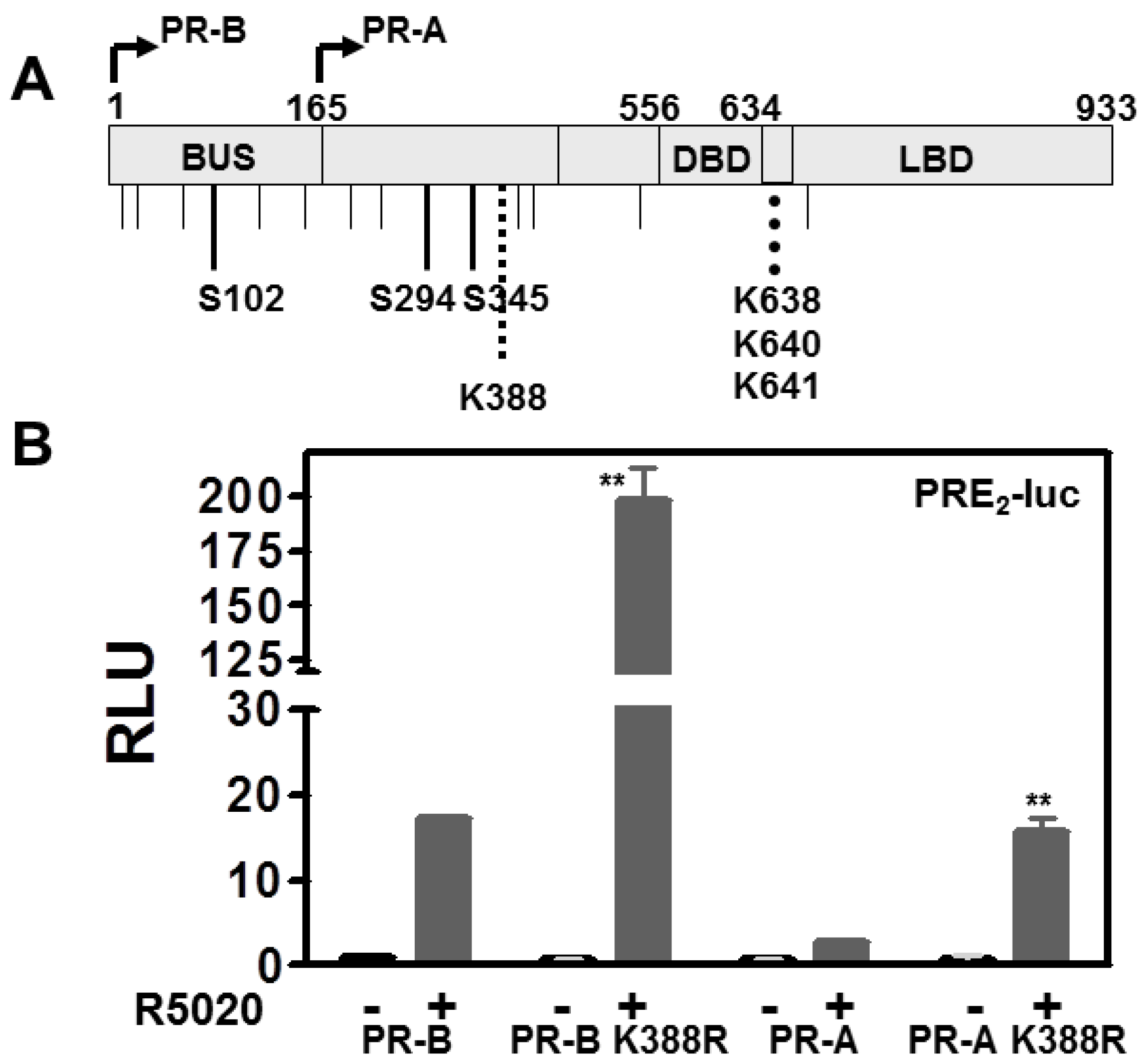
3.3. The PR DNA Binding Domain (DBD) Dimerization Interface Is Unnecessary for SUMOylation or Transcriptional Control
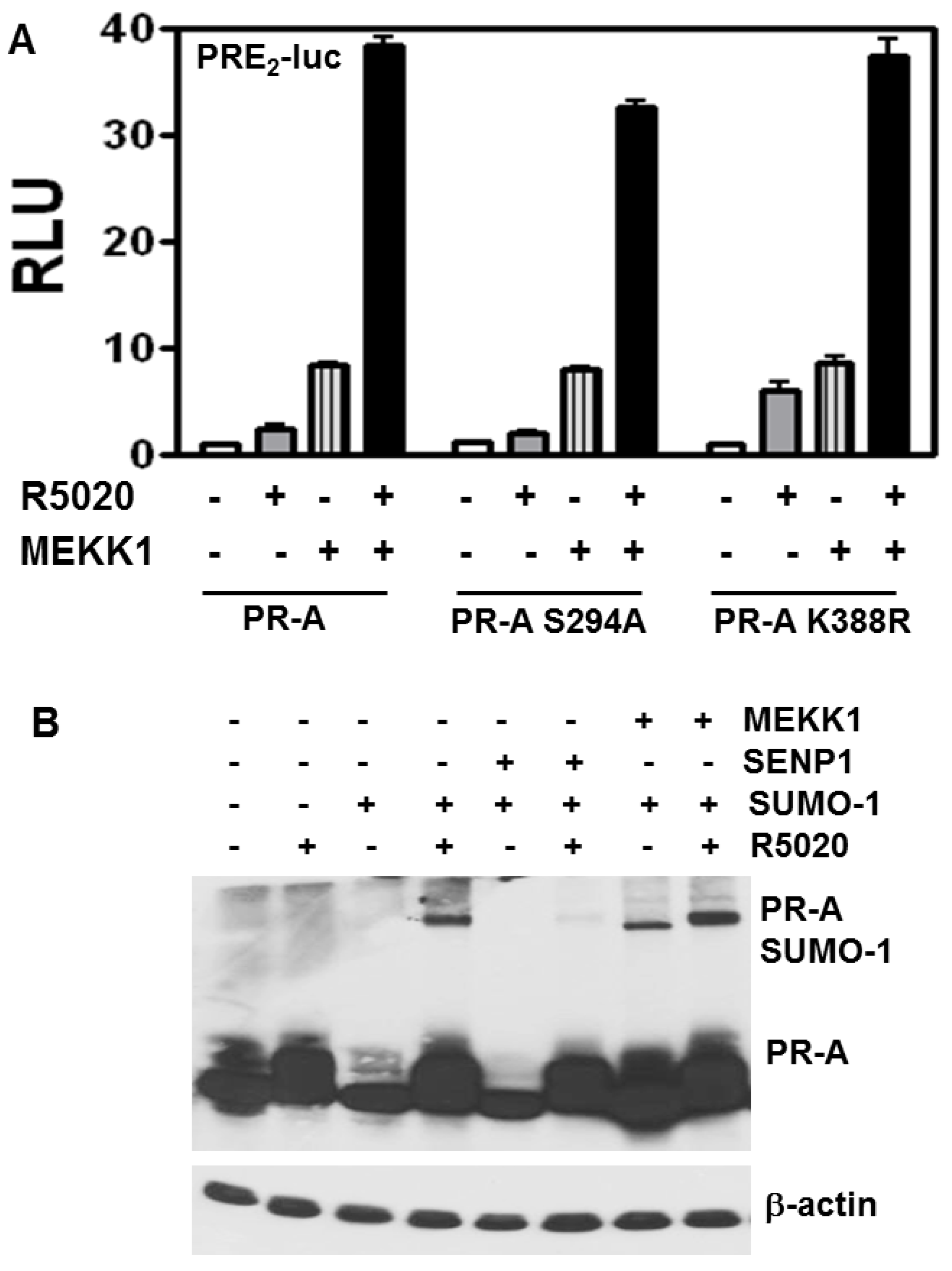
3.4. Stimulation of PR-A Ligand-Independent Transcription by MEKK Is Independent of SUMOylation
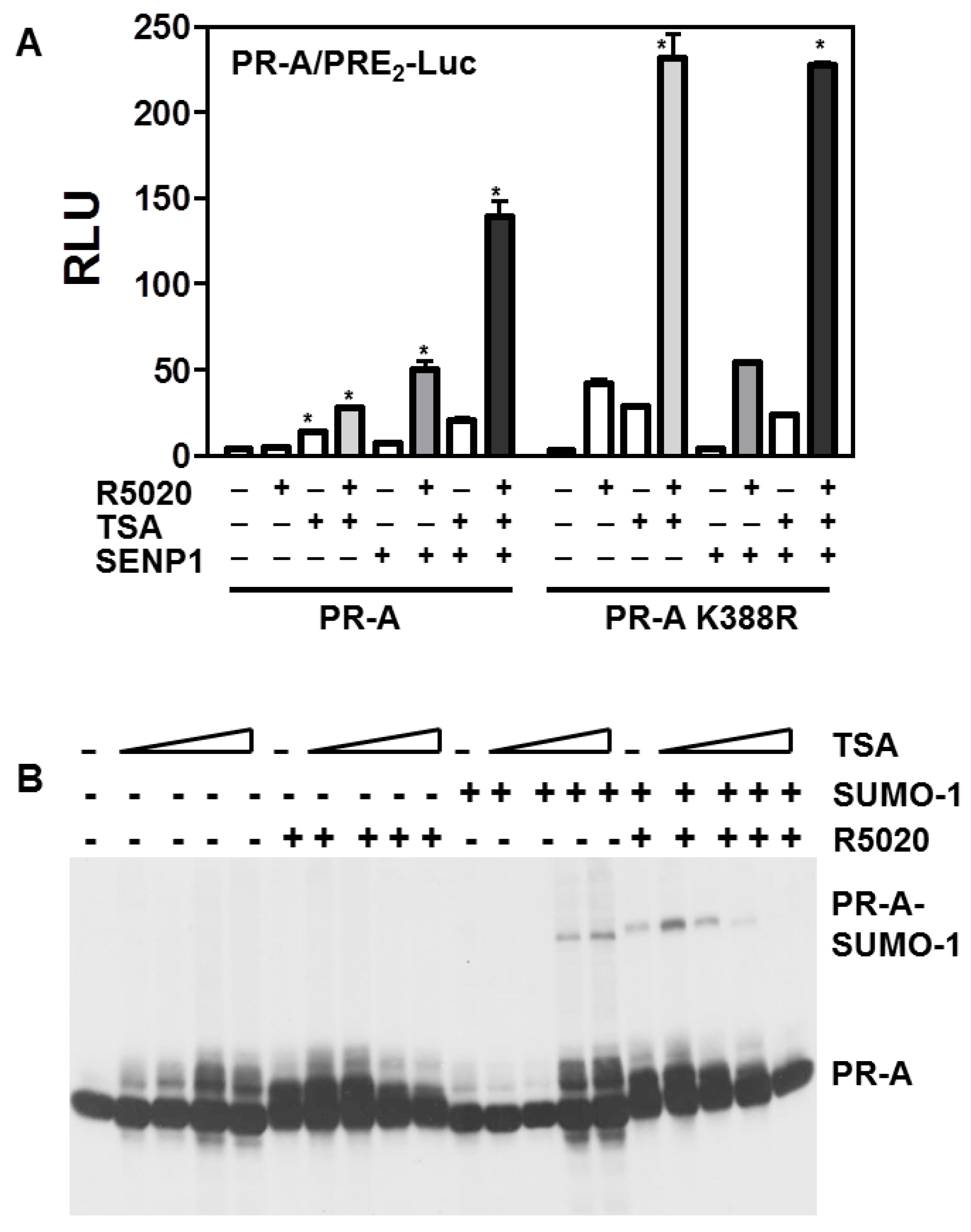
3.5. Histone Acetylation, SUMOylation and Transcription by PR-A
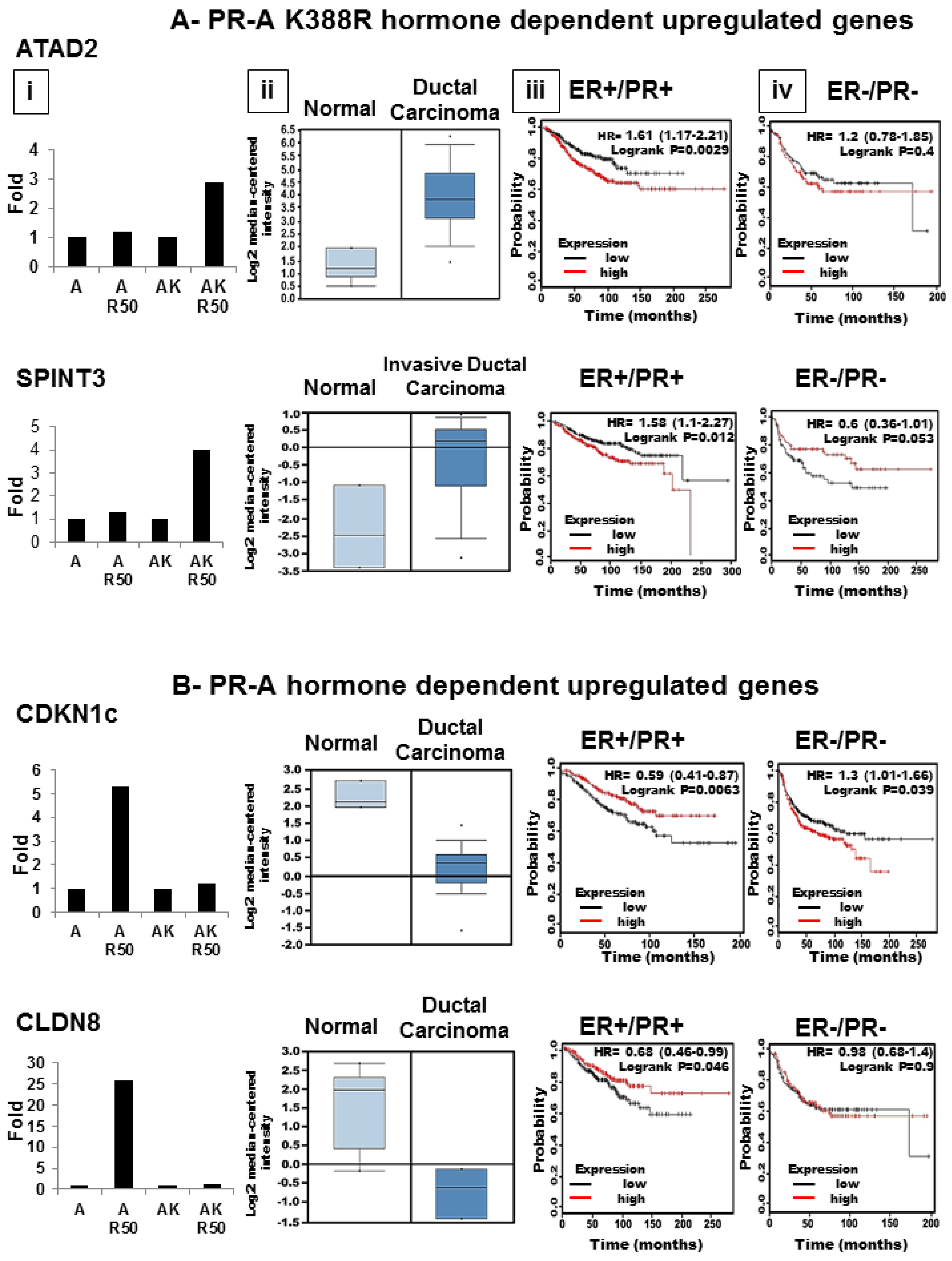
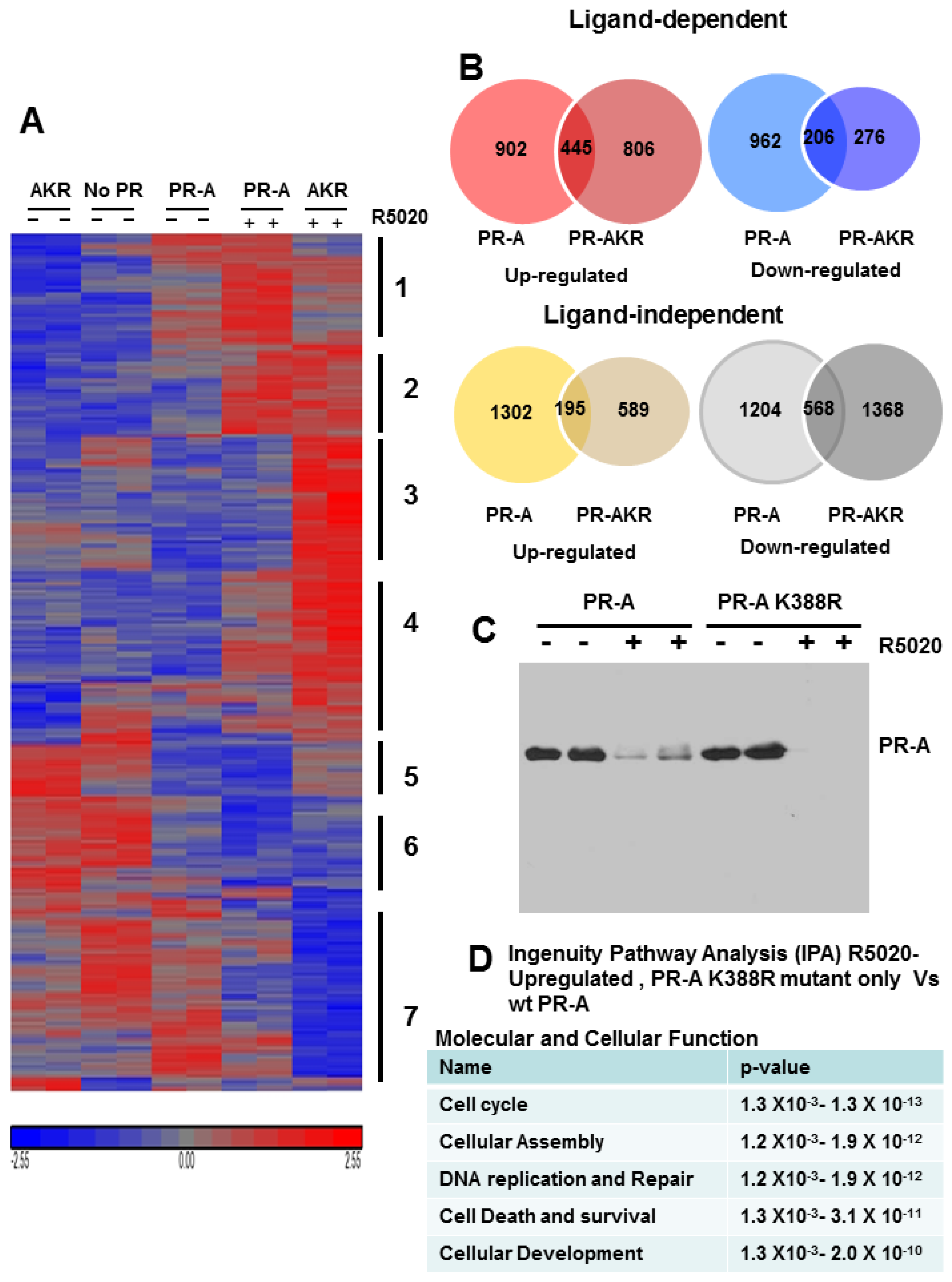
3.6. SUMOylation Differentially Regulates Endogenous Progestin Target Genes in Breast Cancer Cells
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mote, P.A.; Gompel, A.; Howe, C.; Hilton, H.N.; Sestak, I.; Cuzick, J.; Dowsett, M.; Hugol, D.; Forgez, P.; Byth, K.; et al. Progesterone receptor A predominance is a discriminator of benefit from endocrine therapy in the ATAC trial. Breast Cancer Res. Treat. 2015, 151, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Scarpin, K.M.; Graham, J.D.; Mote, P.A.; Clarke, C.L. Progesterone action in human tissues: Regulation by progesterone receptor (PR) isoform expression, nuclear positioning and coregulator expression. Nucl. Recept. Signal. 2009, 7, e009. [Google Scholar] [CrossRef] [PubMed]
- Shyamala, G.; Yang, X.; Cardiff, R.D.; Dale, E. Impact of progesterone receptor on cell-fate decisions during mammary gland development. Proc. Natl. Acad. Sci. USA 2000, 97, 3044–3049. [Google Scholar] [CrossRef] [PubMed]
- Richer, J.K.; Jacobsen, B.M.; Manning, N.G.; Abel, M.G.; Wolf, D.M.; Horwitz, K.B. Differential gene regulation by the two progesterone receptor isoforms in human breast cancer cells. J. Biol. Chem. 2002, 277, 5209–5218. [Google Scholar] [CrossRef] [PubMed]
- McFall, T.; McKnight, B.; Rosati, R.; Kim, S.; Huang, Y.; Viola-Villegas, N.; Ratnam, M. Progesterone Receptor A Promotes Invasiveness and Metastasis of Luminal Breast Cancer by Suppressing Regulation of Critical Micro RNAs by Estrogen. J. Biol. Chem. 2017. [Google Scholar] [CrossRef] [PubMed]
- Isaksson, E.; Wang, H.; Sahlin, L.; von Schoultz, B.; Cline, J.M.; von Schoultz, E. Effects of long-term HRT and tamoxifen on the expression of progesterone receptors A and B in breast tissue from surgically postmenopausal cynomolgus macaques. Breast Cancer Res. Treat. 2003, 79, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, B.M.; Jambal, P.; Schittone, S.A.; Horwitz, K.B. ALU repeats in promoters are position-dependent co-response elements (coRE) that enhance or repress transcription by dimeric and monomeric progesterone receptors. Mol. Endocrinol 2009, 23, 989–1000. [Google Scholar] [CrossRef] [PubMed]
- Onate, S.A.; Prendergast, P.; Wagner, J.P.; Nissen, M.; Reeves, R.; Pettijohn, D.E.; Edwards, D.P. The DNA-bending protein HMG-1 enhances progesterone receptor binding to its target DNA sequences. Mol. Cell. Biol. 1994, 14, 3376–3391. [Google Scholar] [CrossRef] [PubMed]
- Bain, D.L.; Franden, M.A.; McManaman, J.L.; Takimoto, G.S.; Horwitz, K.B. The N-terminal region of human progesterone B-receptors: Biophysical and biochemical comparison to A-receptors. J. Biol. Chem. 2001, 276, 23825–23831. [Google Scholar] [CrossRef] [PubMed]
- Tetel, M.J.; Giangrande, P.H.; Leonhardt, S.A.; McDonnell, D.P.; Edwards, D.P. Hormone-dependent interaction between the amino- and carboxyl-terminal domains of progesterone receptor in vitro and in vivo. Mol. Endocrinol. 1999, 13, 910–924. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Hafiz, H.; Takimoto, G.S.; Tung, L.; Horwitz, K.B. The inhibitory function in human progesterone receptor N termini binds SUMO-1 protein to regulate autoinhibition and transrepression. J. Biol. Chem. 2002, 277, 33950–33956. [Google Scholar] [CrossRef] [PubMed]
- Tung, L.; Abdel-Hafiz, H.; Shen, T.; Harvell, D.M.; Nitao, L.K.; Richer, J.K.; Sartorius, C.A.; Takimoto, G.S.; Horwitz, K.B. Progesterone receptors (PR)-B and -A regulate transcription by different mechanisms: AF-3 exerts regulatory control over coactivator binding to PR-B. Mol. Endocrinol. 2006, 20, 2656–2670. [Google Scholar] [CrossRef] [PubMed]
- Daniel, A.R.; Gaviglio, A.L.; Czaplicki, L.M.; Hillard, C.J.; Housa, D.; Lange, C.A. The progesterone receptor hinge region regulates the kinetics of transcriptional responses through acetylation, phosphorylation, and nuclear retention. Mol. Endocrinol. 2010, 24, 2126–2138. [Google Scholar] [CrossRef] [PubMed]
- Knutson, T.P.; Daniel, A.R.; Fan, D.; Silverstein, K.A.; Covington, K.R.; Fuqua, S.A.; Lange, C.A. Phosphorylated and sumoylation-deficient progesterone receptors drive proliferative gene signatures during breast cancer progression. Breast Cancer Res. 2012, 14, R95. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Hafiz, H.A.; Horwitz, K.B. Control of progesterone receptor transcriptional synergy by SUMOylation and deSUMOylation. BMC Mol. Biol. 2012, 13, 10. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Hafiz, H.; Dudevoir, M.L.; Horwitz, K.B. Mechanisms underlying the control of progesterone receptor transcriptional activity by SUMOylation. J. Biol. Chem. 2009, 284, 9099–9108. [Google Scholar] [CrossRef] [PubMed]
- Sartorius, C.A.; Groshong, S.D.; Miller, L.A.; Powell, R.L.; Tung, L.; Takimoto, G.S.; Horwitz, K.B. New T47D breast cancer cell lines for the independent study of progesterone B- and A-receptors: Only antiprogestin-occupied B-receptors are switched to transcriptional agonists by cAMP. Cancer Res. 1994, 54, 3868–3877. [Google Scholar] [PubMed]
- Badtke, M.M.; Jambal, P.; Dye, W.W.; Spillman, M.A.; Post, M.D.; Horwitz, K.B.; Jacobsen, B.M. Unliganded progesterone receptors attenuate taxane-induced breast cancer cell death by modulating the spindle assembly checkpoint. Breast Cancer Res. Treat. 2012, 131, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Wang, D.; Wang, Z.; Yeh, E.T. SENP1 enhances androgen receptor-dependent transcription through desumoylation of histone deacetylase 1. Mol. Cell. Biol. 2004, 24, 6021–6028. [Google Scholar] [CrossRef] [PubMed]
- Tung, L.; Mohamed, M.K.; Hoeffler, J.P.; Takimoto, G.S.; Horwitz, K.B. Antagonist-occupied human progesterone B-receptors activate transcription without binding to progesterone response elements and are dominantly inhibited by A-receptors. Mol. Endocrinol. 1993, 7, 1256–1265. [Google Scholar] [CrossRef] [PubMed]
- Iniguez-Lluhi, J.A.; Pearce, D. A common motif within the negative regulatory regions of multiple factors inhibits their transcriptional synergy. Mol. Cell. Biol. 2000, 20, 6040–6050. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.H.; Sharrocks, A.D. SUMO promotes HDAC-mediated transcriptional repression. Mol. Cell 2004, 13, 611–617. [Google Scholar] [CrossRef]
- Harvell, D.M.; Kim, J.; O’Brien, J.; Tan, A.C.; Borges, V.F.; Schedin, P.; Jacobsen, B.M.; Horwitz, K.B. Genomic signatures of pregnancy-associated breast cancer epithelia and stroma and their regulation by estrogens and progesterone. Horm. Cancer 2013, 4, 140–153. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, H.; Russell, I.A.; Stark, R.; Rueda, O.M.; Hickey, T.E.; Tarulli, G.A.; Serandour, A.A.; Birrell, S.N.; Bruna, A.; Saadi, A.; et al. Progesterone receptor modulates ERalpha action in breast cancer. Nature 2015, 523, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Finlay-Schultz, J.; Gillen, A.E.; Brechbuhl, H.M.; Ivie, J.J.; Matthews, S.B.; Jacobsen, B.M.; Bentley, D.L.; Kabos, P.; Sartorius, C.A. Breast Cancer Suppression by Progesterone Receptors Is Mediated by Their Modulation of Estrogen Receptors and RNA Polymerase III. Cancer Res. 2017, 77, 4934–4946. [Google Scholar] [CrossRef] [PubMed]
- Conneely, O.M.; Mulac-Jericevic, B.; Lydon, J.P.; De Mayo, F.J. Reproductive functions of the progesterone receptor isoforms: Lessons from knock-out mice. Mol. Cell. Endocrinol. 2001, 179, 97–103. [Google Scholar] [CrossRef]
- Mulac-Jericevic, B.; Lydon, J.P.; DeMayo, F.J.; Conneely, O.M. Defective mammary gland morphogenesis in mice lacking the progesterone receptor B isoform. Proc. Natl. Acad. Sci. USA 2003, 100, 9744–9749. [Google Scholar] [CrossRef] [PubMed]
- Azim, H.A., Jr.; Peccatori, F.A.; Brohee, S.; Branstetter, D.; Loi, S.; Viale, G.; Piccart, M.; Dougall, W.C.; Pruneri, G.; Sotiriou, C. RANK-ligand (RANKL) expression in young breast cancer patients and during pregnancy. Breast Cancer Res. 2015, 17, 538. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.A.; Jackson, H.W.; Beristain, A.G.; Di Grappa, M.A.; Mote, P.A.; Clarke, C.L.; Stingl, J.; Waterhouse, P.D.; Khokha, R. Progesterone induces adult mammary stem cell expansion. Nature 2010, 465, 803–807. [Google Scholar] [CrossRef] [PubMed]
- Roemer, S.C.; Donham, D.C.; Sherman, L.; Pon, V.H.; Edwards, D.P.; Churchill, M.E. Structure of the progesterone receptor-deoxyribonucleic acid complex: Novel interactions required for binding to half-site response elements. Mol. Endocrinol. 2006, 20, 3042–3052. [Google Scholar] [CrossRef] [PubMed]
- Sutinen, P.; Malinen, M.; Heikkinen, S.; Palvimo, J.J. SUMOylation modulates the transcriptional activity of androgen receptor in a target gene and pathway selective manner. Nucleic Acids Res. 2014, 42, 8310–8319. [Google Scholar] [CrossRef] [PubMed]
- Salhia, B.; Kiefer, J.; Ross, J.T.; Metapally, R.; Martinez, R.A.; Johnson, K.N.; DiPerna, D.M.; Paquette, K.M.; Jung, S.; Nasser, S.; et al. Integrated genomic and epigenomic analysis of breast cancer brain metastasis. PLoS ONE 2014, 9, e85448. [Google Scholar] [CrossRef] [PubMed]
- Kalashnikova, E.V.; Revenko, A.S.; Gemo, A.T.; Andrews, N.P.; Tepper, C.G.; Zou, J.X.; Cardiff, R.D.; Borowsky, A.D.; Chen, H.W. ANCCA/ATAD2 overexpression identifies breast cancer patients with poor prognosis, acting to drive proliferation and survival of triple-negative cells through control of B-Myb and EZH2. Cancer Res. 2010, 70, 9402–9412. [Google Scholar] [CrossRef] [PubMed]
- Hsia, E.Y.; Kalashnikova, E.V.; Revenko, A.S.; Zou, J.X.; Borowsky, A.D.; Chen, H.W. Deregulated E2F and the AAA+ coregulator ANCCA drive proto-oncogene ACTR/AIB1 overexpression in breast cancer. Mol. Cancer Res. 2010, 8, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Porter, S.; Span, P.N.; Sweep, F.C.; Tjan-Heijnen, V.C.; Pennington, C.J.; Pedersen, T.X.; Johnsen, M.; Lund, L.R.; Romer, J.; Edwards, D.R. ADAMTS8 and ADAMTS15 expression predicts survival in human breast carcinoma. Int. J. Cancer 2006, 118, 1241–1247. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Wang, J.; Gupta, A.; Shidfar, A.; Branstetter, D.; Lee, O.; Ivancic, D.; Sullivan, M.; Chatterton, R.T., Jr.; Dougall, W.C.; et al. RANKL expression in normal and malignant breast tissue responds to progesterone and is up-regulated during the luteal phase. Breast Cancer Res. Treat. 2014, 146, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Oeggerli, M.; Tian, Y.; Ruiz, C.; Wijker, B.; Sauter, G.; Obermann, E.; Guth, U.; Zlobec, I.; Sausbier, M.; Kunzelmann, K.; et al. Role of KCNMA1 in breast cancer. PLoS ONE 2012, 7, e41664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, A.; He, F.; Gu, X. Identification and characterization of tyrosine kinases in anole lizard indicate the conserved tyrosine kinase repertoire in vertebrates. Mol. Genet. Genom. 2017, 292, 1405–1418. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yang, L.; He, J.; Yang, H. STYK1 promotes Warburg effect through PI3K/AKT signaling and predicts a poor prognosis in nasopharyngeal carcinoma. Tumour Biol. 2017, 39, 1010428317711644. [Google Scholar] [CrossRef] [PubMed]
- Bujko, M.; Kober, P.; Mikula, M.; Ligaj, M.; Ostrowski, J.; Siedlecki, J.A. Expression changes of cell-cell adhesion-related genes in colorectal tumors. Oncol. Lett. 2015, 9, 2463–2470. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, C.; Monteverde, M.; Lattanzio, L.; Gojis, O.; Rudraraju, B.; Fortunato, M.; Syed, N.; Thompson, A.; Garrone, O.; Merlano, M.; et al. Site-specific CpG methylation in the CCAAT/enhancer binding protein delta (CEBPdelta) CpG island in breast cancer is associated with metastatic relapse. Br. J. Cancer 2012, 107, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; He, Y.; Wang, X.; Liang, Z.; He, G.; Zhang, P.; Zhu, H.; Xu, N.; Liang, S. Protein SUMOylation modification and its associations with disease. Open Biol. 2017, 7, 170167. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdel-Hafiz, H.A.; Dudevoir, M.L.; Perez, D.; Abdel-Hafiz, M.; Horwitz, K.B. SUMOylation Regulates Transcription by the Progesterone Receptor A Isoform in a Target Gene Selective Manner. Diseases 2018, 6, 5. https://doi.org/10.3390/diseases6010005
Abdel-Hafiz HA, Dudevoir ML, Perez D, Abdel-Hafiz M, Horwitz KB. SUMOylation Regulates Transcription by the Progesterone Receptor A Isoform in a Target Gene Selective Manner. Diseases. 2018; 6(1):5. https://doi.org/10.3390/diseases6010005
Chicago/Turabian StyleAbdel-Hafiz, Hany A., Michelle L. Dudevoir, Daniel Perez, Mohamed Abdel-Hafiz, and Kathryn B. Horwitz. 2018. "SUMOylation Regulates Transcription by the Progesterone Receptor A Isoform in a Target Gene Selective Manner" Diseases 6, no. 1: 5. https://doi.org/10.3390/diseases6010005