
Interaction between DNA and Drugs Having Protonable Basic Groups: Characterization through Affinity Constants, Drug Release Kinetics, and Conformational Changes

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Analytical Determinations
2.3. Preparation of DNA–Drug Complexes
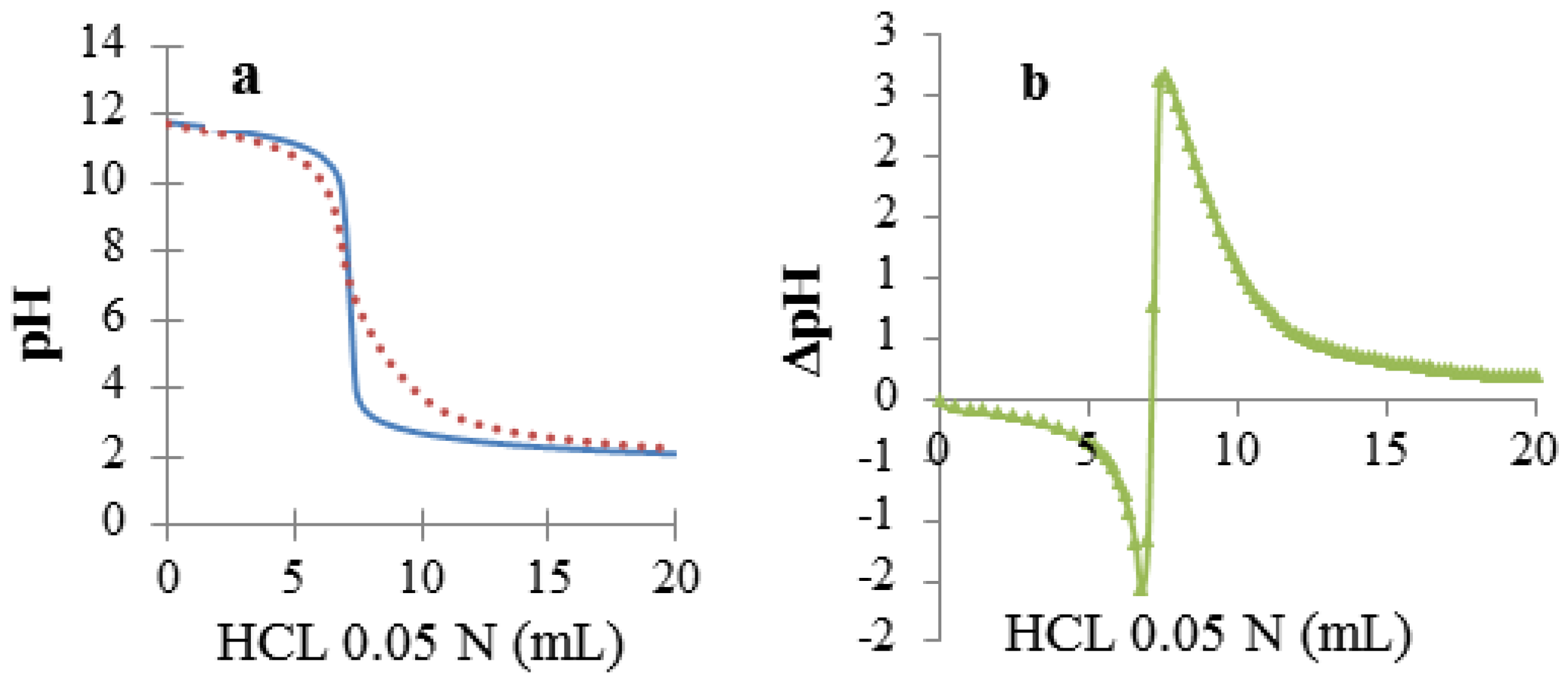
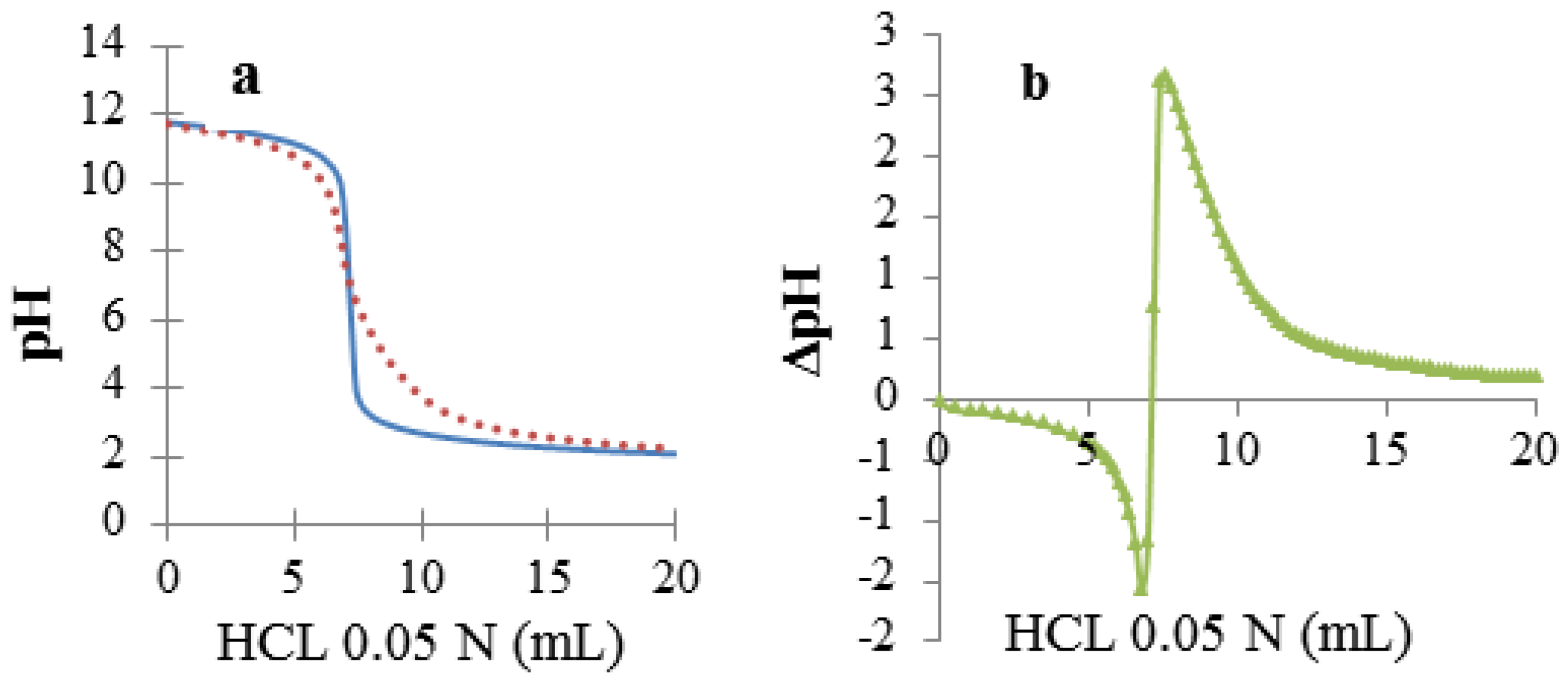
2.4. Determination of Species Distribution and Affinity Constants (K)
2.5. Drug Release from DNA–Dx Complexes
2.6. Reversibility of the Interaction
3. Results and Discussion
3.1. Preparation of Complexes of DNA with Model Drugs
3.2. Species Distribution in the DNA–D Dispersions
3.3. Electrokinetic Potential (ζ)
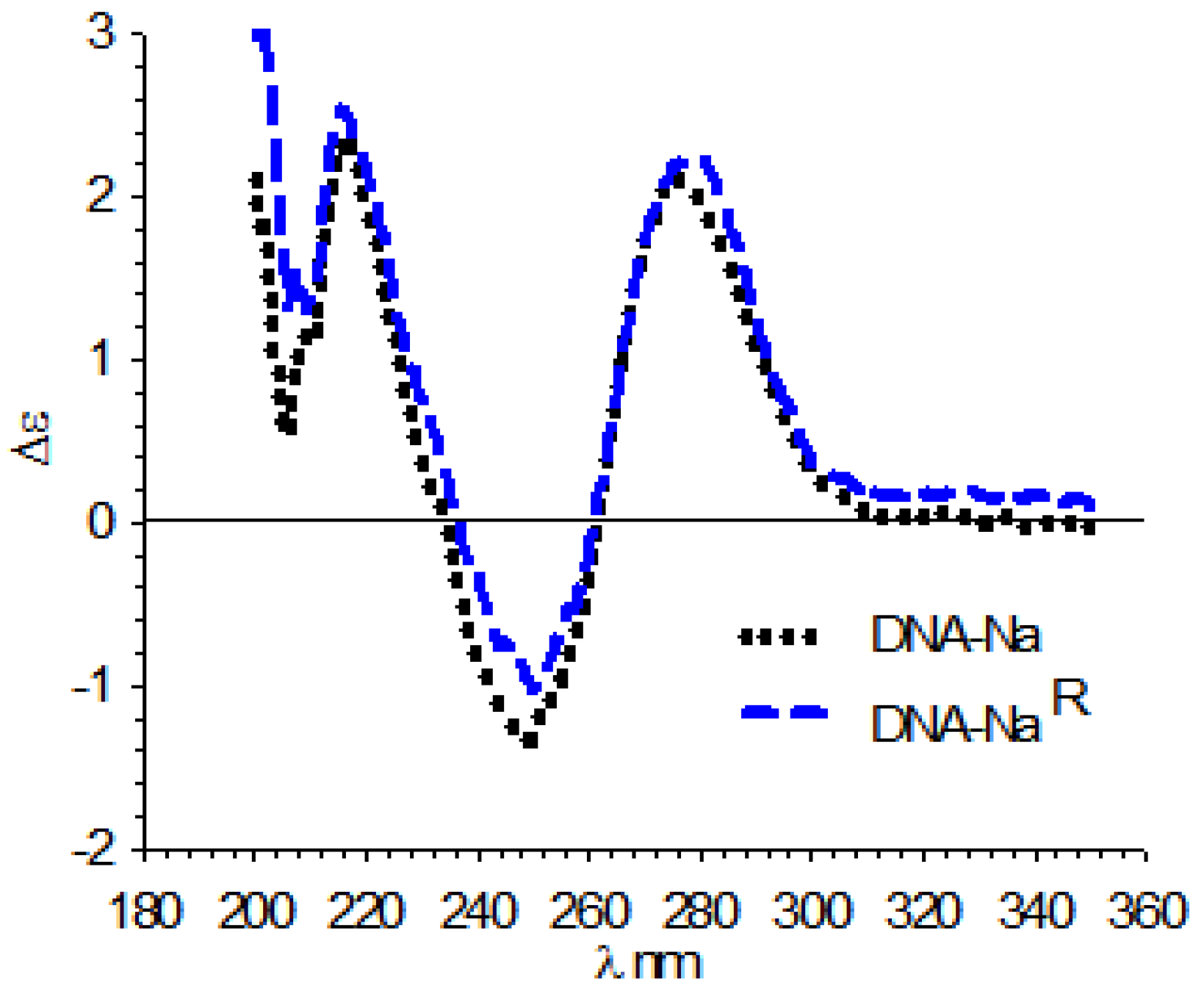
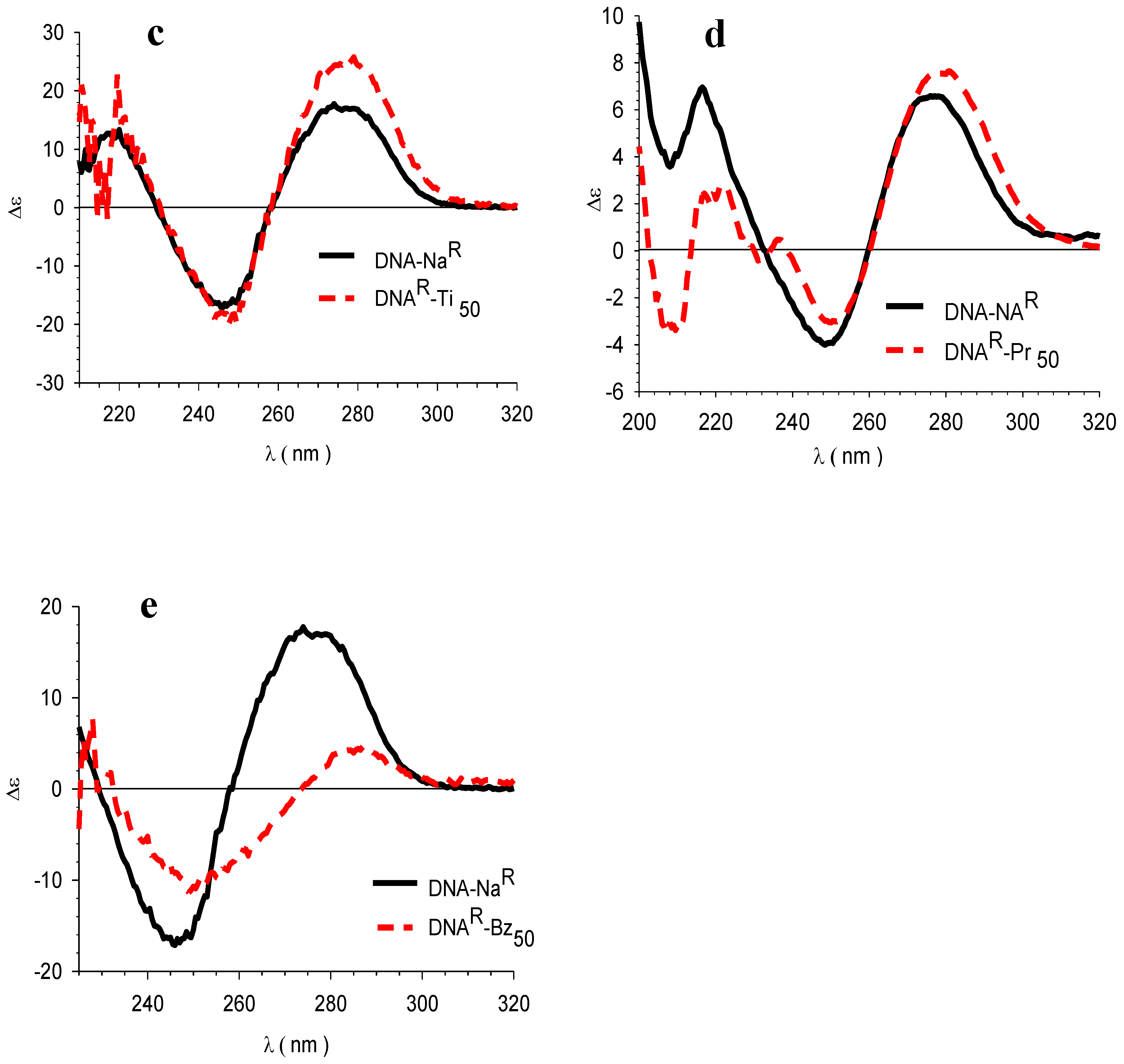
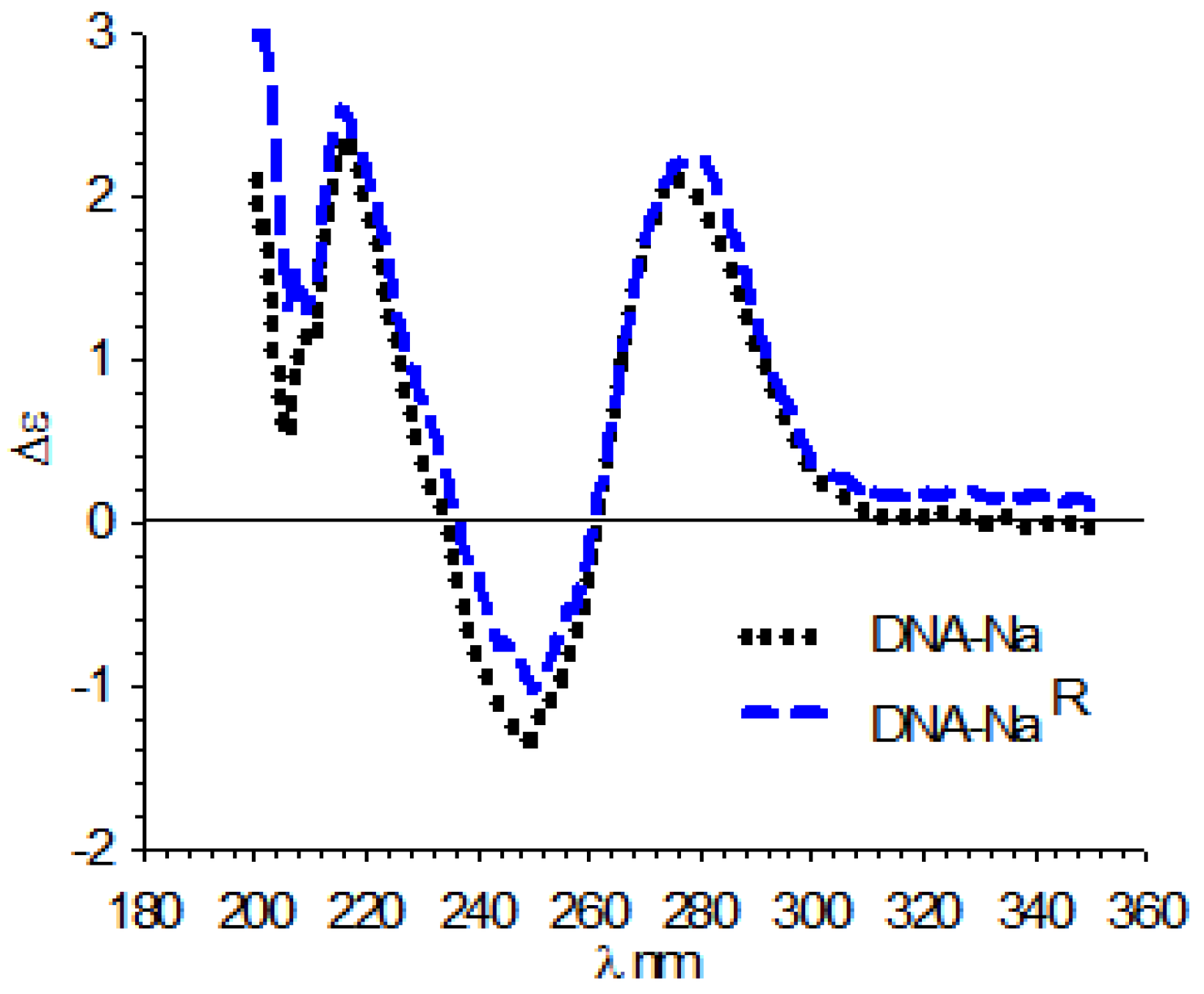
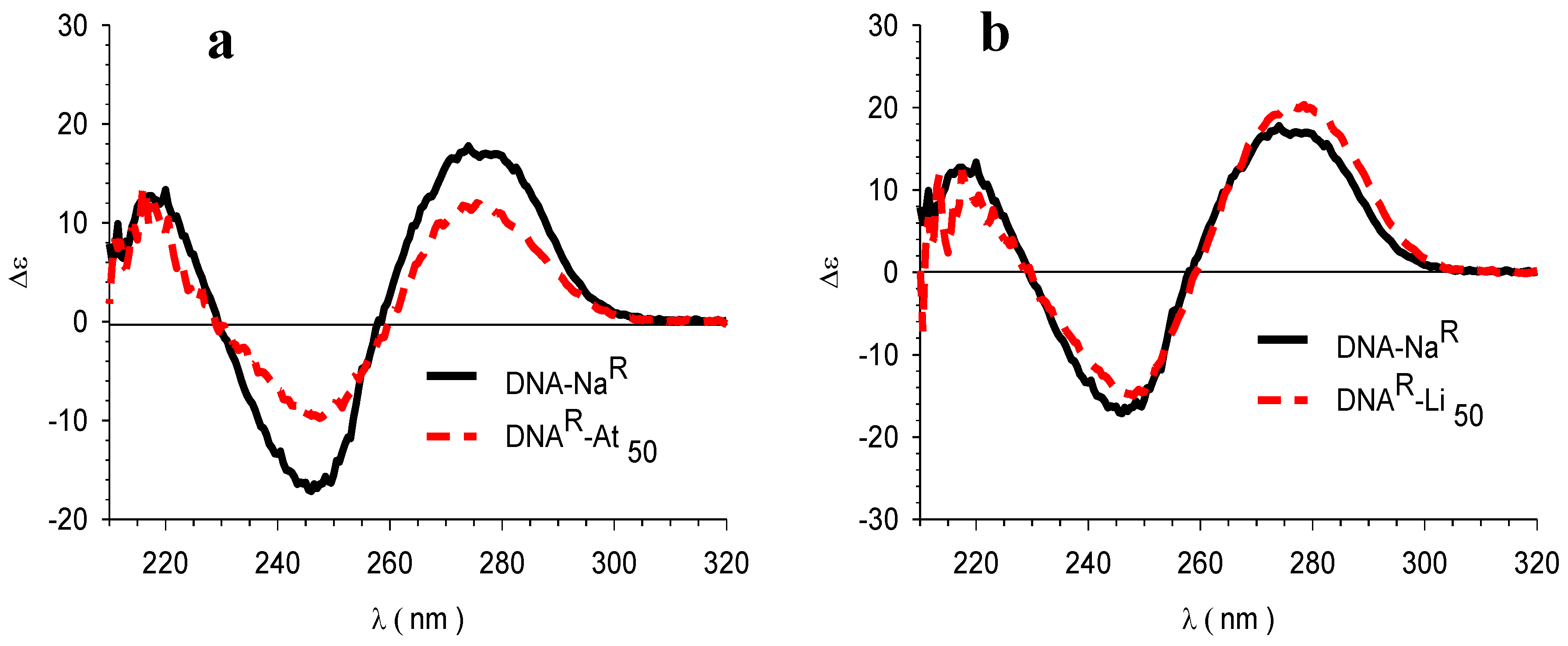
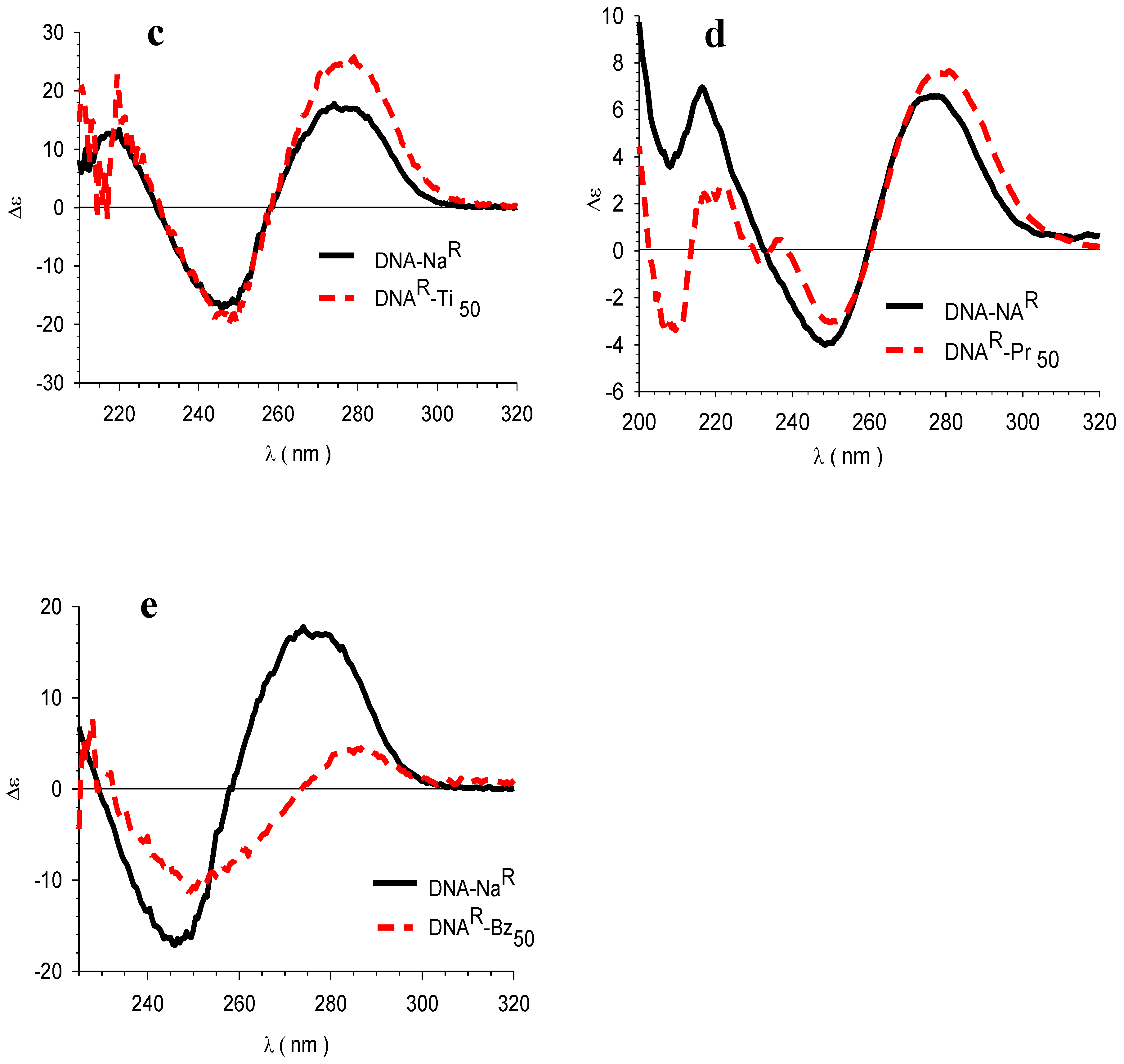
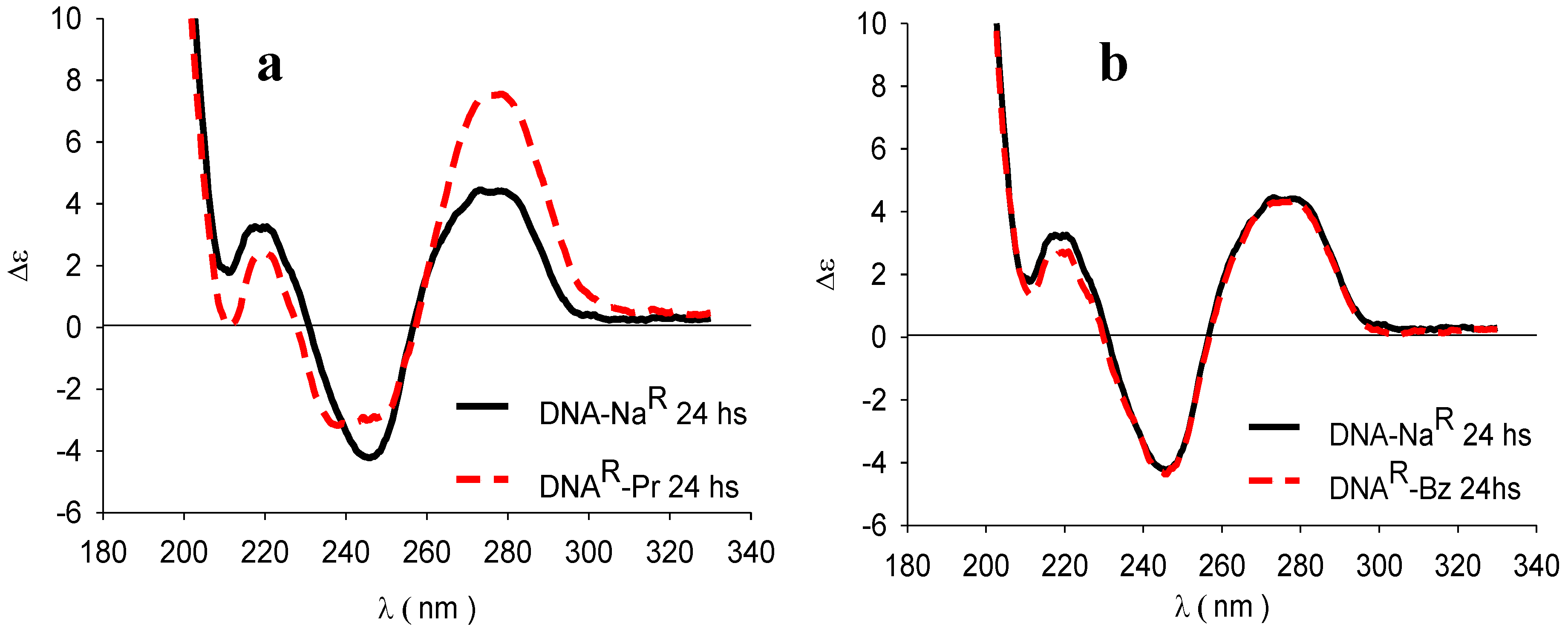
3.4. Circular Dichroism (CD)
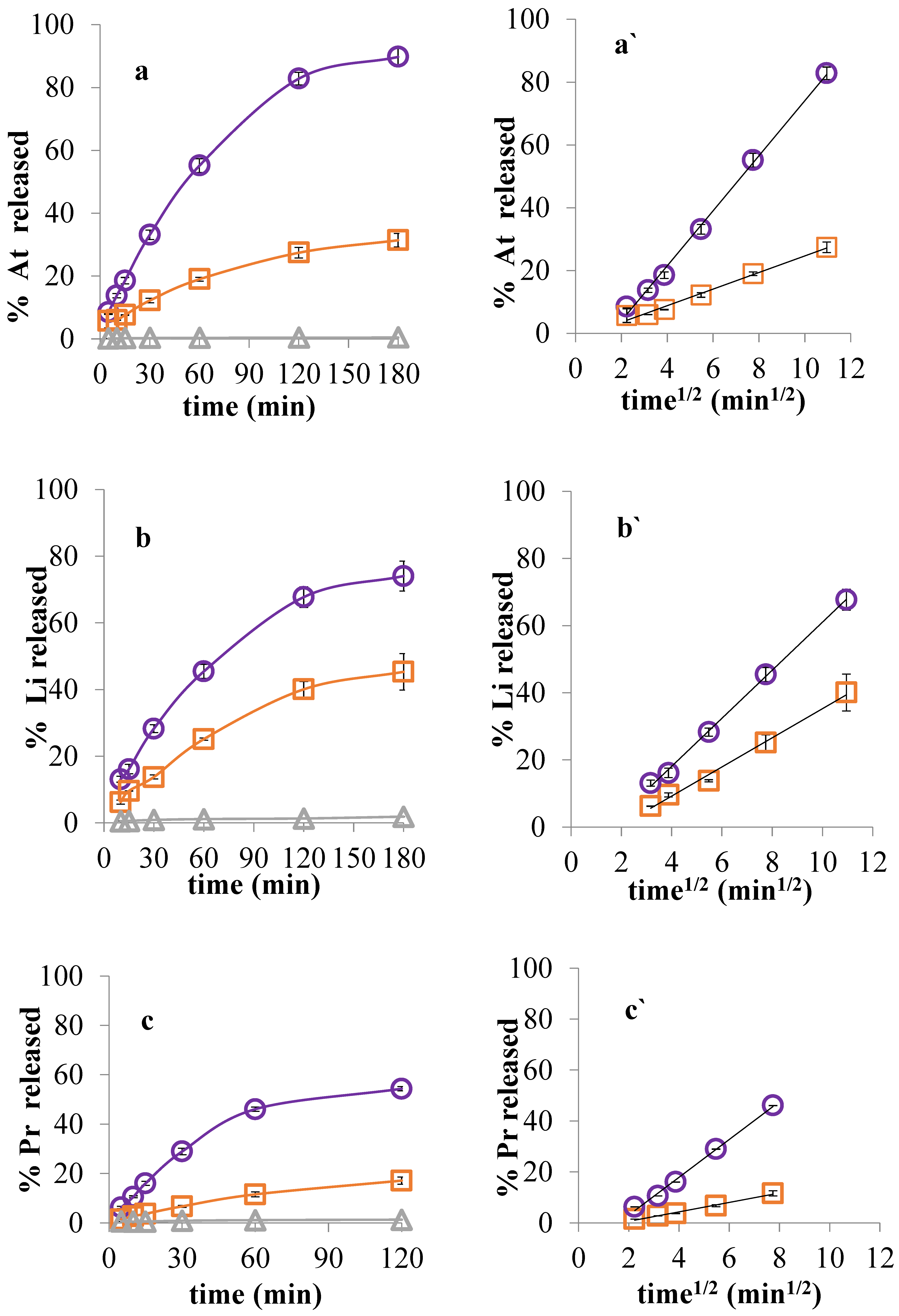
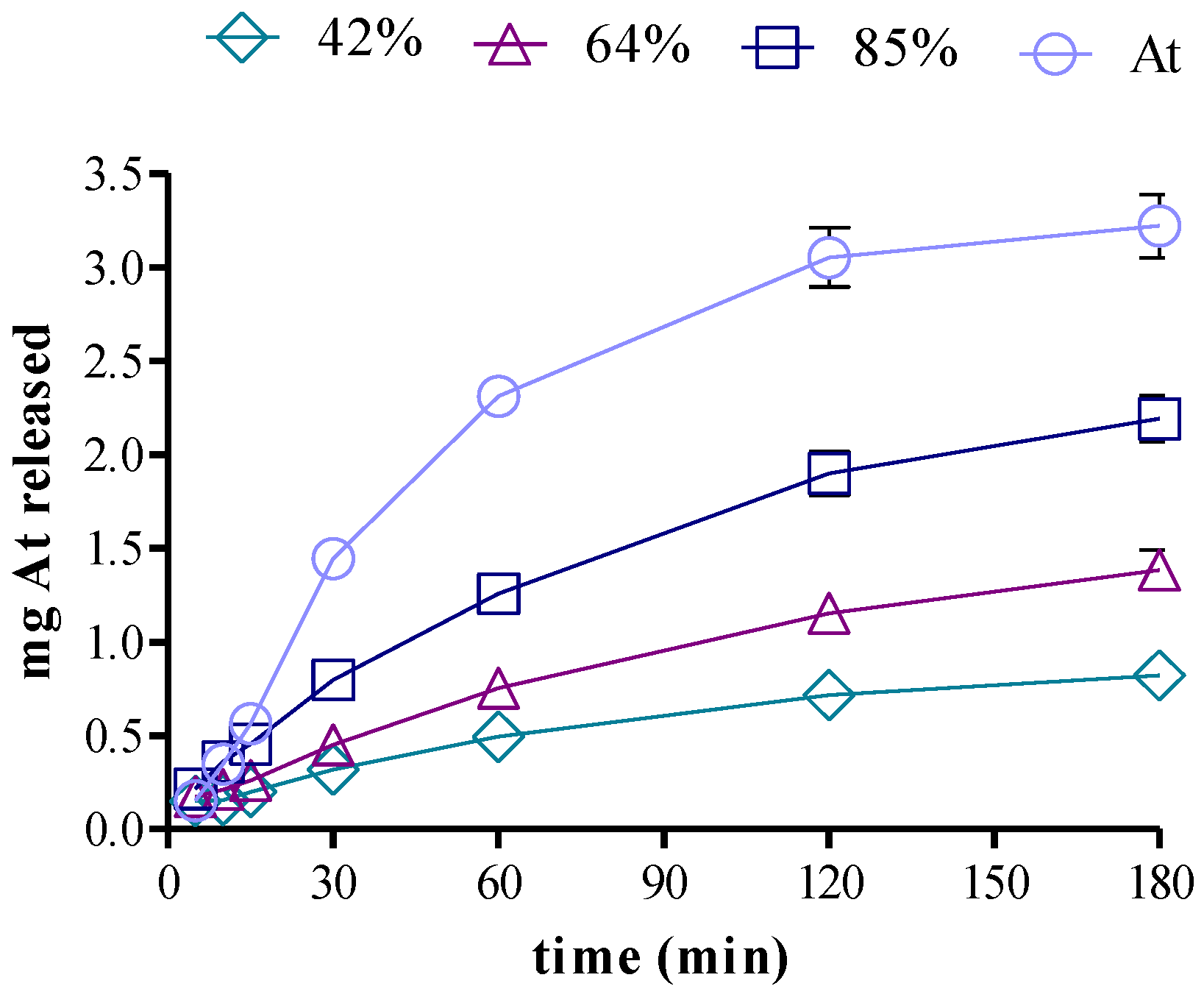
3.5. Drug Release in Two-Compartment Diffusion Cells
Supplementary Materials
Acknowledgments
Author contributions
Conflicts of Interest
References
- Acharya, S. Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Science and Technology 164. Some Aspects of Physicochemical Properties of DNA and RNA; Acta Universitatis Upsaliensis: Uppsala, Sweden, 2006. [Google Scholar]
- Horton, H.R.; Moran, L.A.; Scimgeour, K.G.; Perry, M.D.; Rawn, J.D. Principios de Bioquímica, 4th ed.; Pearson Educación: Naucalpan de Juárez, Mexico, 2008; Chapter 19. [Google Scholar]
- Um, S.H.; Lee, J.B.; Park, N.; Kwon, S.Y.; Umbach, C.C.; Luo, D. Enzyme-catalysed assembly of DNA hydrogel. Nat. Mater. 2006, 5, 97–801. [Google Scholar] [CrossRef] [PubMed]
- Wahyuni, E.T.; Tjahjono, D.H.; Yoshioka, N.; Inoue, H. Spectroscopic studies on the thermodynamic and thermal denaturation of the ct-DNA binding of methylene blue. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2010, 77, 528–534. [Google Scholar]
- Hajian, R.; Shams, N.; Mohagheghian, M. Study on the interaction between doxorubicin and deoxyribonucleic acid with the use of methylene blue as a probe. J. Braz. Chem. Soc. 2009, 20, 1399–1405. [Google Scholar] [CrossRef]
- Shah, A.; Khan, A.M.; Usman, M.; Siddiq, M. Plex with DNA as Studied by Conductometric and Spectroscopic Techniques. J. Chil. Chem. Soc. 2009, 2, 134–137. [Google Scholar]
- Mizuno, Y.; Naoi, T.; Nishikawa, M.; Rattanakiat, S.; Hamaguchi, N.; Hashida, M.; Takakur, Y. Simultaneous delivery of doxorubicin and immunostimulatory CpG motif to tumors using a plasmid DNA/doxorubicin complex in mice. J. Control. Release 2010, 141, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Stepankova, J.; Studenovsky, M.; Malina, J.; Kasparkova, J.; Liskova, B.; Novakova, O.; Ulbrich, K.; Brabec, V. DNA interactions of 2-pyrrolinodoxorubicin, a distinctively more potent daunosamine-modified analogue of doxorubicin. Biochem. Pharmacol. 2011, 82, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Rolland, A.; Sullivan, S.M. Pharmaceutical Gene Delivery Systems. In Drugs and the Pharmaceutical Science; Taylor & Francis Group: Boca Raton, FL, USA, 2003; Volume 131. [Google Scholar]
- Feuerstein, B.G.; Pattabiraman, N.; Marton, L.J. Molecular mechanics of the interactions of spermine with DNA: DNA bending as a result of ligand binding. Nucleic Acids Res. 1990, 18, 1271–1282. [Google Scholar] [CrossRef] [PubMed]
- Utsuno, K.; Uluda, H. Thermodynamics of polyethylenimine-DNA binding and DNA condensation. Biophys. J. 2010, 99, 201–207. [Google Scholar] [CrossRef] [PubMed]
- McGregor, T.D.; Bousfield, W.; Qu, Y.; Farrell, N. Circular dichroism study of the irreversibility of conformational changes induced by polyamine-linked dinuclear platinum compounds. J. Inorg. Biochem. 2002, 91, 212–219. [Google Scholar] [CrossRef]
- Hackl, E.V.; Kornilova, S.V.; Blagoi, Y.P. DNA structural transitions induced by divalent metal ions in aqueous solutions. Int. J. Biol. Macromol. 2005, 35, 175–191. [Google Scholar] [CrossRef] [PubMed]
- Battistini, F.D.; Olivera, M.E.; Manzo, R.H. Equilibrium and release properties of hyaluronic acid-drug complexes. Eur. J. Pharm. Sci. 2013, 49, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Ramírez Rigo, M.V.; Allemandi, D.A.; Manzo, R.H. A Linear Free Energy Relationship Treatment of the Affinity between Carboxymethylcellulose and Basic Drugs. Mol. Pharm. 2004, 1, 383–386. [Google Scholar] [CrossRef] [PubMed]
- Navarro, J.; Maldonado, J.; Narbona, E.; Ruiz-Bravo, A.; García Salmerón, J.L.; Molina, J.A.; Gil, A. Influence of dietary nucleotides on plasma immunoglobulin levels and lymphocyte subsets of preterm infants. Biofactors 1999, 10, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Diao, H.; Nishi, N. Applied chemistry of natural DNA. Chem. Soc. Rev. 2008, 37, 2745–2757. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Nagai, N.; Murata, M.; Nishimura, D.; Sugi, M.; Munekata, M. Development of salmon milt DNA/salmon collagen composite for wound dressing. J. Mater. Sci. Mater. Med. 2008, 19, 3473–3479. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, Y.; Miyoshi, D.; Sugimoto, N. Utilization of salmon milt DNA against UV damage. Appl. Biochem. Biotechnol. 2010, 160, 2458–2466. [Google Scholar] [CrossRef] [PubMed]
- Van Den Beucken, J.J.J.P.; Vos, M.R.J.; Thüne, P.C.; Hayakawa, T.; Fukushima, T.; Okahata, Y.; Walboomers, X.F.; Sommerdijk, N.A.J.M.; Nolte, R.J.M.; Jansen, J.A. Fabrication, characterization, and biological assessment of multilayered DNA-coatings for biomaterial purposes. Biomaterials 2006, 27, 691–701. [Google Scholar] [CrossRef] [PubMed]
- Avdeef, A. Absorption and Drug Development: Solubility, Permeability, and Charge State; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2003. [Google Scholar]
- PubChem Compound Database; Lidocaíne CID 3676. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/3676#section=Solubility (accessed on 28 December 2016).
- PubChem Compound Database; racemic-Timolol CID 5478. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/5478#section=Solubility (accessed on 28 December 2016).
- PubChem Compound Database; Benzydamine CID 12555. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/12555#section=Chemical-and-Physical-Properties (accessed on 28 December 2016).
- Description, Propranolol. Clinical Pharmacology. Available online: http://www.clinicalpharmacology.com/Forms/drugoptions.aspx?cpnum=1083&n=Hydrochlorothiazide%2c+HCTZ%3b+Propranolol&t=0 (accessed on 28 December 2016).
- Description, Atenolol. Clinical Pharmacology. Available online: http://www.clinicalpharmacology.com/Forms/drugoptions.aspx?cpnum=46&n=Atenolol&t=0 (accessed on 28 December 2016).
- Deoxyribonucleic Acid (DNA), Sodium Salt from Salmon Testes, Catalog Number D1626. Sigma-Aldrich: Saint Louis, MO, USA, 28 December 2016.
- USP 34-NF 29. The United States Pharmacopoeia, 34th ed.; U.S. Pharmacopoeial Convention., Inc.: Rockville, MD, USA, 2011. [Google Scholar]
- Luna, E. Algunas Aplicaciones de la Potenciometría Diferencial de Bamdo (PDB) en el Campo de las Ciencias Farmacéuticas. Acta Farm. Bonaer. 1989, 8, 75–82. [Google Scholar]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory: New York, NY, USA, 1989; p. 626. [Google Scholar]
- Battistini, F.D.; Flores-Martin, J.; Olivera, M.E.; Genti-Raimondi, S.; Manzo, R.H. Hyaluronan as drug carrier. The in vitro efficacy and selectivity of Hyaluronan-Doxorubicin complexes to affect the viability of overexpressing CD44 receptor cells. Eur. J. Pharm. Sci. 2014, 65, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Ardusso, M.S.; Manzo, R.H.; Jimenez-Kairuz, A.F. Comparative study of three structurally related acid polyelectrolytes as carriers of basic drugs: Carbomer, Eudragit L-100 and S-100. Supramol. Chem. 2010, 22, 289–296. [Google Scholar] [CrossRef]
- Zhong, W.; Yu, J.-S.; Liang, Y.; Fan, K.; Lai, L. Chlorobenzylidine-calf thymus DNA interaction II: Circular dichroism and nuclear magnetic resonance studies. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2004, 60, 2985–2992. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.C. Determination of the Conformation of Nucleic Acids by Electronic CD. In Circular Dichroism and the Conformational Analysis of Biomolecules; Springer: Heidelberg, Germany, 1996; pp. 433–465. [Google Scholar]
- Sousa, F.; Prazeres, D.M.F.; Queiroz, J.A. Circular dichroism investigation of the effect of plasmid DNA structure on retention in histidine chromatography. Arch. Biochem. Biophys. 2007, 467, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Zsila, F. Apparent circular dichroism signature of stirring-oriented DNA and drug-DNA complexes. Int. J. Biol. Macromol. 2015, 72, 1034–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palena, M.C.; Ardusso, M.S.; Manzo, R.H.; Jimenez-kairuz, A.F. Polyelectrolytes as Drug Carriers. Analysis by Dynamic Light Scattering of Reconstituted and in-situ Prepared Model Polymethacrylate-Drug Aqueous Dispersions. Open Nanosci. J. 2010, 4, 1–7. [Google Scholar] [CrossRef]
- Benegas, J.C.; Paoletti, S.; Van Den Hoop, M.A.G.T. Affinity interactions in counterion-polyelectrolyte systems: Competition between different counterions. Macromol. Theory Simul. 1999, 8, 61–64. [Google Scholar] [CrossRef]
- Porasso, R.D.; Benegas, J.C.; Van Den Hoop, A.G.T.; Paoletti, S. Chemical bonding of divalent counterions to linear polyelectrolytes: Theoretical treatment within the counterion condensation theory. Phys. Chem. Chem. Phys. 2001, 3, 1057–1062. [Google Scholar] [CrossRef]
- Drifford, M.; Delsanti, M. Physical Chemistry of Polyelectrolytes; Surfactant Science Series; Radeva, T., Ed.; Marcel Dekker: New York, NY, USA, 2001; Volume 99, p. 135. [Google Scholar]
- Xi, Z.; Ouyang, D.; Mu, H.T. Interaction of bulged DNA with leucine-containing mimics of NCS-chrom. Bioorg. Med. Chem. Lett. 2006, 16, 1185–1190. [Google Scholar] [CrossRef] [PubMed]
- Visakh, P.M.; Bayraktar, O. Polyelectrolytes Thermodynamics and Rheology; Springer: Heidelberg, Germany, 2014. [Google Scholar]
- Jimenez-Kairuz, A.F.; Allemandi, D.A.; Manzo, R.H. Equilibrium properties and mechanism of kinetic release of metoclopramide from carbomer hydrogels. Int. J. Pharm. 2003, 250, 129–136. [Google Scholar] [CrossRef]
- Jimenez-kairuz, A.; Allemandi, D.; Manzo, R.H. Mechanism of Lidocaine Release from Carbomer-Lidocaine Hydrogels. J. Pharm. Sci. 2002, 91, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.-X.; Shaw, A.; Zeng, X.; Benson, E.; Nyström, A.M.; Högberg, B. DNA origami delivery system for cancer therapy with tunable release properties. ACS Nano 2012, 6, 8684–8691. [Google Scholar] [CrossRef] [PubMed]
- Ishii-Mizuno, Y.; Umeki, Y.; Takahashi, Y.; Kato, Y.; Takabayashi, T.; Fujieda, S.; Takakura, Y.; Nishikawa, M. Nasal delivery of Japanese cedar pollen Cryj1 by using self-gelling immunostimulatory DNA for effective induction of immune responses in mice. J. Control. Release 2015, 200, 52–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, N.; Tao, J.; Hua, H.; Sun, P.; Zhao, Y. Low-density lipoprotein peptide-combined DNA nanocomplex as an efficient anticancer drug delivery vehicle. Eur. J. Pharm. Biopharm. 2015, 94, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, M.; Ogawa, K.; Umeki, Y.; Mohri, K.; Kawasaki, Y.; Watanabe, H.; Takahashi, N.; Kusuki, E.; Takahashi, R.; Takahashi, Y.; et al. Injectable, self-gelling, biodegradable, and immunomodulatory DNA hydrogel for antigen delivery. J. Control. Release 2014, 180, 25–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug (D) | MM a (g/mol) | Type of Amine | pKa | Log PC o/w b | Solubility (mg/mL) |
|---|---|---|---|---|---|
| Atenolol (At) | 266.34 | Secondary | 9.54 | 0.22 | 13.3 |
| Propranolol (Pr) | 295.81 | Secondary | 9.53 | 3.48 | 7.09E−2 |
| Timolol (Ti) | 432.50 | Secondary | 9.53 | 2.12 | 2.74 c |
| Lidocaine (Li) | 234.34 | Ternary | 7.95 | 2.44 | 4.10 c |
| Benzydamine (Bz) | 345.90 | Ternary | 9.27 | 3.71 c | 5.11E−2 |
| Ratio Vd/Vr a | Complex | D b Concentration (mg/mL) | Dx (mol %) | pH Donor Equilibrium | Species distribution (%) | Log Kcc | ||
|---|---|---|---|---|---|---|---|---|
| [R−DH+] | DH+ | D | ||||||
| 1/40 | DNA–Atx | 2.20 | 9.38 | 6.21 | 83.98 | 16.01 | 0.010 | 5.86 |
| 3.33 | 18.34 | 6.31 | 83.28 | 16.70 | 0.010 | 5.79 | ||
| 4.49 | 29.30 | 6.44 | 96.70 | 3.30 | 0.003 | 6.49 | ||
| 6.60 | 28.99 | 6.19 | 91.49 | 8.51 | 0.004 | 6.30 | ||
| DNA–Prx | 1.25 | 11.25 | 5.66 | 76.06 | 23.94 | 0.004 | 6.06 | |
| 2.45 | 20.71 | 5.73 | 74.47 | 25.53 | 0.004 | 6.00 | ||
| 3.72 | 24.51 | 5.72 | 69.78 | 30.21 | 0.005 | 5.92 | ||
| 4.90 | 26.19 | 5.94 | 82.65 | 17.35 | 0.005 | 6.03 | ||
| DNA–Lix | 2.24 | 9.42 | 5.91 | 85.01 | 14.75 | 0.230 | 4.51 | |
| 3.39 | 12.65 | 5.91 | 86.44 | 13.34 | 0.225 | 4.58 | ||
| 4.48 | 20.59 | 5.81 | 84.94 | 14.87 | 0.191 | 4.66 | ||
| DNA–At85 | 4.48 | 28.79 | 6.44 | 96.70 | 3.30 | 0.003 | 6.49 | |
| DNA–At85–NaCl25 | 4.48 | 22.67 | 6.18 | 91.19 | 8.81 | 0.004 | 6.26 | |
| DNA–At85–NaCl50 | 4.48 | 14.08 | 6.29 | 89.98 | 10.01 | 0.006 | 6.04 | |
| DNA–At85–NaCl100 | 4.48 | 13.32 | 5.99 | 49.35 | 50.63 | 0.018 | 5.37 | |
| 1/10 | DNA–Atx | 2.20 | 36.53 | 6.43 | 98.67 | 1.33 | 0.001 | 6.95 |
| 3.33 | 59.44 | 6.41 | 99.27 | 0.73 | 0.001 | 7.42 | ||
| 4.49 | 75.37 | 6.44 | 98.80 | 1.20 | 0.001 | 7.40 | ||
| 5.52 | 92.46 | 7.00 | 97.22 | 2.77 | 0.009 | 6.98 | ||
| DNAR–Atx | 1.29 | 40.77 | 6.71 | 93.19 | 6.80 | 0.011 | 6.66 | |
| 1.57 | 41.95 | 6.63 | 86.60 | 13.39 | 0.018 | 6.43 | ||
| DNAR–Prx | 0.71 | 30.77 | 6.28 | 88.45 | 11.54 | 0.01 | 6.63 | |
| 1.45 | 43.60 | 6.25 | 63.24 | 36.74 | 0.02 | 6.09 | ||
| 2.10 | 55.13 | 6.26 | 68.25 | 31.74 | 0.02 | 6.28 | ||
| DNAR–Lix | 1.44 | 30.74 | 6.13 | 53.04 | 45.73 | 1.21 | 4.40 | |
| 2.13 | 50.93 | 6.07 | 70.29 | 29.03 | 0.69 | 4.93 | ||
| D Proportion (X mol %) | DNA–Na | DNA–NaR | |||||||
|---|---|---|---|---|---|---|---|---|---|
| At | Pr | Li | Me | At | Pr | Li | Ti | Bz | |
| 0 | −28.04 | −28.04 | −28.04 | −28.04 | −35.86 | −35.86 | −35.86 | −35.86 | −35.86 |
| 25 | −21.53 | −21.24 | −20.63 | −18.96 | - | - | - | - | - |
| 45 | −21.17 | −19.62 | −18.15 | −18.50 | - | - | - | - | - |
| 65 | −19.38 | −19.16 | −16.13 | −14.53 | −26.68 | −34.91 | −26.70 | −28.06 | −28.38 |
| 85 | −16.59 | −16.88 | −15.60 | −13.36 | - | - | - | - | - |
| System | Slope | Δ Slope | y-Intercept | r2 |
|---|---|---|---|---|
| At | 8.76 | 6.12 | −13.56 | 1.000 |
| DNA–At | 2.64 | −1.77 | 0.989 | |
| Li | 7.15 | 2.82 | −10.52 | 0.999 |
| DNA–Li | 4.33 | −8.11 | 0.993 | |
| Pr | 7.42 | 5.57 | −11.80 | 0.996 |
| DNA–Pr | 1.85 | −3.07 | 0.992 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alarcón, L.P.; Baena, Y.; Manzo, R.H. Interaction between DNA and Drugs Having Protonable Basic Groups: Characterization through Affinity Constants, Drug Release Kinetics, and Conformational Changes. Sci. Pharm. 2017, 85, 1. https://doi.org/10.3390/scipharm85010001
Alarcón LP, Baena Y, Manzo RH. Interaction between DNA and Drugs Having Protonable Basic Groups: Characterization through Affinity Constants, Drug Release Kinetics, and Conformational Changes. Scientia Pharmaceutica. 2017; 85(1):1. https://doi.org/10.3390/scipharm85010001
Chicago/Turabian StyleAlarcón, Liliana P., Yolima Baena, and Rubén H. Manzo. 2017. "Interaction between DNA and Drugs Having Protonable Basic Groups: Characterization through Affinity Constants, Drug Release Kinetics, and Conformational Changes" Scientia Pharmaceutica 85, no. 1: 1. https://doi.org/10.3390/scipharm85010001