
Development of RP-HPLC, Stability Indicating Method for Degradation Products of Linagliptin in Presence of Metformin HCl by Applying 2 Level Factorial Design; and Identification of Impurity-VII, VIII and IX and Synthesis of Impurity-VII

Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Chemicals
2.2. Instrumentation and Chromatographic Conditions
2.3. UPLC-TOF/MS, LC-MS and NMR Conditions
2.4. Preparation of Standard, System Suitability, and Sample Solution
3. Results and Discussion
3.1. Method Development and Optimization
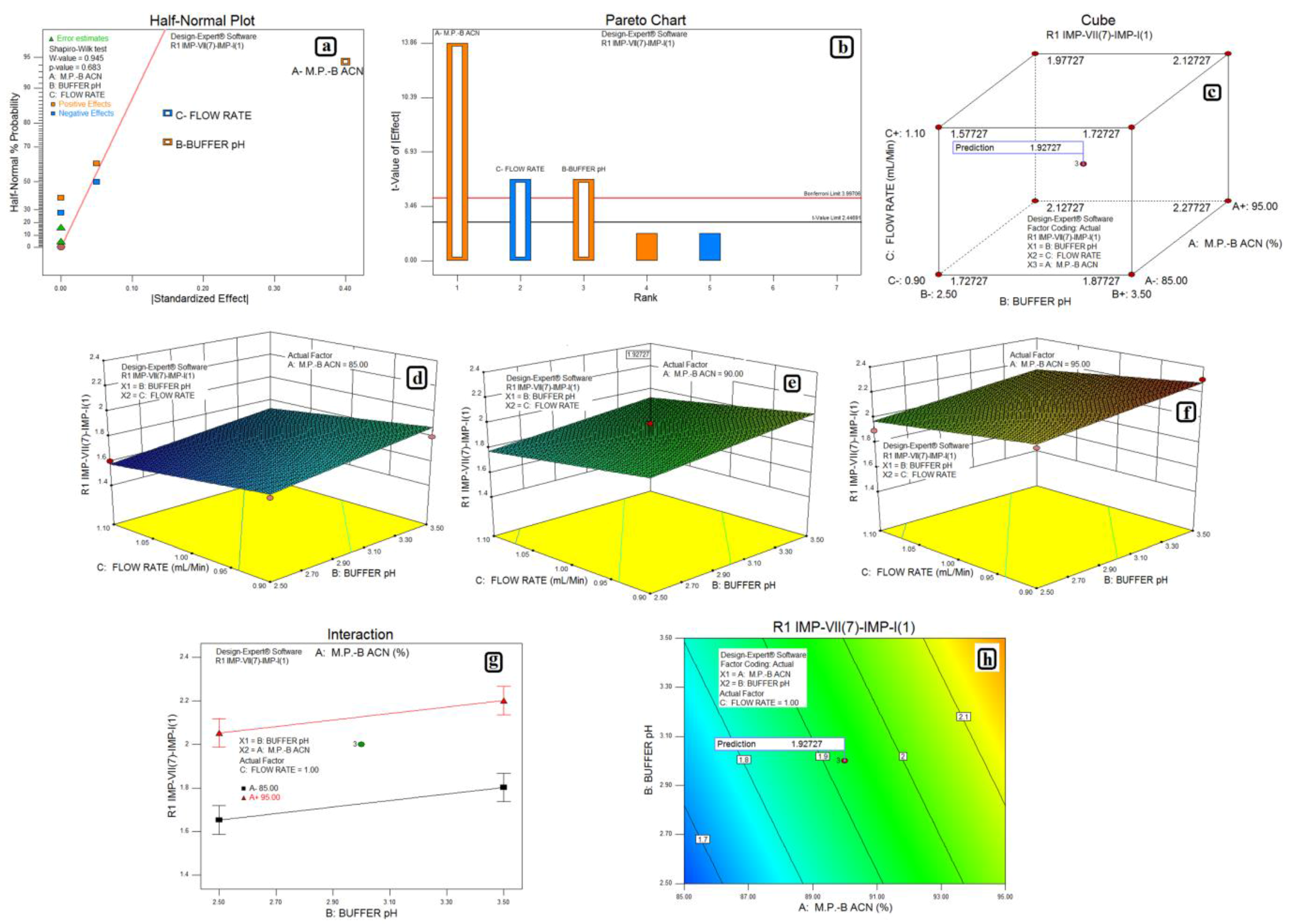
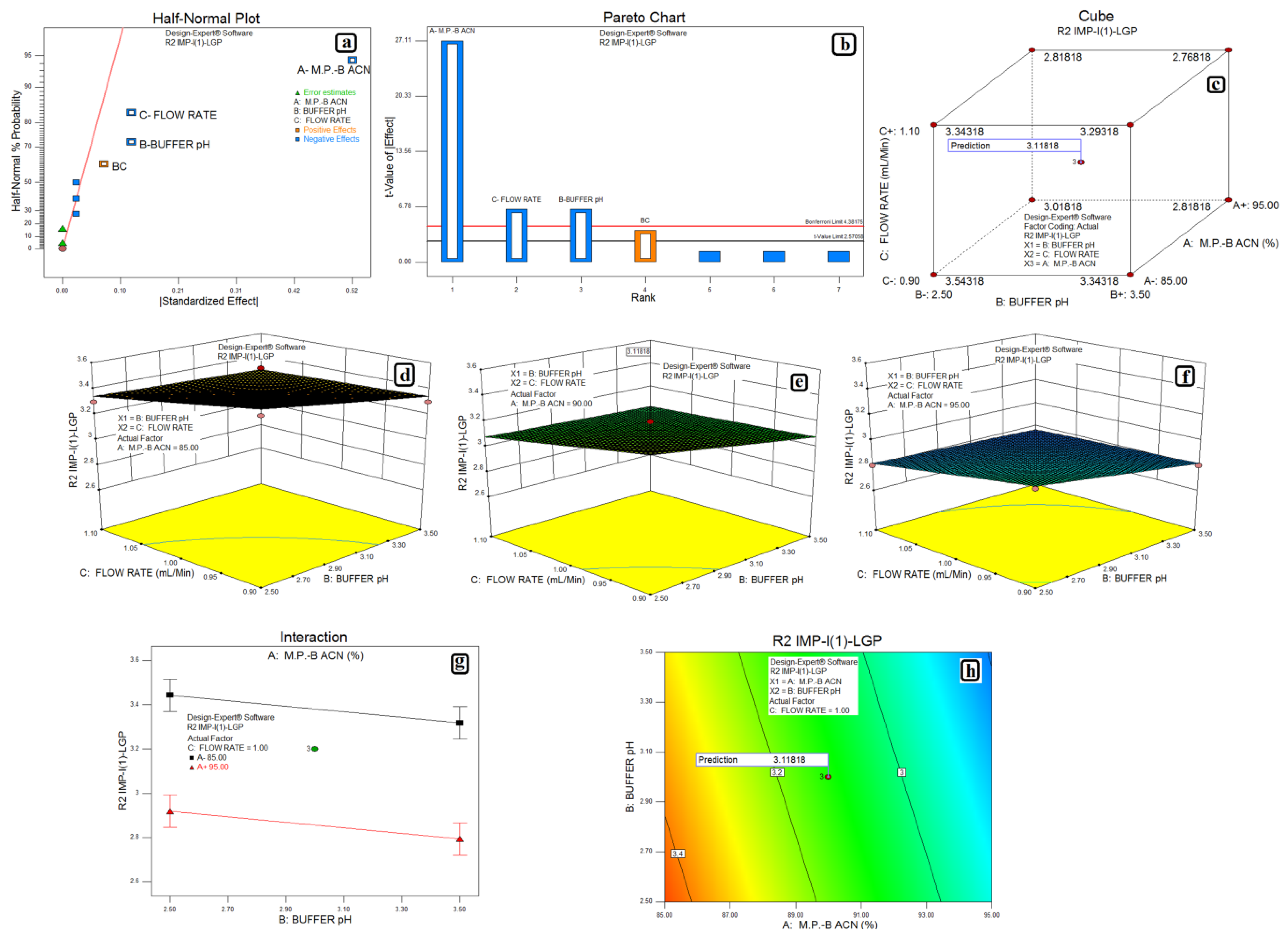
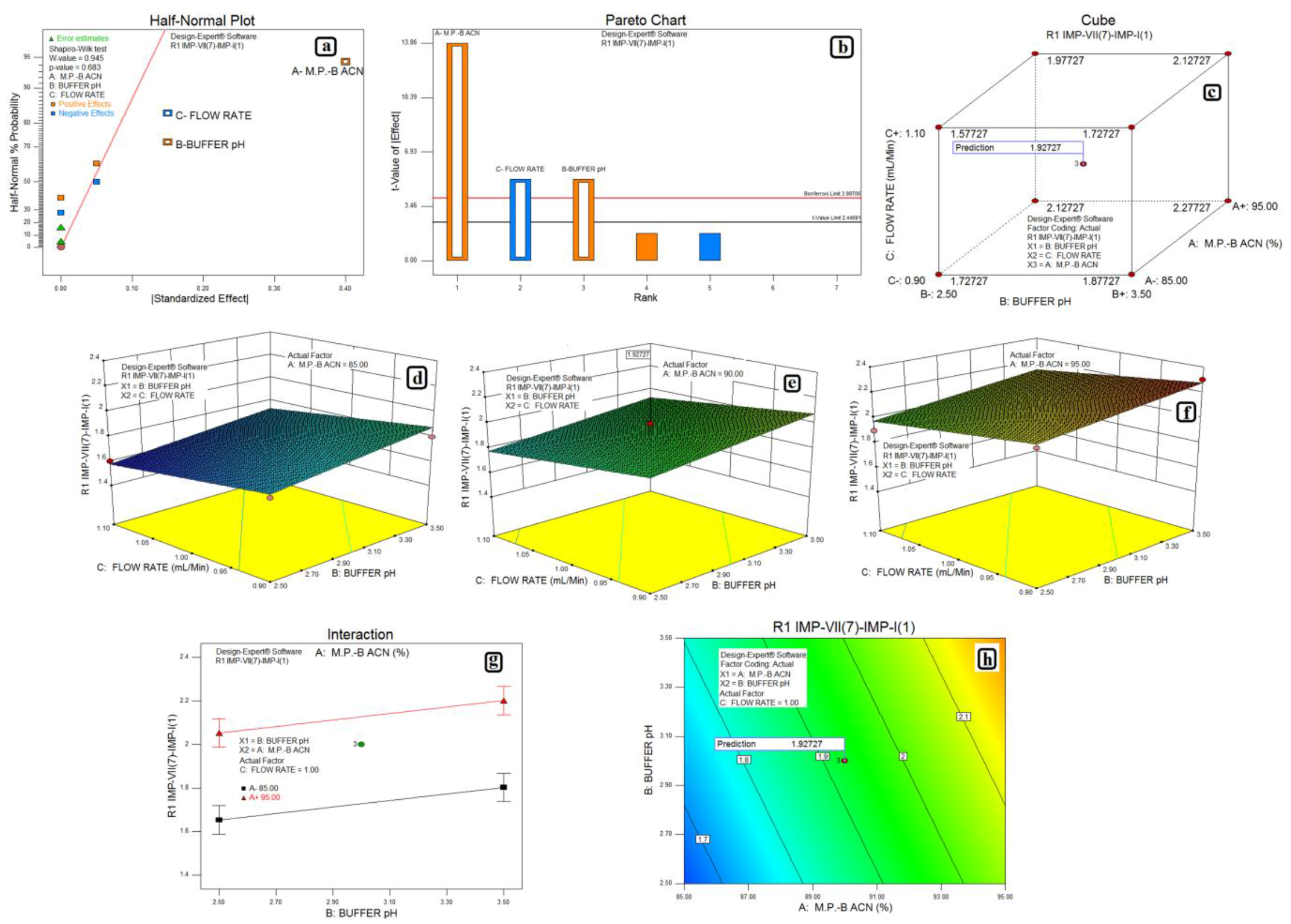
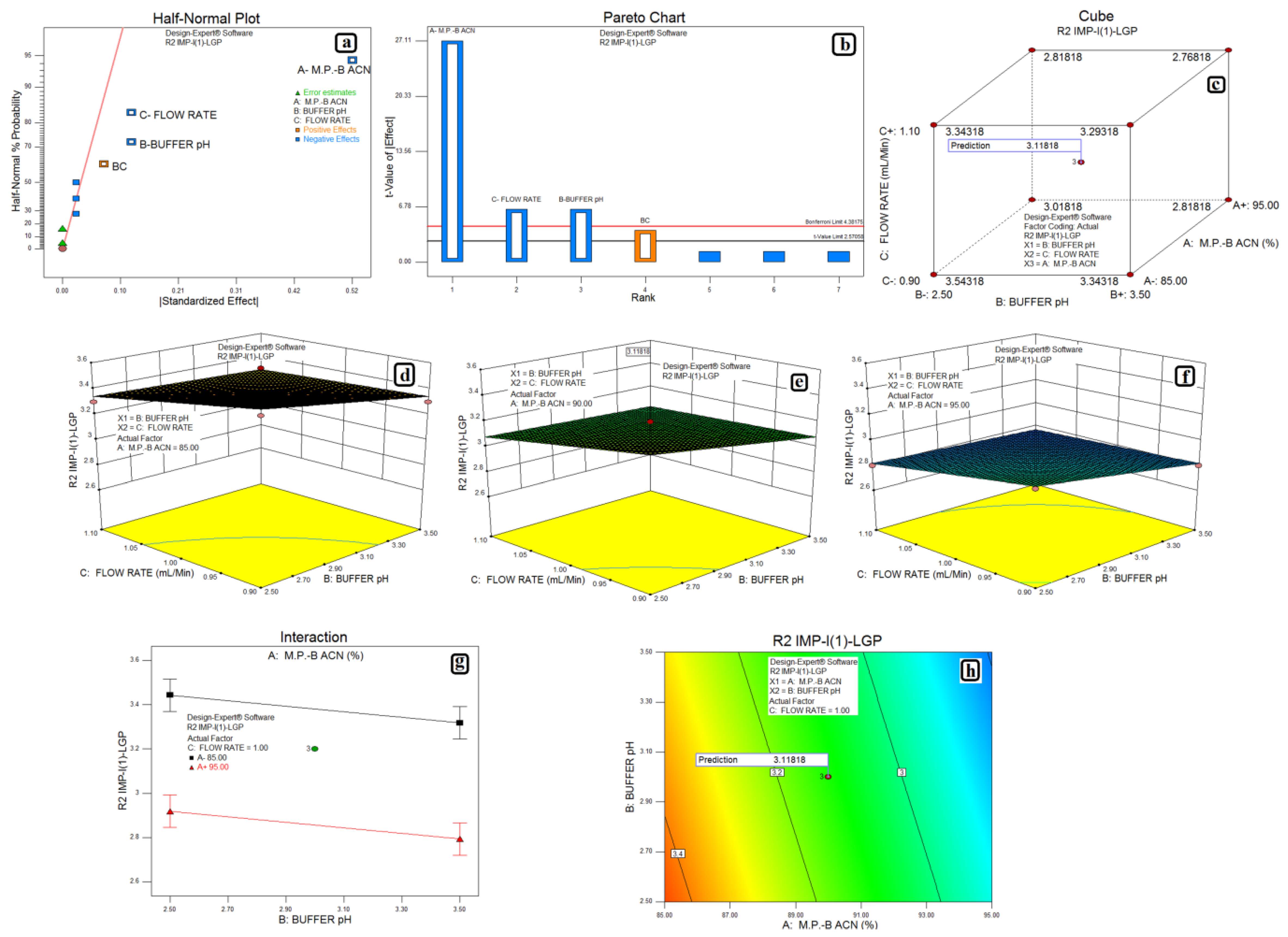
3.2. Design of Experiments and Data Analysis
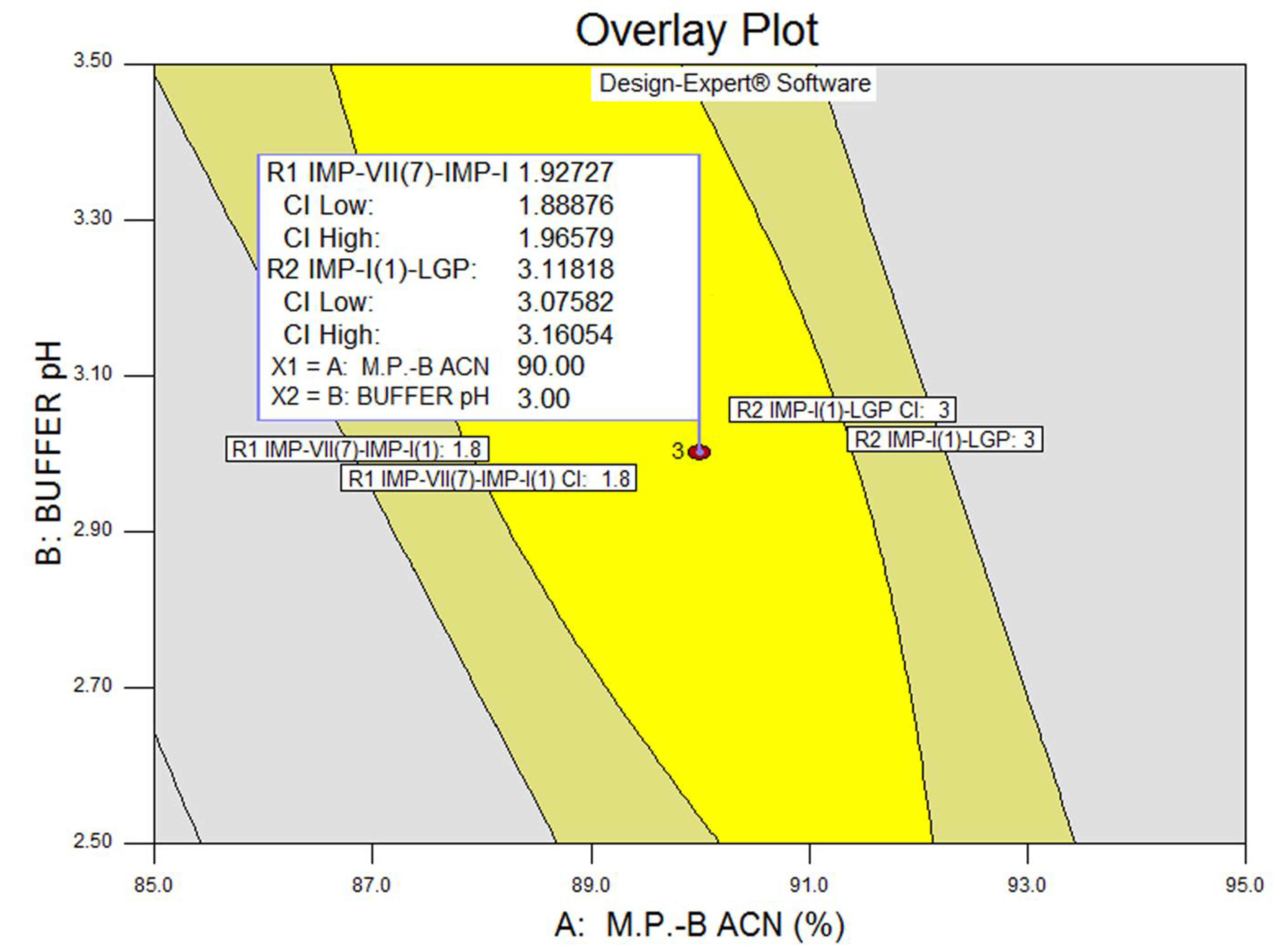
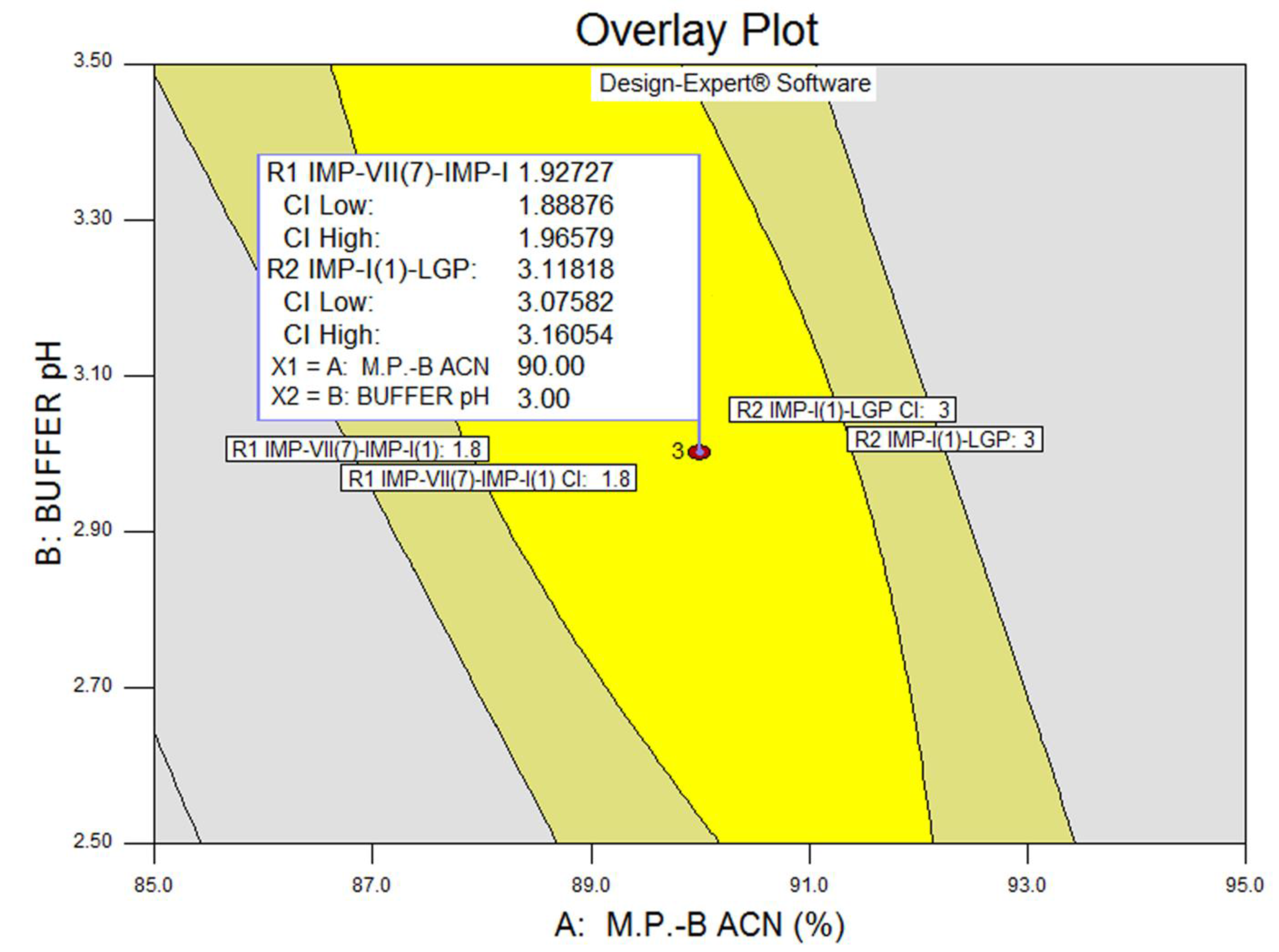
3.3. Statistical Analysis: ANOVA Analysis, Desirability and Overlay Plot
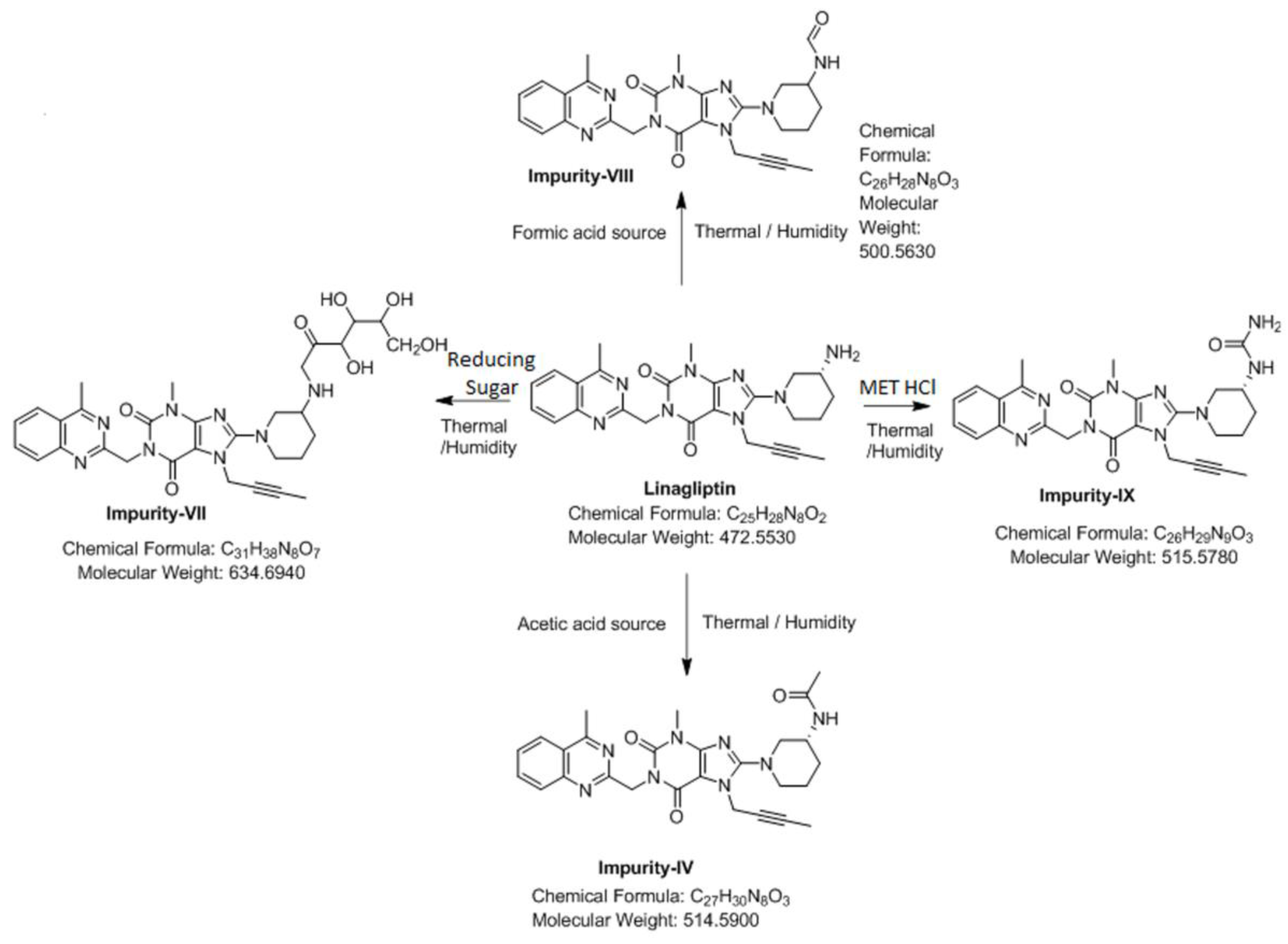
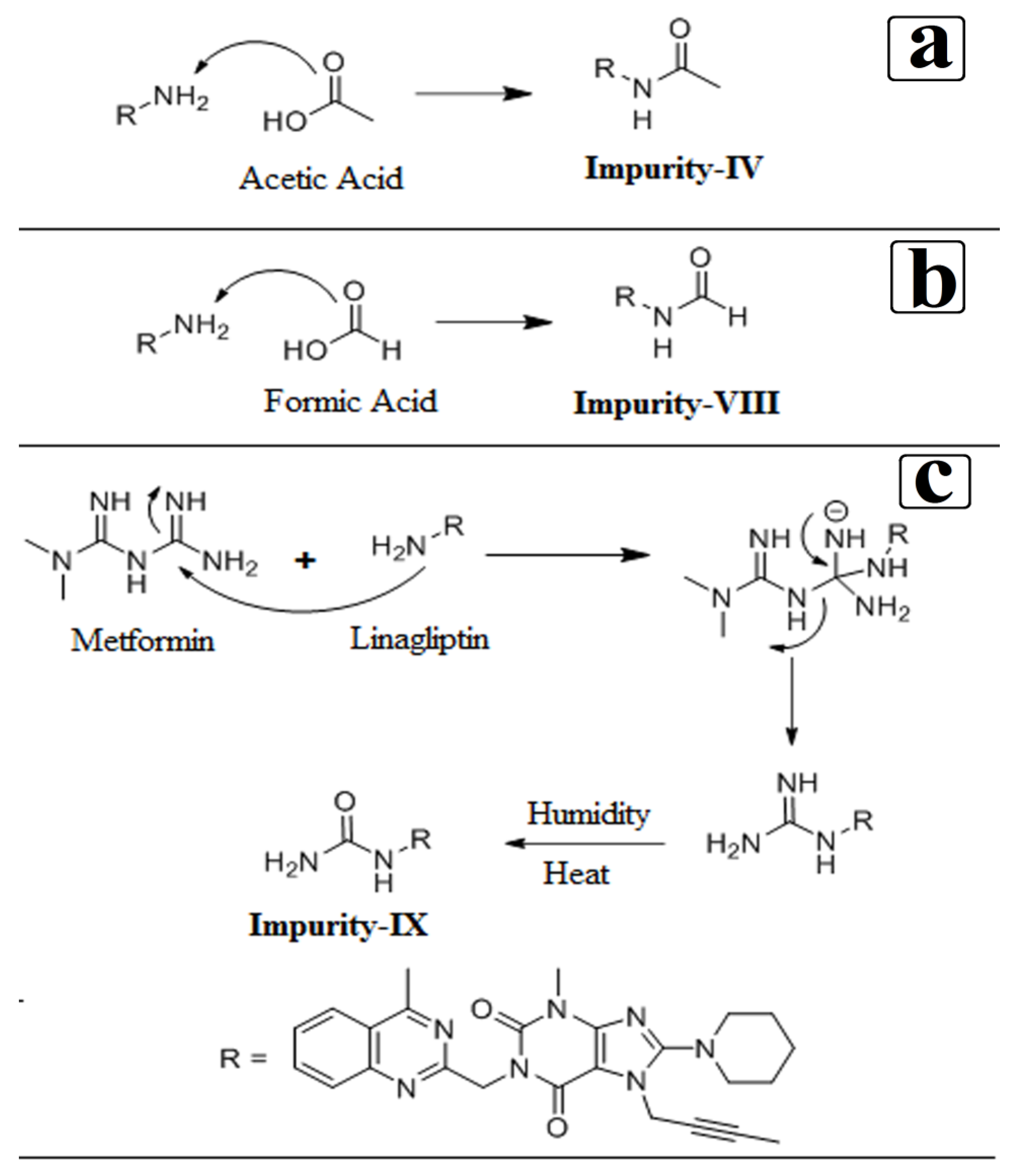
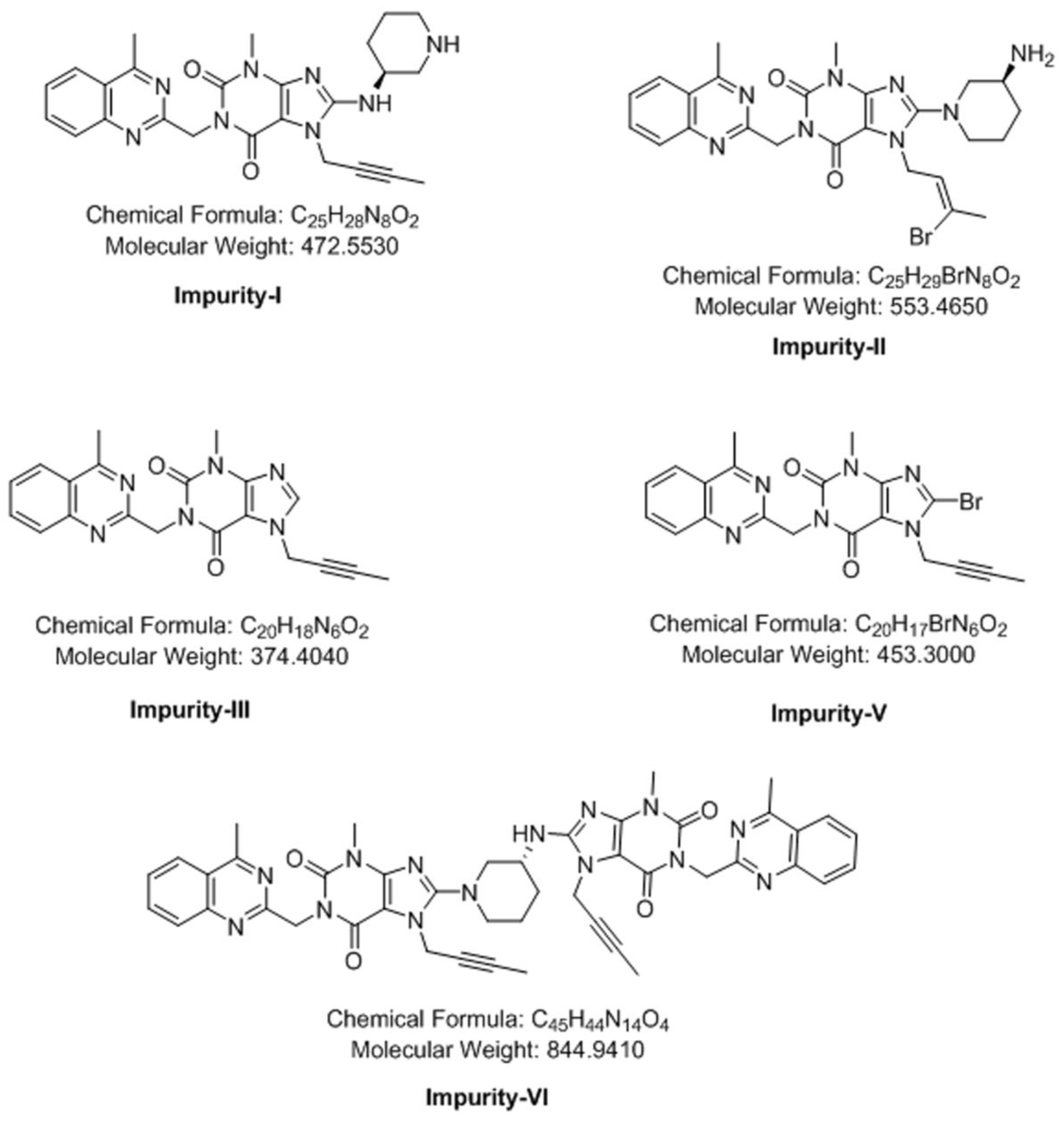
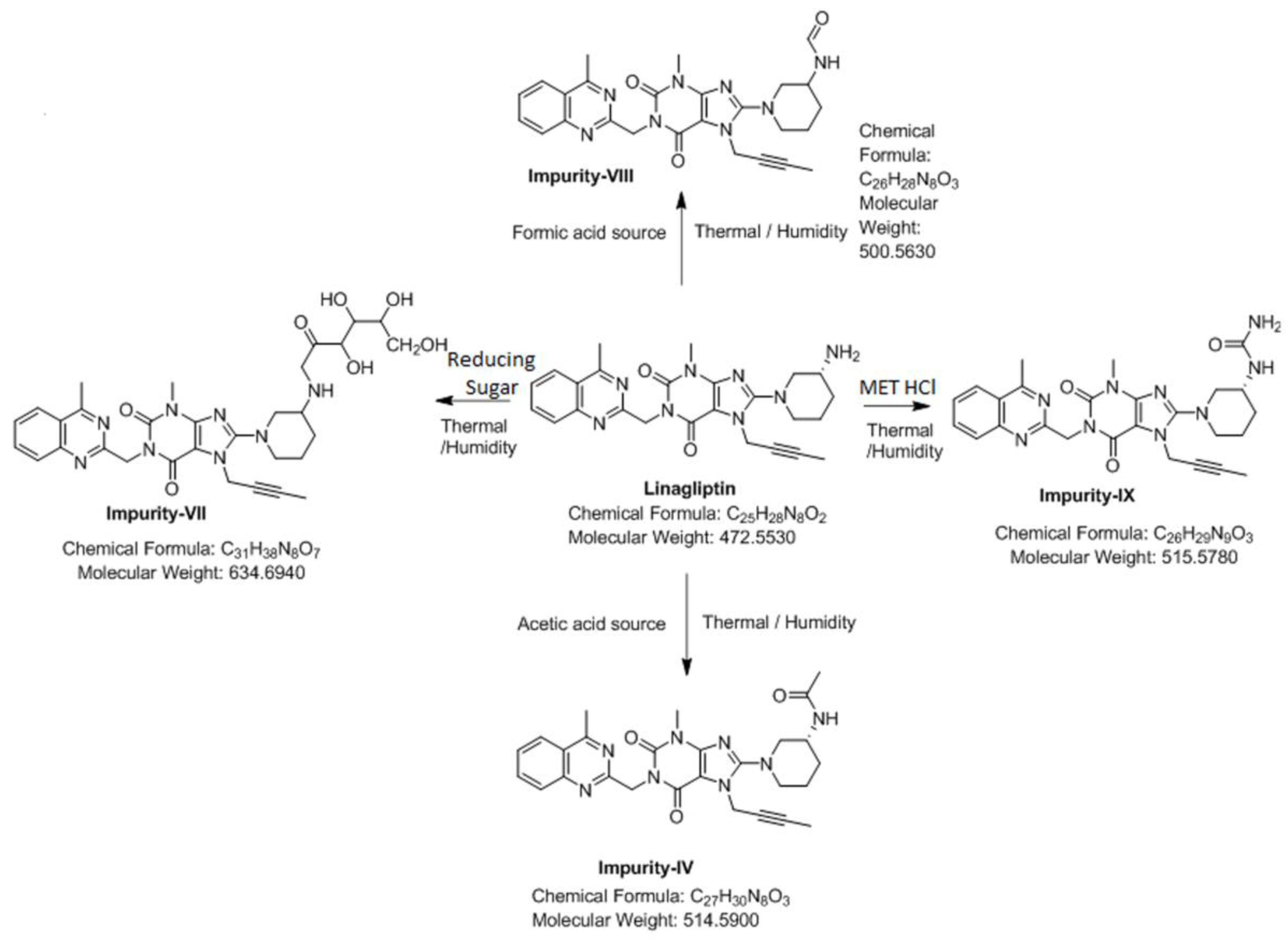
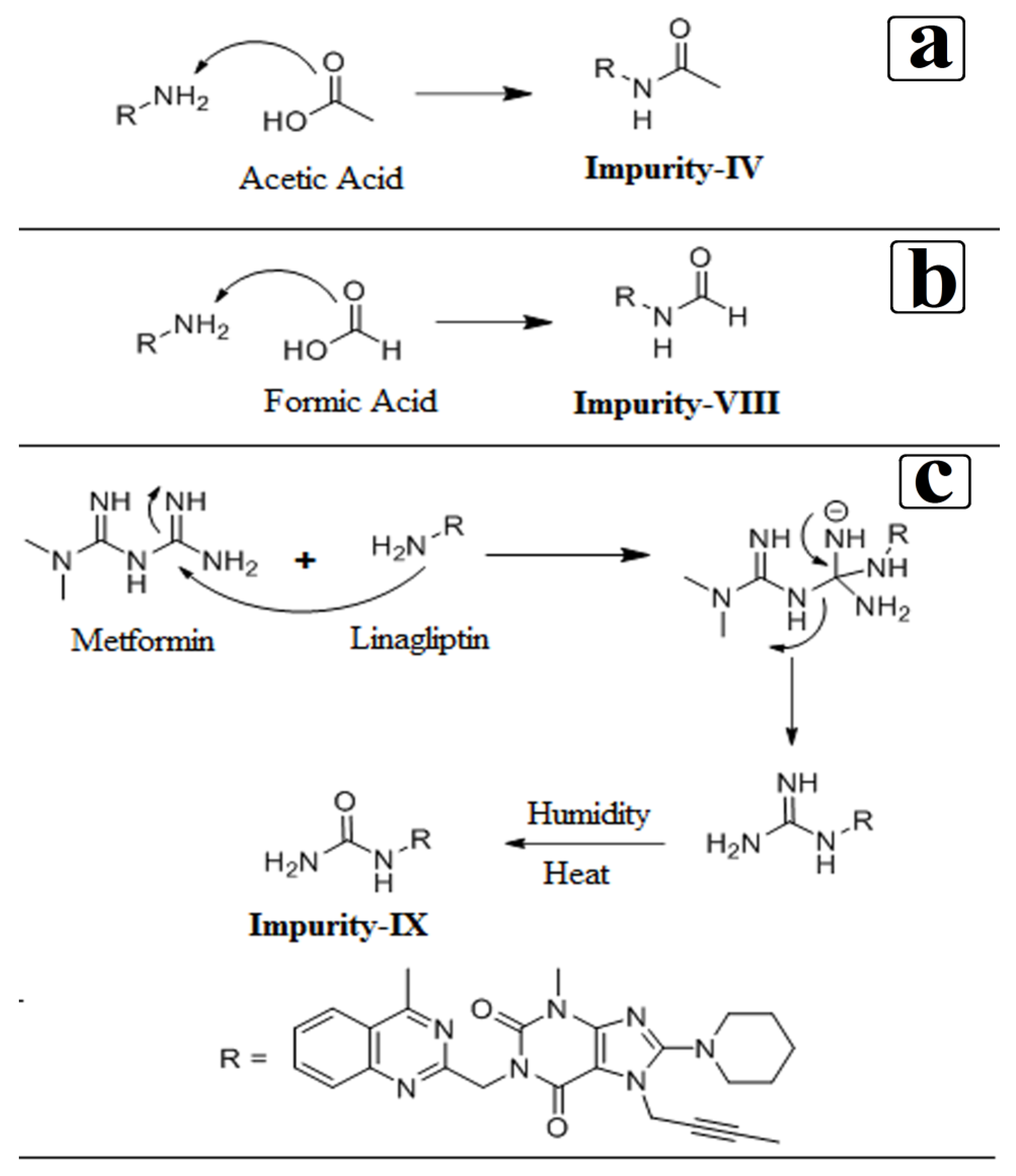
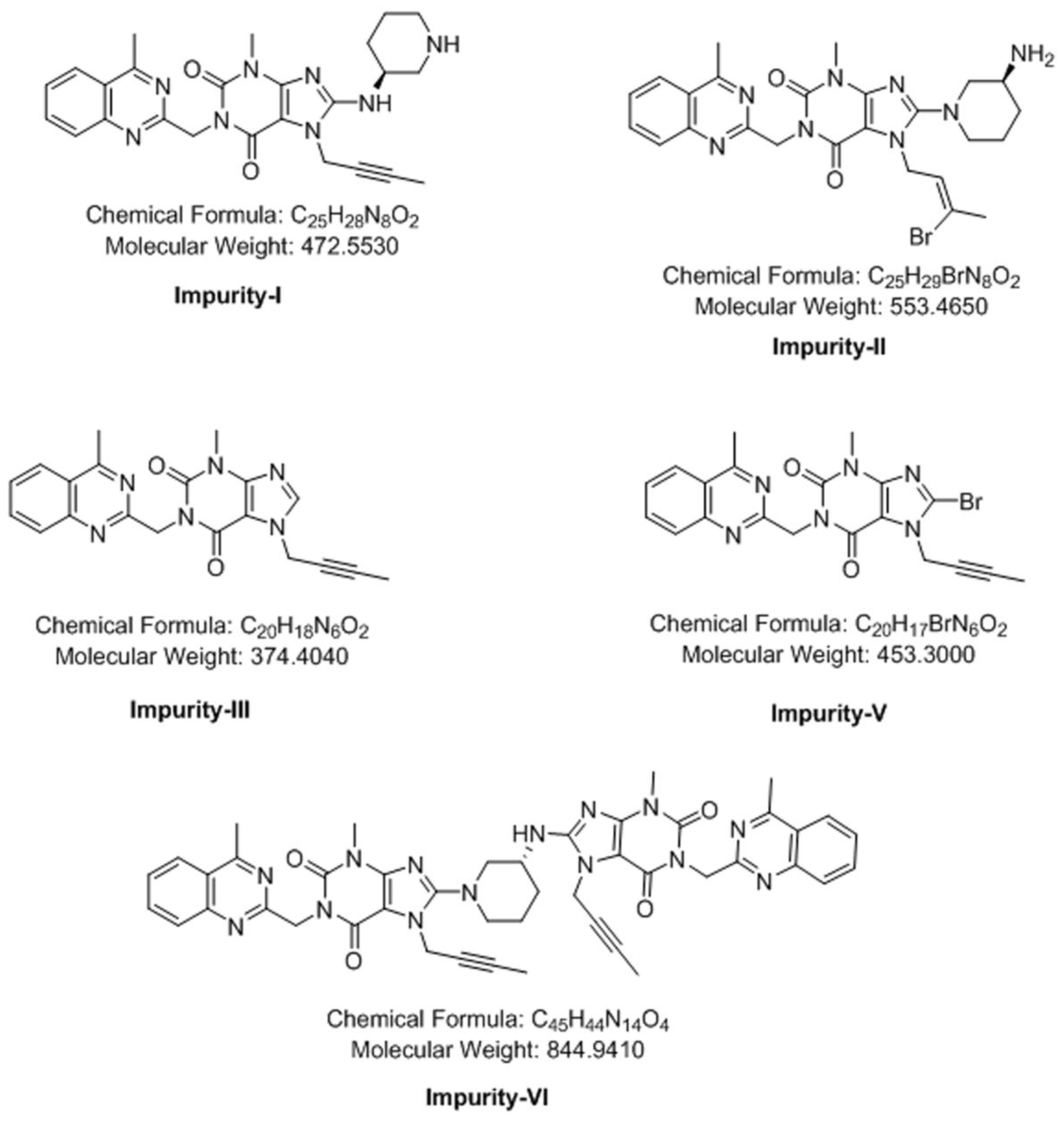
3.4. Synthesis of Impurity-VII (7) and Structural Elucidation of Unspecified, Degradation Impurities
3.4.1. Synthesis and Structural Elucidation of Impurity-VII (7)
3.4.2. Structural Elucidation of Impurity-VIII (8)
3.4.3. Structural Elucidation of Impurity-IX (9)
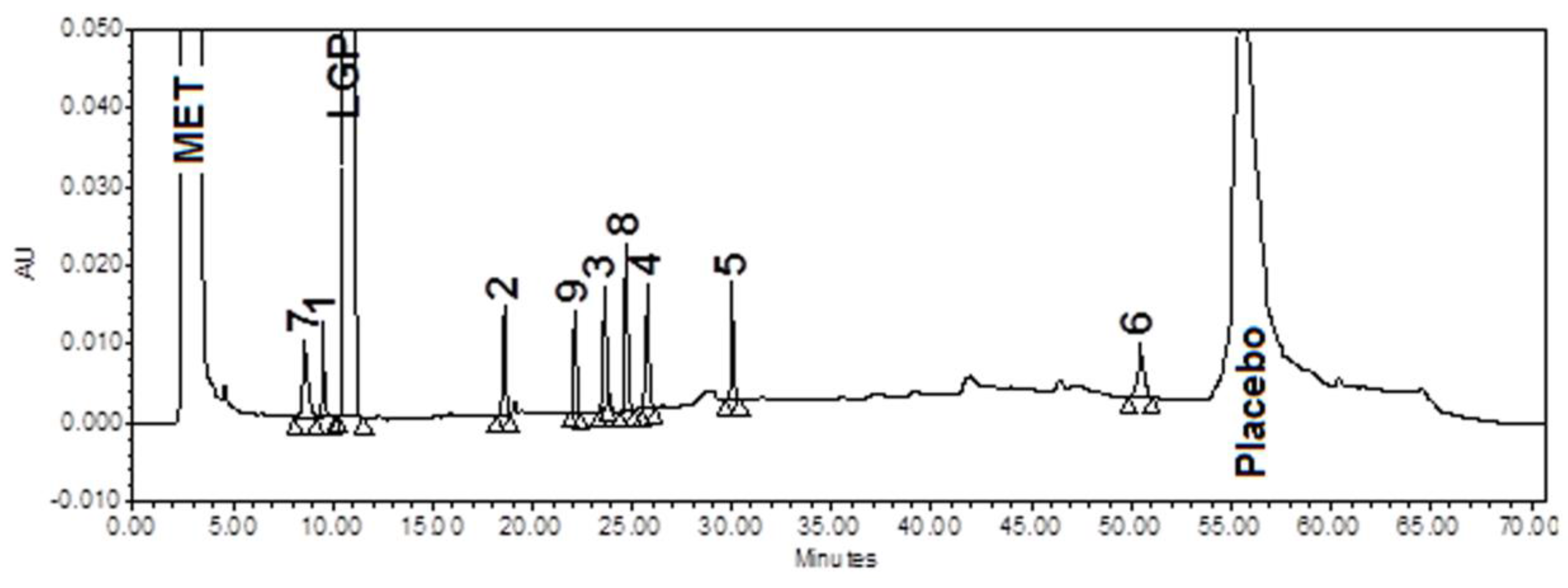
3.5. Stress (Forced Degradation) Study/Specificity
3.6. Validation
3.6.1. Precision
3.6.2. Limit of Detection and Limit of Quantitation
3.6.3. Accuracy
3.6.4. Linearity and Range
3.6.5. Robustness
3.6.6. Solution and Mobile Phase Stability
3.6.7. Filter Compatibility
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Deacon, C.F.; Holst, J.J. Linagliptin, a xanthine-based dipeptidyl peptidase-4 inhibitor with an unusual profile for the treatment of type 2 diabetes. Expert Opin. Investig. Drugs 2010, 1, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Guedes, E.P.; Hohl, A.; de Melo, T.G.; Lauand, F. Linagliptin: Pharmacology, efficacy and safety in type 2 diabetes treatment. Diabetol. Metab. Syndr. 2013, 5, 25. [Google Scholar] [CrossRef] [PubMed]
- Barnett, A.H. Linagliptin: A novel dipeptidyl peptidase 4 inhibitor with a unique place in therapy. Adv. Ther. 2011, 28, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Sachs, G.; Chang, H.H.; Rabon, E.; Schackman, R.; Lewin, M.; Saccomani, G. A nonelectrogenic H+ pump in plasma membrane of hog stomach. J. Biol. Chem. 1976, 251, 7690–7698. [Google Scholar] [PubMed]
- Dibona, D.R.; Ito, S.; Berglindh, T.; Sachs, G. Cellular site of acid secretion. Proc. Natl. Acad. Sci. USA 1979, 76, 6689–6693. [Google Scholar] [CrossRef] [PubMed]
- Fellenius, E.; Berglindh, T.; Sachs, G.; Olbe, L.; Elander, B.; Sjöstrand, S.E.; Wallmark, B. Substituted benzimidazoles inhibit gastric acid secretion by blocking H+, K+ ATPase. Nature 1981, 290, 156–161. [Google Scholar] [CrossRef]
- Del Prato, S.; Barnett, A.H.; Huisman, H.; Neubacher, D.; Woerle, H.J.; Dugi, K.A. Effect of Linagliptin monotherapy on glycaemic control and markers of β-cell function in patients with inadequately controlled type 2 diabetes: A randomized controlled trial. Diabetes Obes. Metab. 2011, 3, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Jentadeuto—Mechanism of Action. Available online: http://www.rxlist.com/jentadueto-drug/clinical-pharmacology.htm (accessed on 6 December 2016).
- Phung, O.J.; Scholle, J.M.; Talwar, M.; Coleman, C.L. Effect of noninsulin antidiabetic drugs added to metformin therapy on glycemic control, weight gain, and hypoglycemia in type 2 diabetes. JAMA 2010, 303, 1410–1418. [Google Scholar] [CrossRef] [PubMed]
- Karagiannis, T.; Paschos, P.; Paletas, K.; Matthews, D.R.; Tsapas, A. Dipeptidyl peptidase-4 inhibitors for treatment of type 2 diabetes mellitus in the clinical setting: Systematic review and meta-analysis. BMJ 2012, 344, e1369. [Google Scholar] [CrossRef] [PubMed]
- Amiel, S.A.; Dixon, T.; Mann, R.; Jameson, K. Hypoglycaemia in Type 2 diabetes. Diabet. Med. 2008, 25, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.C.; Tu, Y.K.; Chien, M.N.; Chien, K.L. Effect of antidiabetic agents added to metformin on glycemic control, hypoglycemia and weight change in patients with type 2 diabetes: A network meta-analysis. Diabetes Obes. Metab. 2012, 14, 810–820. [Google Scholar] [CrossRef] [PubMed]
- Hermansen, K.; Mortensen, L.S. Bodyweight changes associated with antihyperglycemic agents in type 2 diabetes mellitus. Drug Saf. 2007, 30, 1127–1142. [Google Scholar] [CrossRef] [PubMed]
- Bonora, E. Antidiabetic medications in overweight/obese patients with type 2 diabetes: Drawbacks of current drugs and potential advantages of incretin-based treatment on body weight. Int. J. Clin. Pract. Suppl. 2007, 154, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Scheen, A.J. Drug interactions of clinical importance with antihyperglycemic agents: An update. Drug Saf. 2005, 28, 601–631. [Google Scholar] [CrossRef] [PubMed]
- Metformin Hydrochloride U.S. FDA. Available online: http://www.fda.gov/ohrms/dockets/dailys/02/May02/053102/800471e6.pdf (accessed on 6 December 2016).
- Jadhav, S.B.; Mane, R.M.; Narayanan, K.L.; Bhosale, P.N. Analytical Enantio-Separation of Linagliptin in Linagliptin and Metformin HCl Dosage Forms by Applying Two-Level Factorial Design. Sci. Pharm. 2016, 84, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Mitri, J.; Hamdy, O. Diabetes medications and body weight. Expert Opin. Drug Saf. 2009, 8, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Glodek, M.; Liebowitz, S.; McCarthy, R.; McNally, G.; Oksanen, C.; Schultz, T.; Sundararajan, M.; Vorkapich, R.; Vukovinsky, K.; Watts, C.; et al. Process robustness—A PQRI White paper. Pharm. Eng. 2006, 26, 1–11. [Google Scholar]
- Jadhav, S.B.; Kumar, C.K.; Bandichhor, R.; Bhosale, P.N. Development of RP UPLC-TOF/MS, stability indicating method for omeprazole and its related substances by applying two level factorial design; and identification and synthesis of non-pharmacopoeial impurities. J. Pharm Biomed. Anal. 2016, 118, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Metformin Hydrochloride. In Indian Pharmacopoeia; Government of India, Ministry of Health and Family Welfare: Ghaziabad, India, 2014; pp. 2186–2188.
- Metformin Hydrochloride. In British Pharmacopoeia; Medicines and Healthcare Products Regulatory Agency: London, UK, 2016; Volume 2, pp. 233–235.
- Metformin Hydrochloride. In European Pharmacopoeia, 9th ed.; European Pharmacopoeia Commission, Council of Europe European Directorate for the Quality of Medicines (EDQM): Strasbourg, France, 2016; pp. 4764–4766.
- Metformin Hydrochloride. In United States Pharmacopoeia, 39th ed.; United States Pharmacopeial Convention: Rockville, MD, USA, 2016; pp. 4764–4766, 5416–5419, 4119–4122, 8070–8074, 8092–8100.
- Huang, Y.; He, X.; Wu, T.; Zhang, F. Synthesis and Characterization of Process-Related Impurities of Antidiabetic Drug Linagliptin. Molecules 2016, 21, 1041. [Google Scholar] [CrossRef] [PubMed]
- Nandi, S.; Naresh, A.; Reddy, G.B.N.; Venkateswarlu, P.; Reddy, A.S.P.; Reddy, K.S.K. Process Related Impurities in Anti-Diabetic Drug Linagliptin. J. Pharm. Res. Opin. 2016, 5. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Response | Source | Sum of Squares | Degrees of Freedom | Mean Square | F Value | p-Value Prob > F | Model Status | R2 | Adjusted R2 | Predicted R2 |
|---|---|---|---|---|---|---|---|---|---|---|
| Impurity-VII (7) and Impurity-I (1) | Model | 0.41 | 3 | 0.14 | 82.00 | <0.0001 | Significant | 0.9280 | 0.8971 | 0.8300 |
| A-M.P.-B:ACN | 0.32 | 1 | 0.32 | 192.00 | <0.0001 | |||||
| B-Buffer pH | 0.045 | 1 | 0.045 | 27.00 | 0.0020 | |||||
| C-Flow Rate | 0.045 | 1 | 0.045 | 27.00 | 0.0020 | |||||
| Residual | 0.010 | 6 | 0.00166 | |||||||
| Lack of Fit | 0.010 | 4 | 0.00250 | |||||||
| Pure Error | 0.000 | 2 | 0.000 | |||||||
| Cor total | 0.44 | 10 | ||||||||
| Impurity-I (1) and LGP | Model | 0.62 | 4 | 0.16 | 208.33 | <0.0001 | Significant | 0.9522 | 0.9204 | 0.8603 |
| A-M.P.-B:ACN | 0.55 | 1 | 0.55 | 735.00 | <0.0001 | |||||
| B-Buffer pH | 0.031 | 1 | 0.031 | 41.67 | 0.0013 | |||||
| C-Flow Rate | 0.031 | 1 | 0.031 | 41.67 | 0.0013 | |||||
| Residual | 0.00375 | 5 | 0.00075 | |||||||
| Lack of Fit | 0.0037 | 3 | 0.00125 | |||||||
| Pure Error | 0.000 | 2 | 0.000 | |||||||
| Cor total | 0.66 | 10 |
| Nature of Stress | Stress Condition | Purity Angle | Purity Threshold | %Degradation | Mass Balance | Purity Flag |
|---|---|---|---|---|---|---|
| Acid | 10 mL-5N HCl- for 23 h on bench top | 0.146 | 0.267 | 0.1117 | 99.7 | No |
| Base | 10 mL-5N NaOH- for 25 h on bench top | 0.192 | 0.320 | 3.3189 | 96.2 | No |
| Peroxide | 20 mL 30% H2O2-2 hon bench top | 0.137 | 0.319 | 1.6259 | 95.8 | No |
| Water | 10 mL of water 5 days on bench top | 0.144 | 0.262 | Nil | 99.7 | No |
| Thermal | At 105 °C for 6 h in an oven | 0.145 | 0.267 | 1.8186 | 96.3 | No |
| Photostabilty | 200 watt hours/square meter and 1.2 million lux hours | 0.143 | 0.271 | 0.0528 | 101.3 | No |
| Humidity | 90%RH at 25 °C for 7 days | 0.140 | 0.275 | 0.1550 | 100.5 | No |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jadhav, S.B.; Reddy, P.S.; Narayanan, K.L.; Bhosale, P.N. Development of RP-HPLC, Stability Indicating Method for Degradation Products of Linagliptin in Presence of Metformin HCl by Applying 2 Level Factorial Design; and Identification of Impurity-VII, VIII and IX and Synthesis of Impurity-VII. Sci. Pharm. 2017, 85, 25. https://doi.org/10.3390/scipharm85030025
Jadhav SB, Reddy PS, Narayanan KL, Bhosale PN. Development of RP-HPLC, Stability Indicating Method for Degradation Products of Linagliptin in Presence of Metformin HCl by Applying 2 Level Factorial Design; and Identification of Impurity-VII, VIII and IX and Synthesis of Impurity-VII. Scientia Pharmaceutica. 2017; 85(3):25. https://doi.org/10.3390/scipharm85030025
Chicago/Turabian StyleJadhav, Sushant B., P. Sunil Reddy, Kalyanaraman L. Narayanan, and Popatrao N. Bhosale. 2017. "Development of RP-HPLC, Stability Indicating Method for Degradation Products of Linagliptin in Presence of Metformin HCl by Applying 2 Level Factorial Design; and Identification of Impurity-VII, VIII and IX and Synthesis of Impurity-VII" Scientia Pharmaceutica 85, no. 3: 25. https://doi.org/10.3390/scipharm85030025