Systematic Applications of Metabolomics in Metabolic Engineering

Abstract
:Abbreviations

{kind=link}
{kind=link}
| Abbreviation | Meaning | Abbreviation | Meaning |
|---|---|---|---|
| CE-MS | Capillary Electrophoresis-Mass Spectrometry | MOMA | Minimization Of Metabolic Adjustment |
| CHO | Chinese Hamster Ovary | NET | Network-Embedded Thermodynamic |
| COBRA | Constraints Based Reconstruction and Analysis | NMR | Nuclear Magnetic Resonance |
| dFBA | Dynamic Flux Balance Analysis | OMNI | Optimal Metabolic Network Identification |
| EMUs | Elementary Metabolite Units | ODE | Ordinary Differential Equation |
| FBA | Flux Balance Analysis | PLS | Partial Least Squares |
| GC-MS | Gas Chromatography-Mass Spectrometry | PLS-DA | Partial Least Squares Discriminant Analysis |
| HCA | Hierarchical Clustering Analysis | PCA | Principal Components Analysis |
| HPLC | High-Performance Liquid Chromatography | QP | Quadratic Programming |
| idFBA | Integrated-Dynamic Flux Balance Analysis | rFBA | Regulatory Flux Balance Analysis |
| iFBA | Integrated Flux Balance Analysis | SBRT | Systems Biology Research Tool |
| IOMA | Integrative “Omics”-Metabolic Analysis | TMFA | Thermodynamic Metabolic Flux Analysis |
| LP | Linear Programming | TAL | Transaldolase |
| LC-MS | Liquid Chromatography-Mass Spectrometry | TKL | Transketolase |
| MASS | Mass Action Stoichiometric Simulation | TCA | Tricarboxylic Acid |
| MCA | Metabolic Control Analysis | VIP | Variable Importance in the Projection |
| MFA | Metabolic Flux Analysis | VHG | Very High Gravity |
1. Introduction
2. Metabolomics Background
2.1. Analytical Platforms
2.2. Data Analysis
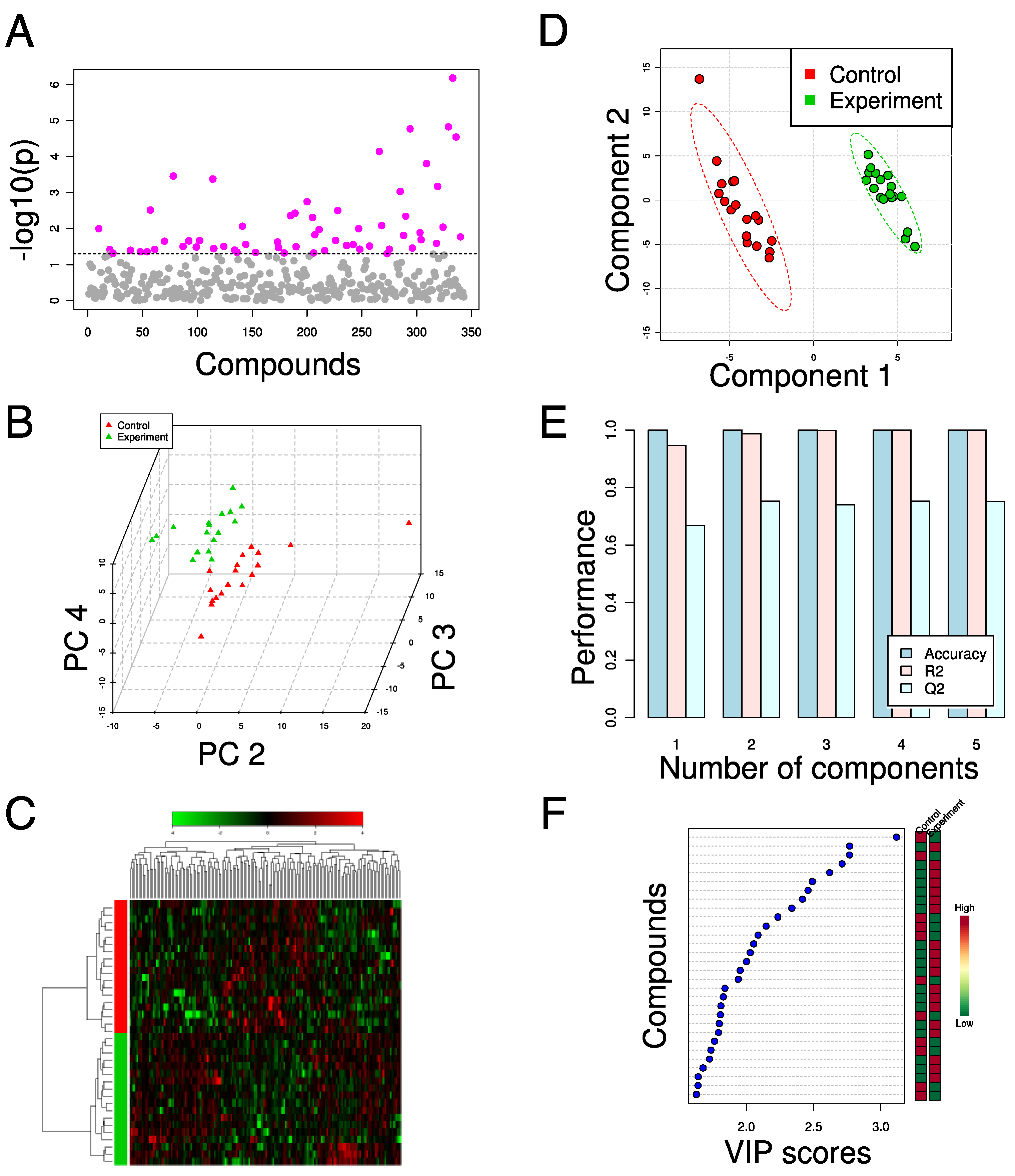
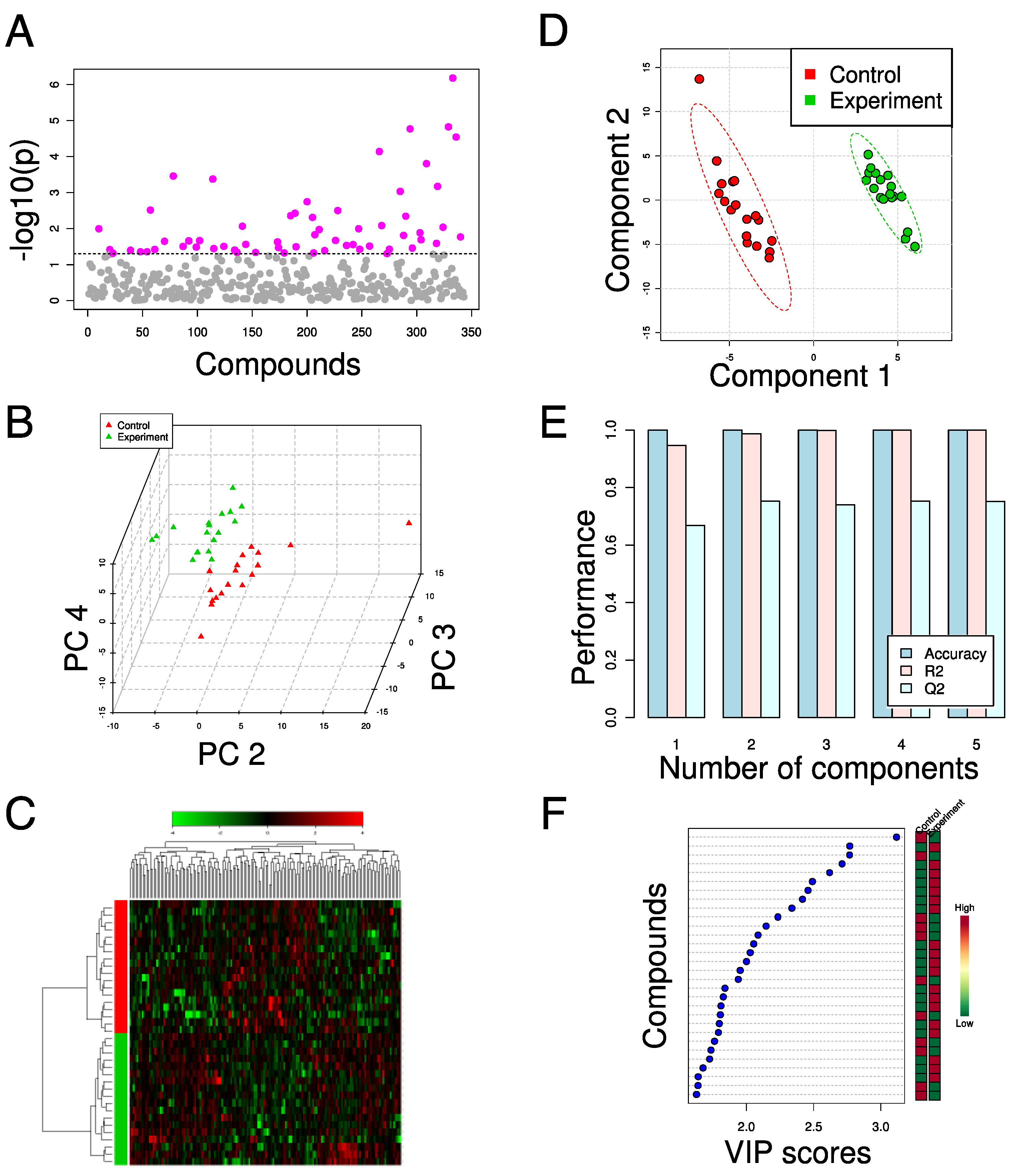
3. Applications of Metabolomics in Metabolic Engineering
3.1. Metabolomics Data as an Extension of Small-Scale, Targeted Analysis
3.2. General Strategies for Integrating Metabolomics into Metabolic Engineering
3.2.1. Adaptive Evolution and High Throughput Libraries: Locating the Cause of Improved Phenotypes
3.2.2. Other Global Analysis Approaches: Harnessing Proteomics, Transcriptomics, and Genomics for Metabolic Engineering
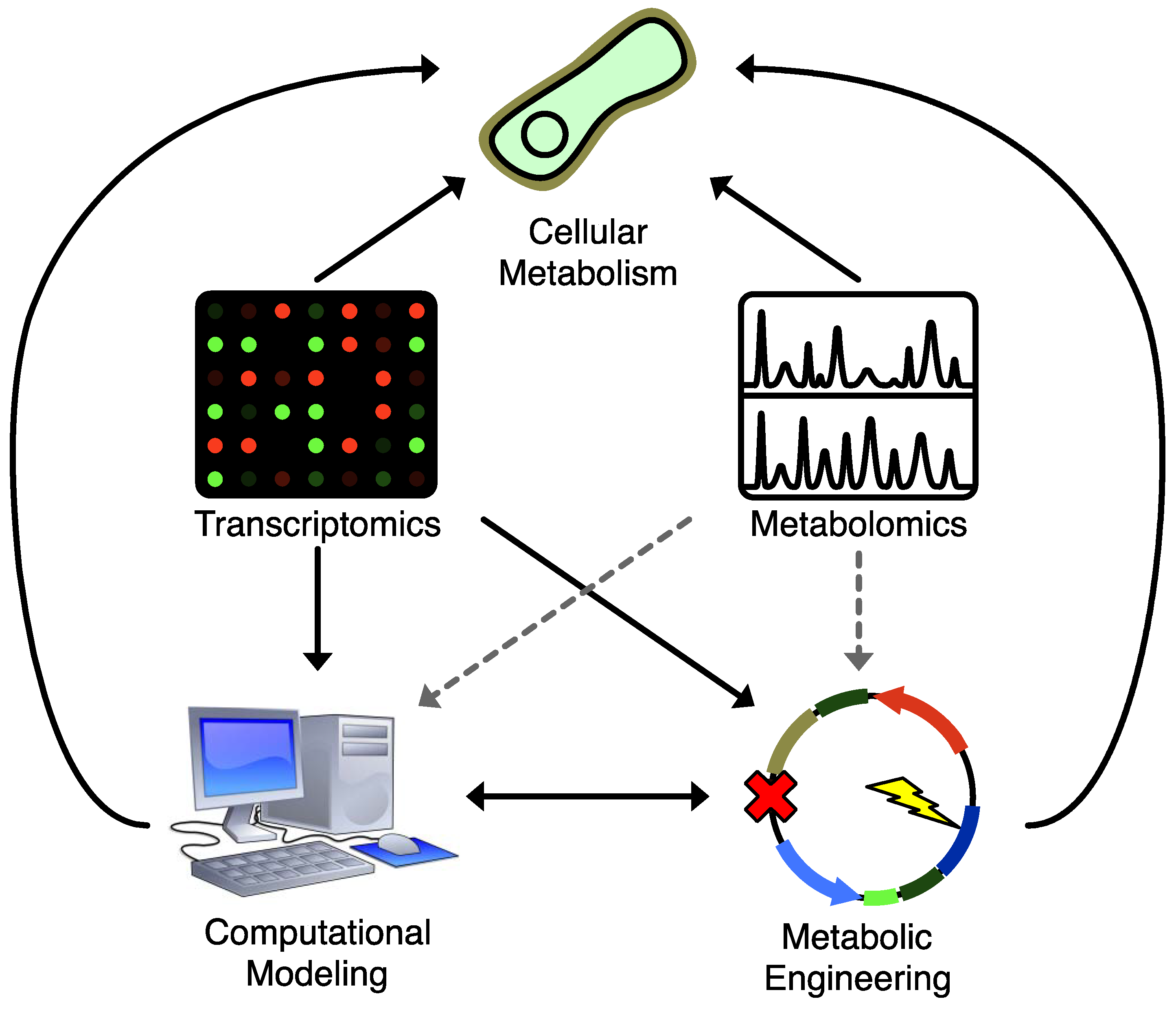
4. Computational Methods for Combining Metabolomics and Metabolic Engineering
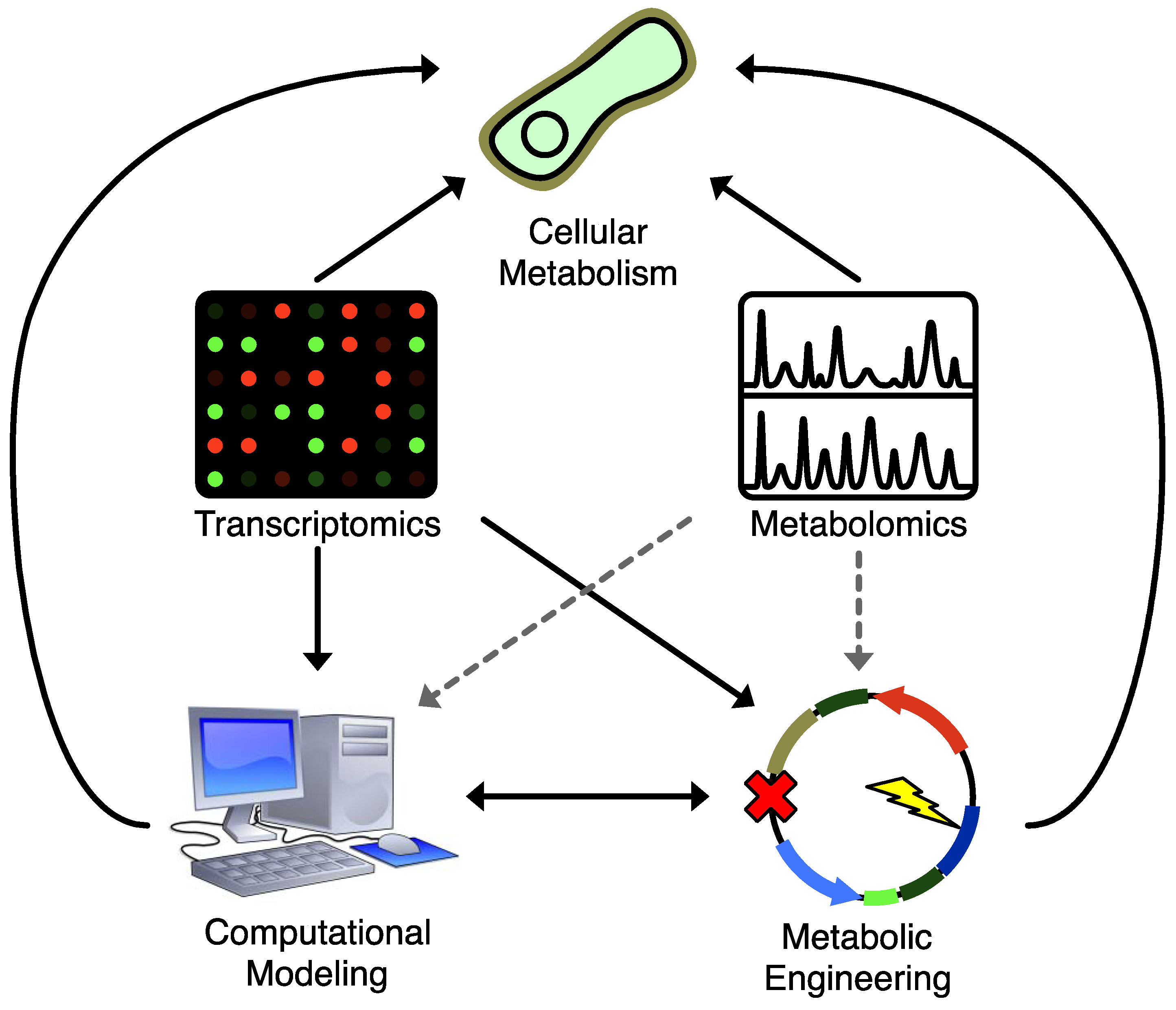
4.1. Constraint-Based Models
4.1.1. Flux Balance Analysis: The Prototypical Constraint-based Model
4.1.2. Model Reconstructions
4.1.3. Applications of Constraint-based Models in General to Metabolic Engineering
4.1.4. Thermodynamic Constraints: Integrating Metabolomics Data into Constraint-based Models
| Tool Name | Reference | Description |
|---|---|---|
| Metabolomics Data Processing | ||
| ChromA | [72] | GC-MS Peak Alignment |
| Metab | [75] | GC-MS Data Statistical Analysis Package |
| MetaboAnalyst 2.0 | [78] | Web-based Metabolomics Data Processing Pipeline |
| MetAlign | [76] | GC-MS and LC-MS Data Processing Pipeline |
| Mzmine 2 | [74] | MS Data Processing Pipeline |
| SpectConnect | [71] | GC-MS Peak Alignment |
| Xalign | [69] | LC-MS Data Pre-processing |
| XCMS Online | [77] | Web-based Untargeted Metabolomics Pipeline |
| Constraint-Based Modeling | ||
| anNET | [168] | MATLAB-based NET analysis |
| CellNetAnalyzer | [132] | MATLAB-based Metabolic and Signal Network Analysis |
| COBRA Toolbox | [131] | MATLAB-based FBA Toolbox Suite |
| OptFlux | [134] | Open Source, Modular Constraint-based Model Strain Design Software Toolbox |
| Systems Biology Research Tool | [133] | Open Source, Modular Systems Biology Computational Tool |
| Network Reconstruction | ||
| GapFind, GapFill | [137] | Automated Network Gap Identification and Hypothesis Generation |
| GeneForce | [139] | Regulatory Rule Correction for Integrated Metabolic and Regulatory Models |
| MetRxn | [147] | Web-based Knowledgebase Comparison Tool |
| Model SEED | [140] | Web-based Generation, Optimization and Analysis of Genome-scale Metabolic Models |
| Databases | ||
| BioCyc | [169] | Genome and pathway database for >2000 organisms |
| BRENDA | [170] | Comprehensive enzyme database, ~5000 enzymes |
| ChEBI | [171] | Biologically relevant small molecules and their properties |
| KEGG | [172] | Genomes, enzymatic pathways, and biological chemicals |
| MetaCyc | [173] | >1,900 metabolic pathways from >2,200 different organisms |
| PubChem | [174] | Biological activity and structures of small molecules |
4.2. Kinetic Models
4.2.1. Recent Developments in Kinetic Modeling Strategies
4.2.2. Examples of Kinetic Models for Metabolic Engineering
4.2.3. Integrating Metabolomics Datasets into Kinetic Models
5. Conclusions
Acknowledgments
Conflict of Interest
References
- Stephanopoulos, G. Metabolic Fluxes and Metabolic Engineering. Metab. Eng. 1999, 1, 1–11. [Google Scholar] [CrossRef]
- Zaldivar, J.Z.; Borges, A.B.; Johansson, B.J.; Smits, H.S.; Villas-Bôas, S.V.-B.; Nielsen, J.N.; Olsson, L.O. Fermentation performance and intracellular metabolite patterns in laboratory and industrial xylose-fermenting Saccharomyces cerevisiae. Appl. Microbiol. Biot. 2002, 59, 436–442. [Google Scholar] [CrossRef]
- Sonderegger, M.; Sauer, U. Evolutionary Engineering of Saccharomyces cerevisiae for Anaerobic Growth on Xylose. Appl. Environ. Microb. 2003, 69, 1990–1998. [Google Scholar] [CrossRef]
- Sonderegger, M.; Jeppsson, M.; Hahn-Hägerdal, B.; Sauer, U. Molecular Basis for Anaerobic Growth of Saccharomyces cerevisiae on Xylose, Investigated by Global Gene Expression and Metabolic Flux Analysis. Appl. Environ. Microb. 2004, 70, 2307–2317. [Google Scholar] [CrossRef]
- Bro, C.; Regenberg, B.; Förster, J.; Nielsen, J. In silico aided metabolic engineering of Saccharomyces cerevisiae for improved bioethanol production. Metab. Eng. 2006, 8, 102–111. [Google Scholar] [CrossRef]
- Meijer, S.; Panagiotou, G.; Olsson, L.; Nielsen, J. Physiological characterization of xylose metabolism in Aspergillus niger under oxygen-limited conditions. Biotechnol. Bioeng. 2007, 98, 462–475. [Google Scholar] [CrossRef]
- Panagiotou, G.; Andersen, M.R.; Grotkjær, T.; Regueira, T.B.; Hofmann, G.; Nielsen, J.; Olsson, L. Systems Analysis Unfolds the Relationship between the Phosphoketolase Pathway and Growth in Aspergillus nidulans. PLoS ONE 2008, 3, e3847. [Google Scholar] [Green Version]
- Wisselink, H.W.; Toirkens, M.J.; Wu, Q.; Pronk, J.T.; van Maris, A.J.A. Novel Evolutionary Engineering Approach for Accelerated Utilization of Glucose, Xylose, and Arabinose Mixtures by Engineered Saccharomyces cerevisiae Strains. Appl. Environ. Microb. 2009, 75, 907–914. [Google Scholar] [CrossRef]
- Klimacek, M.; Krahulec, S.; Sauer, U.; Nidetzky, B. Limitations in Xylose-Fermenting Saccharomyces cerevisiae, Made Evident through Comprehensive Metabolite Profiling and Thermodynamic Analysis. Appl. Environ. Microb. 2010, 76, 7566–7574. [Google Scholar] [CrossRef]
- Hasunuma, T.; Sanda, T.; Yamada, R.; Yoshimura, K.; Ishii, J.; Kondo, A. Metabolic pathway engineering based on metabolomics confers acetic and formic acid tolerance to a recombinant xylose-fermenting strain of Saccharomyces cerevisiae. Microb. Cell Fact. 2011, 10, 2. [Google Scholar] [CrossRef]
- Koppram, R.; Albers, E.; Olsson, L. Evolutionary engineering strategies to enhance tolerance of xylose utilizing recombinant yeast to inhibitors derived from spruce biomass. Biotechnol. Biofuels 2012, 5, 32. [Google Scholar] [CrossRef]
- Zhang, W.; Geng, A. Improved ethanol production by a xylose-fermenting recombinant yeast strain constructed through a modified genome shuffling method. Biotechnol. Biofuels 2012, 5, 46. [Google Scholar] [CrossRef]
- Kresnowati, M.T.A.P.; van Winden, W.A.; van Gulik, W.M.; Heijnen, J.J. Dynamic in vivo metabolome response of Saccharomyces cerevisiae to a stepwise perturbation of the ATP requirement for benzoate export. Biotechnol. Bioeng. 2008, 99, 421–441. [Google Scholar] [CrossRef]
- Sillers, R.; Chow, A.; Tracy, B.; Papoutsakis, E.T. Metabolic engineering of the non-sporulating, non-solventogenic Clostridium acetobutylicum strain M5 to produce butanol without acetone demonstrate the robustness of the acid-formation pathways and the importance of the electron balance. Metab. Eng. 2008, 10, 321–332. [Google Scholar] [CrossRef]
- Song, H.; Jang, S.H.; Park, J.M.; Lee, S.Y. Modeling of batch fermentation kinetics for succinic acid production by Mannheimia succiniciproducens. Biochem. Eng. J. 2008, 40, 107–115. [Google Scholar] [CrossRef]
- Zhang, K.; Sawaya, M.R.; Eisenberg, D.S.; Liao, J.C. Expanding metabolism for biosynthesis of nonnatural alcohols. Proc. Natl. Acad. Sci. USA 2008, 105, 20653–20658. [Google Scholar]
- Hou, J.; Lages, N.F.; Oldiges, M.; Vemuri, G.N. Metabolic impact of redox cofactor perturbations in Saccharomyces cerevisiae. Metab. Eng. 2009, 11, 253–261. [Google Scholar] [CrossRef]
- Sillers, R.; Al-Hinai, M.A.; Papoutsakis, E.T. Aldehyde-alcohol dehydrogenase and/or thiolase overexpression coupled with CoA transferase downregulation lead to higher alcohol titers and selectivity in Clostridium acetobutylicum fermentations. Biotechnol. Bioeng. 2009, 102, 38–49. [Google Scholar] [CrossRef]
- Trinh, C.T.; Huffer, S.; Clark, M.E.; Blanch, H.W.; Clark, D.S. Elucidating mechanisms of solvent toxicity in ethanologenic Escherichia coli. Biotechnol. Bioeng. 2010, 106, 721–730. [Google Scholar] [CrossRef]
- Oh, E.L.; Mingshou; Park, Changhun; Park, Changhun; Oh, Han Bin; Lee, Sang Yup; Lee, Jinwon. Dynamic Modeling of Lactic Acid Fermentation Metabolism with Lactococcus lactis. J. Microbiol. Biotechn. 2011, 21, 162–169. [Google Scholar] [CrossRef]
- Pereira, F.B.; Guimarães, P.M.R.; Teixeira, J.A.; Domingues, L. Robust industrial Saccharomyces cerevisiae strains for very high gravity bio-ethanol fermentations. J. Biosci. Bioeng. 2011, 112, 130–136. [Google Scholar] [CrossRef] [Green Version]
- Trinh, C.T.; Li, J.; Blanch, H.W.; Clark, D.S. Redesigning Escherichia coli Metabolism for Anaerobic Production of Isobutanol. Appl. Environ. Microb. 2011, 77, 4894–4904. [Google Scholar] [CrossRef]
- Aboka, F.O.; van Winden, W.A.; Reginald, M.M.; van Gulik, W.M.; van de Berg, M.; Oudshoorn, A.; Heijnen, J.J. Identification of informative metabolic responses using a minibioreactor: a small step change in the glucose supply rate creates a large metabolic response in Saccharomyces cerevisiae. Yeast 2012, 29, 95–110. [Google Scholar] [CrossRef]
- Lu, M.; Lee, S.; Kim, B.; Park, C.; Oh, M.; Park, K.; Lee, S.Y.; Lee, J. Identification of Factors Regulating Escherichia coli 2,3-Butanediol Production by Continuous Culture and Metabolic Flux Analysis. J. Microbiol. Biotechn. 2012, 22, 659–667. [Google Scholar] [CrossRef]
- Villas-Bôas, S.G.; Åkesson, M.; Nielsen, J. Biosynthesis of glyoxylate from glycine in Saccharomyces cerevisiae. FEMS Yeast Res. 2005, 5, 703–709. [Google Scholar] [CrossRef]
- Wu, L.; van Dam, J.; Schipper, D.; Kresnowati, M.T.A.P.; Proell, A.M.; Ras, C.; van Winden, W.A.; van Gulik, W.M.; Heijnen, J.J. Short-Term Metabolome Dynamics and Carbon, Electron, and ATP Balances in Chemostat-Grown Saccharomyces cerevisiae CEN.PK 113-7D following a Glucose Pulse. Appl. Environ. Microb. 2006, 72, 3566–3577. [Google Scholar]
- Costenoble, R.; Müller, D.; Barl, T.; Van Gulik, W.M.; Van Winden, W.A.; Reuss, M.; Heijnen, J.J. 13C-Labeled metabolic flux analysis of a fed-batch culture of elutriated Saccharomyces cerevisiae. FEMS Yeast Res. 2007, 7, 511–526. [Google Scholar] [CrossRef]
- Kleijn, R.J.; Geertman, J.-M.A.; Nfor, B.K.; Ras, C.; Schipper, D.; Pronk, J.T.; Heijnen, J.J.; Van Maris, A.J.A.; Van Winden, W.A. Metabolic flux analysis of a glycerol-overproducing Saccharomyces cerevisiae strain based on GC-MS, LC-MS and NMR-derived 13C-labelling data. FEMS Yeast Res. 2007, 7, 216–231. [Google Scholar] [CrossRef]
- Nasution, U.; van Gulik, W.M.; Ras, C.; Proell, A.; Heijnen, J.J. A metabolome study of the steady-state relation between central metabolism, amino acid biosynthesis and penicillin production in Penicillium chrysogenum. Metab. Eng. 2008, 10, 10–23. [Google Scholar] [CrossRef]
- Moxley, J.F.; Jewett, M.C.; Antoniewicz, M.R.; Villas-Boas, S.G.; Alper, H.; Wheeler, R.T.; Tong, L.; Hinnebusch, A.G.; Ideker, T.; Nielsen, J.; Stephanopoulos, G. Linking high-resolution metabolic flux phenotypes and transcriptional regulation in yeast modulated by the global regulator Gcn4p. Proc. Natl. Acad. Sci. USA 2009, 106, 6477–6482. [Google Scholar]
- Suthers, P.F.; Chang, Y.J.; Maranas, C.D. Improved computational performance of MFA using elementary metabolite units and flux coupling. Metab. Eng. 2010, 12, 123–128. [Google Scholar] [CrossRef]
- Ravikirthi, P.; Suthers, P.F.; Maranas, C.D. Construction of an E. Coli genome-scale atom mapping model for MFA calculations. Biotechnol. Bioeng. 2011, 108, 1372–1382. [Google Scholar] [CrossRef]
- Wu, L.; Mashego, M.R.; van Dam, J.C.; Proell, A.M.; Vinke, J.L.; Ras, C.; van Winden, W.A.; van Gulik, W.M.; Heijnen, J.J. Quantitative analysis of the microbial metabolome by isotope dilution mass spectrometry using uniformly 13C-labeled cell extracts as internal standards. Anal. Biochem. 2005, 336, 164–171. [Google Scholar]
- Büscher, J.r.M.; Czernik, D.; Ewald, J.C.; Sauer, U.; Zamboni, N. Cross-Platform Comparison of Methods for Quantitative Metabolomics of Primary Metabolism. Anal. Chem. 2009, 81, 2135–2143. [Google Scholar] [CrossRef]
- Choi, J.; Antoniewicz, M.R. Tandem mass spectrometry: A novel approach for metabolic flux analysis. Metab. Eng. 2011, 13, 225–233. [Google Scholar] [CrossRef]
- Choi, J.; Grossbach, M.T.; Antoniewicz, M.R. Measuring Complete Isotopomer Distribution of Aspartate Using Gas Chromatography/Tandem Mass Spectrometry. Anal. Chem. 2012, 84, 4628–4632. [Google Scholar]
- Srour, O.; Young, J.; Eldar, Y. Fluxomers: a new approach for 13C metabolic flux analysis. BMC Syst. Biol. 2011, 5, 129. [Google Scholar] [CrossRef]
- Chang, Y.; Suthers, P.F.; Maranas, C.D. Identification of optimal measurement sets for complete flux elucidation in metabolic flux analysis experiments. Biotechnol. Bioeng. 2008, 100, 1039–1049. [Google Scholar] [CrossRef]
- Antoniewicz, M.R.; Kelleher, J.K.; Stephanopoulos, G. Elementary metabolite units (EMU): A novel framework for modeling isotopic distributions. Metab. Eng. 2007, 9, 68–86. [Google Scholar] [CrossRef]
- Young, J.D.; Walther, J.L.; Antoniewicz, M.R.; Yoo, H.; Stephanopoulos, G. An elementary metabolite unit (EMU) based method of isotopically nonstationary flux analysis. Biotechnol. Bioeng. 2008, 99, 686–699. [Google Scholar] [CrossRef]
- Raamsdonk, L.M.; Teusink, B.; Broadhurst, D.; Zhang, N.; Hayes, A.; Walsh, M.C.; Berden, J.A.; Brindle, K.M.; Kell, D.B.; Rowland, J.J.; Westerhoff, H.V.; van Dam, K.; Oliver, S.G. A functional genomics strategy that uses metabolome data to reveal the phenotype of silent mutations. Nat. Biotech. 2001, 19, 45–50. [Google Scholar] [CrossRef]
- Allen, J.; Davey, H.M.; Broadhurst, D.; Heald, J.K.; Rowland, J.J.; Oliver, S.G.; Kell, D.B. High-throughput classification of yeast mutants for functional genomics using metabolic footprinting. Nat. Biotech. 2003, 21, 692–696. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Pan, Z.; Talaty, N.; Raftery, D.; Cooks, R.G. Combining desorption electrospray ionization mass spectrometry and nuclear magnetic resonance for differential metabolomics without sample preparation. Rapid Commun. Mass Sp. 2006, 20, 1577–1584. [Google Scholar] [CrossRef]
- Sreekumar, A.; Poisson, L.M.; Rajendiran, T.M.; Khan, A.P.; Cao, Q.; Yu, J.; Laxman, B.; Mehra, R.; Lonigro, R.J.; Li, Y.; Nyati, M.K.; Ahsan, A.; Kalyana-Sundaram, S.; Han, B.; Cao, X.; Byun, J.; Omenn, G.S.; Ghosh, D.; Pennathur, S.; Alexander, D.C.; Berger, A.; Shuster, J.R.; Wei, J.T.; Varambally, S.; Beecher, C.; Chinnaiyan, A.M. Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature 2009, 457, 910–914. [Google Scholar]
- Huang, Z.; Chen, Y.; Hang, W.; Gao, Y.; Lin, L.; Li, D.; Xing, J.; Yan, X. Holistic metabonomic profiling of urine affords potential early diagnosis for bladder and kidney cancers. Metabolomics 2012, 1–11. [Google Scholar]
- Krömer, J.O.; Sorgenfrei, O.; Klopprogge, K.; Heinzle, E.; Wittmann, C. In-Depth Profiling of Lysine-Producing Corynebacterium glutamicum by Combined Analysis of the Transcriptome, Metabolome, and Fluxome. J. Bacteriol. 2004, 186, 1769–1784. [Google Scholar] [CrossRef] [Green Version]
- Hua, Q.; Joyce, A.R.; Fong, S.S.; Palsson, B.Ø. Metabolic analysis of adaptive evolution for in silico-designed lactate-producing strains. Biotechnol. Bioeng. 2006, 95, 992–1002. [Google Scholar] [CrossRef]
- Mas, S.; Villas-Bôas, S.G.; Edberg Hansen, M.; Åkesson, M.; Nielsen, J. A comparison of direct infusion MS and GC-MS for metabolic footprinting of yeast mutants. Biotechnol. Bioeng. 2007, 96, 1014–1022. [Google Scholar] [CrossRef]
- van der Werf, M.J.; Overkamp, K.M.; Muilwijk, B.; Koek, M.M.; van der Werff-van der Vat, B.J.C.; Jellema, R.H.; Coulier, L.; Hankemeier, T. Comprehensive analysis of the metabolome of Pseudomonas putida S12 grown on different carbon sources. Mol. Biosyst. 2008, 4, 315–327. [Google Scholar] [CrossRef]
- Kleijn, R.J.; Buescher, J.M.; Le Chat, L.; Jules, M.; Aymerich, S.; Sauer, U. Metabolic Fluxes during Strong Carbon Catabolite Repression by Malate in Bacillus subtilis. J. Biol. Chem. 2010, 285, 1587–1596. [Google Scholar]
- Taymaz-Nikerel, H.; de Mey, M.; Ras, C.; ten Pierick, A.; Seifar, R.M.; van Dam, J.C.; Heijnen, J.J.; van Gulik, W.M. Development and application of a differential method for reliable metabolome analysis in Escherichia coli. Anal. Biochem. 2009, 386, 9–19. [Google Scholar]
- Kümmel, A.; Ewald, J.C.; Fendt, S.-M.; Jol, S.J.; Picotti, P.; Aebersold, R.; Sauer, U.; Zamboni, N.; Heinemann, M. Differential glucose repression in common yeast strains in response to HXK2 deletion. FEMS Yeast Res. 2010, 10, 322–332. [Google Scholar] [CrossRef]
- Taymaz-Nikerel, H.; van Gulik, W.M.; Heijnen, J.J. Escherichia coli responds with a rapid and large change in growth rate upon a shift from glucose-limited to glucose-excess conditions. Metab. Eng. 2011, 13, 307–318. [Google Scholar] [CrossRef]
- Canelas, A.B.; Ras, C.; ten Pierick, A.; van Gulik, W.M.; Heijnen, J.J. An in vivo data-driven framework for classification and quantification of enzyme kinetics and determination of apparent thermodynamic data. Metab. Eng. 2011, 13, 294–306. [Google Scholar] [CrossRef]
- Piddocke, M.; Fazio, A.; Vongsangnak, W.; Wong, M.; Heldt-Hansen, H.; Workman, C.; Nielsen, J.; Olsson, L. Revealing the beneficial effect of protease supplementation to high gravity beer fermentations using "-omics" techniques. Microb. Cell Fact. 2011, 10, 27. [Google Scholar] [CrossRef]
- Carnicer, M.; Canelas, A.; ten Pierick, A.; Zeng, Z.; van Dam, J.; Albiol, J.; Ferrer, P.; Heijnen, J.; van Gulik, W. Development of quantitative metabolomics for Pichia pastoris. Metabolomics 2012, 8, 284–298. [Google Scholar] [CrossRef]
- Carnicer, M.; Pierich, A.; Dam, J.; Heijnen, J.; Albiol, J.; Gulik, W.; Ferrer, P. Quantitative metabolomics analysis of amino acid metabolism in recombinant pichia pastoris under different oxygen availability conditions. Microb. Cell Fact. 2012, 11, 83. [Google Scholar]
- Dikicioglu, D.; Dunn, W.B.; Kell, D.B.; Kirdar, B.; Oliver, S.G. Short- and long-term dynamic responses of the metabolic network and gene expression in yeast to a transient change in the nutrient environment. Mol. Biosyst. 2012, 8, 1760–1774. [Google Scholar] [CrossRef]
- Fendt, S.-M.; Buescher, J.M.; Rudroff, F.; Picotti, P.; Zamboni, N.; Sauer, U. Tradeoff between enzyme and metabolite efficiency maintains metabolic homeostasis upon perturbations in enzyme capacity. Mol. Syst. Biol. 2010, 6, 356. [Google Scholar]
- Yuan, J.; Doucette, C.D.; Fowler, W.U.; Feng, X.-J.; Piazza, M.; Rabitz, H.A.; Wingreen, N.S.; Rabinowitz, J.D. Metabolomics-driven quantitative analysis of ammonia assimilation in E. coli. Mol. Syst. Biol. 2009, 5, 302. [Google Scholar]
- Buescher, J.M.; Moco, S.; Sauer, U.; Zamboni, N. Ultrahigh Performance Liquid Chromatography−Tandem Mass Spectrometry Method for Fast and Robust Quantification of Anionic and Aromatic Metabolites. Anal. Chem. 2010, 82, 4403–4412. [Google Scholar] [CrossRef]
- Christen, S.; Sauer, U. Intracellular characterization of aerobic glucose metabolism in seven yeast species by 13C flux analysis and metabolomics. FEMS Yeast Res. 2011, 11, 263–272. [Google Scholar] [CrossRef]
- McKee, A.; Rutherford, B.; Chivian, D.; Baidoo, E.; Juminaga, D.; Kuo, D.; Benke, P.; Dietrich, J.; Ma, S.; Arkin, A.; Petzold, C.; Adams, P.; Keasling, J.; Chhabra, S. Manipulation of the carbon storage regulator system for metabolite remodeling and biofuel production in Escherichia coli. Microb. Cell Fact. 2012, 11, 79. [Google Scholar] [CrossRef]
- Hirayama, A.; Nakashima, E.; Sugimoto, M.; Akiyama, S.-i.; Sato, W.; Maruyama, S.; Matsuo, S.; Tomita, M.; Yuzawa, Y.; Soga, T. Metabolic profiling reveals new serum biomarkers for differentiating diabetic nephropathy. Anal. Bioanal. Chem. 2012, 404, 1–9. [Google Scholar] [CrossRef]
- Toya, Y.; Nakahigashi, K.; Tomita, M.; Shimizu, K. Metabolic regulation analysis of wild-type and arcA mutant Escherichia coli under nitrate conditions using different levels of omics data. Mol. Biosyst. 2012, 8, 2593–2604. [Google Scholar]
- Canelas, A.B.; Harrison, N.; Fazio, A.; Zhang, J.; Pitkanen, J.-P.; van den Brink, J.; Bakker, B.M.; Bogner, L.; Bouwman, J.; Castrillo, J.I.; Cankorur, A.; Chumnanpuen, P.; Daran-Lapujade, P.; Dikicioglu, D.; van Eunen, K.; Ewald, J.C.; Heijnen, J.J.; Kirdar, B.; Mattila, I.; Mensonides, F.I.C.; Niebel, A.; Penttila, M.; Pronk, J.T.; Reuss, M.; Salusjarvi, L.; Sauer, U.; Sherman, D.; Siemann-Herzberg, M.; Westerhoff, H.; de Winde, J.; Petranovic, D.; Oliver, S.G.; Workman, C.T.; Zamboni, N.; Nielsen, J. Integrated multilaboratory systems biology reveals differences in protein metabolism between two reference yeast strains. Nat. Commun. 2010, 1, 145. [Google Scholar]
- Cajka, T.; Riddellova, K.; Tomaniova, M.; Hajslova, J. Ambient mass spectrometry employing a DART ion source for metabolomic fingerprinting/profiling: a powerful tool for beer origin recognition. Metabolomics 2011, 7, 500–508. [Google Scholar] [CrossRef]
- Villas-Bôas, S.G.; Moxley, J.F.; Akesson, M.; Stephanopoulos, G.; Nielsen, J. High-throughput metabolic state analysis: the missing link in integrated functional genomics of yeasts. Biochem. J. 2005, 388, 669–677. [Google Scholar] [CrossRef]
- Zhang, X.; Asara, J.M.; Adamec, J.; Ouzzani, M.; Elmagarmid, A.K. Data pre-processing in liquid chromatography-mass spectrometry-based proteomics. Bioinformatics 2005, 21, 4054–4059. [Google Scholar] [CrossRef]
- Smith, C.A.; Want, E.J.; O'Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing Mass Spectrometry Data for Metabolite Profiling Using Nonlinear Peak Alignment, Matching, and Identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef]
- Styczynski, M.P.; Moxley, J.F.; Tong, L.V.; Walther, J.L.; Jensen, K.L.; Stephanopoulos, G.N. Systematic Identification of Conserved Metabolites in GC/MS Data for Metabolomics and Biomarker Discovery. Anal. Chem. 2006, 79, 966–973. [Google Scholar]
- Hoffmann, N.; Stoye, J. ChromA: signal-based retention time alignment for chromatography-mass spectrometry data. Bioinformatics 2009, 25, 2080–2081. [Google Scholar] [CrossRef]
- Xia, J.; Psychogios, N.; Young, N.; Wishart, D.S. MetaboAnalyst: a web server for metabolomic data analysis and interpretation. Nucleic Acids Res. 2009, 37, W652–W660. [Google Scholar] [CrossRef]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Oresic, M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinformatics 2010, 11, 395. [Google Scholar] [CrossRef]
- Aggio, R.; Villas−Bôas, S.G.; Ruggiero, K. Metab: an R package for high-throughput analysis of metabolomics data generated by GC-MS. Bioinformatics 2011, 27, 2316–2318. [Google Scholar] [CrossRef]
- Lommen, A.; Kools, H.J. MetAlign 3.0: performance enhancement by efficient use of advances in computer hardware. Metabolomics 2012, 8, 719–726. [Google Scholar] [CrossRef]
- Tautenhahn, R.; Patti, G.J.; Rinehart, D.; Siuzdak, G. XCMS Online: A Web-Based Platform to Process Untargeted Metabolomic Data. Anal. Chem. 2012, 84, 5035–5039. [Google Scholar] [CrossRef]
- Xia, J.; Mandal, R.; Sinelnikov, I.V.; Broadhurst, D.; Wishart, D.S. MetaboAnalyst 2.0—a comprehensive server for metabolomic data analysis. Nucleic Acids Res. 2012, 40, W127–W133. [Google Scholar]
- Dunn, W.B.; Broadhurst, D.I.; Atherton, H.J.; Goodacre, R.; Griffin, J.L. Systems level studies of mammalian metabolomes: the roles of mass spectrometry and nuclear magnetic resonance spectroscopy. Chem. Soc. Rev. 2011, 40, 387–426. [Google Scholar]
- Devantier, R.; Scheithauer, B.; Villas-Bôas, S.G.; Pedersen, S.; Olsson, L. Metabolite profiling for analysis of yeast stress response during very high gravity ethanol fermentations. Biotechnol. Bioeng. 2005, 90, 703–714. [Google Scholar] [CrossRef]
- van den Berg, R.; Hoefsloot, H.; Westerhuis, J.; Smilde, A.; van der Werf, M. Centering, scaling, and transformations: improving the biological information content of metabolomics data. BMC Genomics 2006, 7, 142. [Google Scholar] [CrossRef]
- Højer-Pedersen, J.; Smedsgaard, J.; Nielsen, J. The yeast metabolome addressed by electrospray ionization mass spectrometry: Initiation of a mass spectral library and its applications for metabolic footprinting by direct infusion mass spectrometry. Metabolomics 2008, 4, 393–405. [Google Scholar] [CrossRef]
- MacKenzie, D.A.; Defernez, M.; Dunn, W.B.; Brown, M.; Fuller, L.J.; de Herrera, S.R.M.S.; Günther, A.; James, S.A.; Eagles, J.; Philo, M.; Goodacre, R.; Roberts, I.N. Relatedness of medically important strains of Saccharomyces cerevisiae as revealed by phylogenetics and metabolomics. Yeast 2008, 25, 501–512. [Google Scholar] [CrossRef]
- Barrett, C.; Herrgard, M.; Palsson, B. Decomposing complex reaction networks using random sampling, principal component analysis and basis rotation. BMC Syst. Biol. 2009, 3, 30. [Google Scholar] [CrossRef]
- Chong, W.P.K.; Goh, L.T.; Reddy, S.G.; Yusufi, F.N.K.; Lee, D.Y.; Wong, N.S.C.; Heng, C.K.; Yap, M.G.S.; Ho, Y.S. Metabolomics profiling of extracellular metabolites in recombinant Chinese Hamster Ovary fed-batch culture. Rapid Commun. Mass Sp. 2009, 23, 3763–3771. [Google Scholar] [CrossRef]
- Kamei, Y.; Tamura, T.; Yoshida, R.; Ohta, S.; Fukusaki, E.; Mukai, Y. GABA metabolism pathway genes, UGA1 and GAD1, regulate replicative lifespan in Saccharomyces cerevisia. Biochem. Bioph. Res. Co. 2011, 407, 185–190. [Google Scholar] [CrossRef]
- Peres-Neto, P.R.; Jackson, D.A.; Somers, K.M. How many principal components? Stopping rules for determining the number of non-trivial axes revisited. Comput. Stat. Data. An. 2005, 49, 974–997. [Google Scholar] [CrossRef]
- Wold, S.; Ruhe, A.; Wold, H.; Dunn, I., W. The Collinearity Problem in Linear Regression. The Partial Least Squares (PLS) Approach to Generalized Inverses. SISC 1984, 5, 735–743. [Google Scholar]
- Trygg, J.; Wold, S. Orthogonal projections to latent structures (O-PLS). J. Chemometr. 2002, 16, 119–128. [Google Scholar] [CrossRef]
- Wiklund, S.; Johansson, E.; Sjostrom, L.; Mellerowicz, E.J.; Edlund, U.; Shockcor, J.P.; Gottfries, J.; Moritz, T.; Trygg, J. Visualization of GC/TOF-MS-Based Metabolomics Data for Identification of Biochemically Interesting Compounds Using OPLS Class Models. Anal. Chem. 2007, 80, 115–122. [Google Scholar]
- Musumarra, G.; Barresi, V.; Condorelli, D.F.; Fortuna, C.G.; Scirè, S. Potentialities of multivariate approaches in genome-based cancer research: identification of candidate genes for new diagnostics by PLS discriminant analysis. J. Chemometr. 2004, 18, 125–132. [Google Scholar] [CrossRef]
- Storey, J.D.; Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA 2003, 100, 9440–9445. [Google Scholar] [CrossRef]
- Westerhuis, J.; Hoefsloot, H.; Smit, S.; Vis, D.; Smilde, A.; van Velzen, E.; van Duijnhoven, J.; van Dorsten, F. Assessment of PLSDA cross validation. Metabolomics 2008, 4, 81–89. [Google Scholar] [CrossRef]
- Dunn, W.B.; Broadhurst, D.; Begley, P.; Zelena, E.; Francis-McIntyre, S.; Anderson, N.; Brown, M.; Knowles, J.D.; Halsall, A.; Haselden, J.N.; Nicholls, A.W.; Wilson, I.D.; Kell, D.B.; Goodacre, R. Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat. Protocols 2011, 6, 1060–1083. [Google Scholar] [CrossRef]
- Broadhurst, D.; Kell, D. Statistical strategies for avoiding false discoveries in metabolomics and related experiments. Metabolomics 2006, 2, 171–196. [Google Scholar] [CrossRef]
- Culeddu, N.; Chessa, M.; Porcu, M.; Fresu, P.; Tonolo, G.; Virgilio, G.; Migaleddu, V. NMR-based metabolomic study of type 1 diabetes. Metabolomics 2012, 8, 1–8. [Google Scholar]
- Sonkar, K.; Behari, A.; Kapoor, V.; Sinha, N. 1H NMR metabolic profiling of human serum associated with benign and malignant gallstone diseases. Metabolomics 2012, 1–14. [Google Scholar]
- Blekherman, G.; Laubenbacher, R.; Cortes, D.; Mendes, P.; Torti, F.; Akman, S.; Torti, S.; Shulaev, V. Bioinformatics tools for cancer metabolomics. Metabolomics 2011, 7, 329–343. [Google Scholar] [CrossRef]
- Mashego, M.R.; Van Gulik, W.M.; Heijnen, J.J. Metabolome dynamic responses of Saccharomyces cerevisiae to simultaneous rapid perturbations in external electron acceptor and electron donor. FEMS Yeast Res. 2007, 7, 48–66. [Google Scholar] [CrossRef]
- Kresnowati, M.T.A.P.; van Winden, W.A.; Almering, M.J.H.; ten Pierick, A.; Ras, C.; Knijnenburg, T.A.; Daran-Lapujade, P.; Pronk, J.T.; Heijnen, J.J.; Daran, J.M. When transcriptome meets metabolome: fast cellular responses of yeast to sudden relief of glucose limitation. Mol. Syst. Biol. 2006, 2, 49. [Google Scholar]
- Douma, R.; Batista, J.; Touw, K.; Kiel, J.; Krikken, A.; Zhao, Z.; Veiga, T.; Klaassen, P.; Bovenberg, R.; Daran, J.-M.; Heijnen, J.; van Gulik, W. Degeneration of penicillin production in ethanol-limited chemostat cultivations of Penicillium chrysogenum: A systems biology approach. BMC Syst. Biol. 2011, 5, 132. [Google Scholar] [CrossRef]
- Santos, C.N.S.; Stephanopoulos, G. Melanin-Based High-Throughput Screen for l-Tyrosine Production in Escherichia coli. Appl. Environ. Microb. 2008, 74, 1190–1197. [Google Scholar] [CrossRef]
- Tyo, K.E.; Zhou, H.; Stephanopoulos, G.N. High-Throughput Screen for Poly-3-Hydroxybutyrate in Escherichia coli and Synechocystis sp. Strain PCC6803. Appl. Environ. Microb. 2006, 72, 3412–3417. [Google Scholar] [CrossRef]
- Lee, S.K.; Chou, H.H.; Pfleger, B.F.; Newman, J.D.; Yoshikuni, Y.; Keasling, J.D. Directed Evolution of AraC for Improved Compatibility of Arabinose- and Lactose-Inducible Promoters. Appl. Environ. Microb. 2007, 73, 5711–5715. [Google Scholar]
- Atsumi, S.; Liao, J.C. Directed Evolution of Methanococcus jannaschii Citramalate Synthase for Biosynthesis of 1-Propanol and 1-Butanol by Escherichia coli. Appl. Environ. Microb. 2008, 74, 7802–7808. [Google Scholar] [CrossRef]
- Leonard, E.; Ajikumar, P.K.; Thayer, K.; Xiao, W.-H.; Mo, J.D.; Tidor, B.; Stephanopoulos, G.; Prather, K.L.J. Combining metabolic and protein engineering of a terpenoid biosynthetic pathway for overproduction and selectivity control. Proc. Natl. Acad. Sci. USA 2010, 107, 13654–13659. [Google Scholar]
- Hong, K.-K.; Vongsangnak, W.; Vemuri, G.N.; Nielsen, J. Unravelling evolutionary strategies of yeast for improving galactose utilization through integrated systems level analysis. Proc. Natl. Acad. Sci. USA 2011, 108, 12179–12184. [Google Scholar]
- Shen, C.R.; Lan, E.I.; Dekishima, Y.; Baez, A.; Cho, K.M.; Liao, J.C. Driving Forces Enable High-Titer Anaerobic 1-Butanol Synthesis in Escherichia coli. Appl. Environ. Microb. 2011, 77, 2905–2915. [Google Scholar] [CrossRef]
- Sun, Z.; Ning, Y.; Liu, L.; Liu, Y.; Sun, B.; Jiang, W.; Yang, C.; Yang, S. Metabolic engineering of the L-phenylalanine pathway in Escherichia coli for the production of S- or R-mandelic acid. Microb. Cell Fact. 2011, 10, 71. [Google Scholar] [CrossRef]
- Bailey, J.E.; Sburlati, A.; Hatzimanikatis, V.; Lee, K.; Renner, W.A.; Tsai, P.S. Inverse metabolic engineering: A strategy for directed genetic engineering of useful phenotypes. Biotechnol. Bioeng. 1996, 52, 109–121. [Google Scholar] [CrossRef]
- Yoshida, S.; Imoto, J.; Minato, T.; Oouchi, R.; Sugihara, M.; Imai, T.; Ishiguro, T.; Mizutani, S.; Tomita, M.; Soga, T.; Yoshimoto, H. Development of Bottom-Fermenting Saccharomyces Strains That Produce High SO2 Levels, Using Integrated Metabolome and Transcriptome Analysis. Appl. Environ. Microb. 2008, 74, 2787–2796. [Google Scholar]
- Wisselink, H.W.; Cipollina, C.; Oud, B.; Crimi, B.; Heijnen, J.J.; Pronk, J.T.; van Maris, A.J.A. Metabolome, transcriptome and metabolic flux analysis of arabinose fermentation by engineered Saccharomyces cerevisiae. Metab. Eng. 2010, 12, 537–551. [Google Scholar] [CrossRef]
- Ding, M.-Z.; Zhou, X.; Yuan, Y.-J. Metabolome profiling reveals adaptive evolution of Saccharomyces cerevisiae during repeated vacuum fermentations. Metabolomics 2010, 6, 42–55. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; Mesirov, J.P. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar]
- Xia, J.; Wishart, D.S. MSEA: a web-based tool to identify biologically meaningful patterns in quantitative metabolomic data. Nucleic Acids Res. 2010, 38, W71–W77. [Google Scholar] [CrossRef]
- Kizer, L.; Pitera, D.J.; Pfleger, B.F.; Keasling, J.D. Application of Functional Genomics to Pathway Optimization for Increased Isoprenoid Production. Appl. Environ. Microb. 2008, 74, 3229–3241. [Google Scholar]
- Alsaker, K.V.; Paredes, C.; Papoutsakis, E.T. Metabolite stress and tolerance in the production of biofuels and chemicals: Gene-expression-based systems analysis of butanol, butyrate, and acetate stresses in the anaerobe Clostridium acetobutylicu. Biotechnol. Bioeng. 2010, 105, 1131–1147. [Google Scholar]
- Cakir, T.; Kirdar, B.; Onsan, Z.I.; Ulgen, K.; Nielsen, J. Effect of carbon source perturbations on transcriptional regulation of metabolic fluxes in Saccharomyces cerevisiae. BMC Syst. Biol. 2007, 1, 18. [Google Scholar] [CrossRef]
- Joshi, A.; Palsson, B.O. Metabolic dynamics in the human red cell: Part I—A comprehensive kinetic model. J. Theor. Biol. 1989, 141, 515–528. [Google Scholar] [CrossRef]
- Chassagnole, C.; Noisommit-Rizzi, N.; Schmid, J.W.; Mauch, K.; Reuss, M. Dynamic modeling of the central carbon metabolism of Escherichia coli. Biotechnol. Bioeng. 2002, 79, 53–73. [Google Scholar] [CrossRef]
- van Eunen, K.; Bouwman, J.; Daran-Lapujade, P.; Postmus, J.; Canelas, A.B.; Mensonides, F.I.C.; Orij, R.; Tuzun, I.; van den Brink, J.; Smits, G.J.; van Gulik, W.M.; Brul, S.; Heijnen, J.J.; de Winde, J.H.; Teixeira de Mattos, M.J.; Kettner, C.; Nielsen, J.; Westerhoff, H.V.; Bakker, B.M. Measuring enzyme activities under standardized in vivo-like conditions for systems biology. FEBS J 2010, 277, 749–760. [Google Scholar]
- van Eunen, K.; Kiewiet, J.A.L.; Westerhoff, H.V.; Bakker, B.M. Testing Biochemistry Revisited: How In Vivo Metabolism Can Be Understood from In Vitro Enzyme Kinetics. PLoS Comput. Biol. 2012, 8, e1002483. [Google Scholar] [CrossRef]
- Gutenkunst, R.N.; Waterfall, J.J.; Casey, F.P.; Brown, K.S.; Myers, C.R.; Sethna, J.P. Universally Sloppy Parameter Sensitivities in Systems Biology Models. PLoS Comput. Biol. 2007, 3, e189. [Google Scholar] [CrossRef]
- Savinell, J.M.; Palsson, B.O. Network analysis of intermediary metabolism using linear optimization. I. Development of mathematical formalism. J. Theor. Biol. 1992, 154, 421–454. [Google Scholar] [CrossRef]
- Varma, A.; Palsson, B.O. Metabolic Capabilities of Escherichia coli: I. Synthesis of Biosynthetic Precursors and Cofactors. J. Theor. Biol. 1993, 165, 477–502. [Google Scholar] [CrossRef]
- Suthers, P.F.; Burgard, A.P.; Dasika, M.S.; Nowroozi, F.; Van Dien, S.; Keasling, J.D.; Maranas, C.D. Metabolic flux elucidation for large-scale models using 13C labeled isotopes. Metab. Eng. 2007, 9, 387–405. [Google Scholar] [CrossRef]
- Choi, H.S.; Kim, T.Y.; Lee, D.-Y.; Lee, S.Y. Incorporating metabolic flux ratios into constraint-based flux analysis by using artificial metabolites and converging ratio determinants. J. Biotechnol. 2007, 129, 696–705. [Google Scholar]
- Schuetz, R.; Kuepfer, L.; Sauer, U. Systematic evaluation of objective functions for predicting intracellular fluxes in Escherichia coli. Mol. Syst. Biol. 2007, 3, 119. [Google Scholar]
- Nookaew, I.; Meechai, A.; Thammarongtham, C.; Laoteng, K.; Ruanglek, V.; Cheevadhanarak, S.; Nielsen, J.; Bhumiratana, S. Identification of flux regulation coefficients from elementary flux modes: A systems biology tool for analysis of metabolic networks. Biotechnol. Bioeng. 2007, 97, 1535–1549. [Google Scholar] [CrossRef]
- Chen, X.; Alonso, A.P.; Allen, D.K.; Reed, J.L.; Shachar-Hill, Y. Synergy between 13C-metabolic flux analysis and flux balance analysis for understanding metabolic adaption to anaerobiosis in E. coli. Metab. Eng. 2011, 13, 38–48. [Google Scholar]
- Hyduke, D.R.; Schellenberger, J.; Que, R.; Fleming, R.M.T.; Thiele, I.; Orth, J.D.; Feist, A.M.; Zielinski, D.C.; Bordbar, A.; Lewis, N.E.; Rahmanian, S.; Kang, J.; Palsson, B.O. Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox v2.0. Nat. Protocols 2011, 6, 1290–1307. [Google Scholar] [CrossRef]
- Klamt, S.; Saez-Rodriguez, J.; Gilles, E. Structural and functional analysis of cellular networks with CellNetAnalyzer. BMC Syst. Biol. 2007, 1, 2. [Google Scholar]
- Wright, J.; Wagner, A. The Systems Biology Research Tool: evolvable open-source software. BMC Syst. Biol. 2008, 2, 55. [Google Scholar] [CrossRef] [Green Version]
- Rocha, I.; Maia, P.; Evangelista, P.; Vilaca, P.; Soares, S.; Pinto, J.; Nielsen, J.; Patil, K.; Ferreira, E.; Rocha, M. OptFlux: an open-source software platform for in silico metabolic engineering. BMC Syst. Biol. 2010, 4, 45. [Google Scholar] [CrossRef] [Green Version]
- Feist, A.M.; Herrgard, M.J.; Thiele, I.; Reed, J.L.; Palsson, B.O. Reconstruction of biochemical networks in microorganisms. Nat. Rev. Micro. 2009, 7, 129–143. [Google Scholar]
- Thiele, I.; Palsson, B.O. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protocols 2010, 5, 93–121. [Google Scholar] [CrossRef]
- Satish Kumar, V.; Dasika, M.; Maranas, C. Optimization based automated curation of metabolic reconstructions. BMC Bioinformatics 2007, 8, 212. [Google Scholar] [CrossRef]
- Senger, R.S.; Papoutsakis, E.T. Genome-scale model for Clostridium acetobutylicum: Part I. Metabolic network resolution and analysis. Biotechnol. Bioeng. 2008, 101, 1036–1052. [Google Scholar] [CrossRef]
- Barua, D.; Kim, J.; Reed, J.L. An Automated Phenotype-Driven Approach (GeneForce) for Refining Metabolic and Regulatory Models. PLoS Comput. Biol. 2010, 6, e1000970. [Google Scholar] [CrossRef]
- Henry, C.S.; DeJongh, M.; Best, A.A.; Frybarger, P.M.; Linsay, B.; Stevens, R.L. High-throughput generation, optimization and analysis of genome-scale metabolic models. Nat. Biotech. 2010, 28, 977–982. [Google Scholar]
- Andersen, M.R.; Nielsen, M.L.; Nielsen, J. Metabolic model integration of the bibliome, genome, metabolome and reactome of Aspergillus niger. Mol. Syst. Biol. 2008, 4, 178. [Google Scholar]
- Milne, C.; Eddy, J.; Raju, R.; Ardekani, S.; Kim, P.-J.; Senger, R.; Jin, Y.-S.; Blaschek, H.; Price, N. Metabolic network reconstruction and genome-scale model of butanol-producing strain Clostridium beijerinckii NCIMB 8052. BMC Syst. Biol. 2011, 5, 130. [Google Scholar] [CrossRef]
- Feist, A.M.; Henry, C.S.; Reed, J.L.; Krummenacker, M.; Joyce, A.R.; Karp, P.D.; Broadbelt, L.J.; Hatzimanikatis, V.; Palsson, B.O. A genome-scale metabolic reconstruction for Escherichia coli K-12 MG1655 that accounts for 1260 ORFs and thermodynamic information. Mol. Syst. Biol. 2007, 3, 121. [Google Scholar]
- Orth, J.D.; Conrad, T.M.; Na, J.; Lerman, J.A.; Nam, H.; Feist, A.M.; Palsson, B.O. A comprehensive genome-scale reconstruction of Escherichia coli metabolism-2011. Mol. Syst. Biol. 2011, 7, 535. [Google Scholar]
- Nookaew, I.; Jewett, M.; Meechai, A.; Thammarongtham, C.; Laoteng, K.; Cheevadhanarak, S.; Nielsen, J.; Bhumiratana, S. The genome-scale metabolic model iIN800 of Saccharomyces cerevisiae and its validation: a scaffold to query lipid metabolism. BMC Syst. Biol. 2008, 2, 71. [Google Scholar] [CrossRef]
- Karr, Jonathan R.; Sanghvi, Jayodita C.; Macklin, Derek N.; Gutschow, Miriam V.; Jacobs, Jared M.; Bolival Jr, B.; Assad-Garcia, N.; Glass, John I.; Covert, Markus W. A Whole-Cell Computational Model Predicts Phenotype from Genotype. Cell 2012, 150, 389–401. [Google Scholar] [CrossRef]
- Kumar, A.; Suthers, P.; Maranas, C. MetRxn: a knowledgebase of metabolites and reactions spanning metabolic models and databases. BMC Bioinformatics 2012, 13, 6. [Google Scholar] [CrossRef]
- Lee, J.M.; Gianchandani, E.P.; Papin, J.A. Flux balance analysis in the era of metabolomics. Brief. Bioinf. 2006, 7, 140–150. [Google Scholar] [CrossRef]
- Gianchandani, E.P.; Chavali, A.K.; Papin, J.A. The application of flux balance analysis in systems biology. Wiley Interdiscip. Rev. Syst. Biol. Med. 2010, 2, 372–382. [Google Scholar] [CrossRef]
- Covert, M.W.; Schilling, C.H.; Palsson, B. Regulation of Gene Expression in Flux Balance Models of Metabolism. J. Theor. Biol. 2001, 213, 73–88. [Google Scholar]
- Covert, M.W.; Palsson, B.Ø. Transcriptional Regulation in Constraints-based Metabolic Models of Escherichia coli. J. Biol. Chem. 2002, 277, 28058–28064. [Google Scholar]
- Meadows, A.L.; Karnik, R.; Lam, H.; Forestell, S.; Snedecor, B. Application of dynamic flux balance analysis to an industrial Escherichia coli fermentation. Metab. Eng. 2010, 12, 150–160. [Google Scholar] [CrossRef]
- Mahadevan, R.; Edwards, J.S.; Doyle Iii, F.J. Dynamic Flux Balance Analysis of Diauxic Growth in Escherichia coli. Biophys. J. 2002, 83, 1331–1340. [Google Scholar] [CrossRef]
- Feng, X.; Xu, Y.; Chen, Y.; Tang, Y.J. Integrating Flux Balance Analysis into Kinetic Models to Decipher the Dynamic Metabolism of Shewanella oneidensis MR-1. PLoS Comput. Biol. 2012, 8, e1002376. [Google Scholar] [CrossRef]
- Covert, M.W.; Xiao, N.; Chen, T.J.; Karr, J.R. Integrating metabolic, transcriptional regulatory and signal transduction models in Escherichia coli. Bioinformatics 2008, 24, 2044–2050. [Google Scholar] [CrossRef]
- Lee, J.M.; Gianchandani, E.P.; Eddy, J.A.; Papin, J.A. Dynamic Analysis of Integrated Signaling, Metabolic, and Regulatory Networks. PLoS Comput. Biol. 2008, 4, e1000086. [Google Scholar] [CrossRef]
- Herrgård, M.J.; Fong, S.S.; Palsson, B.Ø. Identification of Genome-Scale Metabolic Network Models Using Experimentally Measured Flux Profiles. PLoS Comput. Biol. 2006, 2, e72. [Google Scholar] [CrossRef]
- Kumar, V.S.; Maranas, C.D. GrowMatch: An Automated Method for Reconciling In Silico/In Vivo Growth Predictions. PLoS Comput. Biol. 2009, 5, e1000308. [Google Scholar] [CrossRef]
- Zomorrodi, A.; Maranas, C. Improving the iMM904 S. cerevisiae metabolic model using essentiality and synthetic lethality data. BMC Syst. Biol. 2010, 4, 178. [Google Scholar] [CrossRef]
- Oh, Y.-G.; Lee, D.-Y.; Lee, S.Y.; Park, S. Multiobjective flux balancing using the NISE method for metabolic network analysis. Biotechnol. Progr. 2009, 25, 999–1008. [Google Scholar] [CrossRef]
- Adadi, R.; Volkmer, B.; Milo, R.; Heinemann, M.; Shlomi, T. Prediction of Microbial Growth Rate versus Biomass Yield by a Metabolic Network with Kinetic Parameters. PLoS Comput. Biol. 2012, 8, e1002575. [Google Scholar] [CrossRef]
- Segrè, D.; Vitkup, D.; Church, G.M. Analysis of optimality in natural and perturbed metabolic networks. Proc. Natl. Acad. Sci. USA 2002, 99, 15112–15117. [Google Scholar]
- Burgard, A.P.; Pharkya, P.; Maranas, C.D. Optknock: A bilevel programming framework for identifying gene knockout strategies for microbial strain optimization. Biotechnol. Bioeng. 2003, 84, 647–657. [Google Scholar] [CrossRef]
- Patil, K.; Rocha, I.; Forster, J.; Nielsen, J. Evolutionary programming as a platform for in silico metabolic engineering. BMC Bioinformatics 2005, 6, 308. [Google Scholar]
- Asadollahi, M.A.; Maury, J.; Patil, K.R.; Schalk, M.; Clark, A.; Nielsen, J. Enhancing sesquiterpene production in Saccharomyces cerevisiae through in silico driven metabolic engineering. Metab. Eng. 2009, 11, 328–334. [Google Scholar] [CrossRef]
- Ranganathan, S.; Suthers, P.F.; Maranas, C.D. OptForce: An Optimization Procedure for Identifying All Genetic Manipulations Leading to Targeted Overproductions. PLoS Comput. Biol. 2010, 6, e1000744. [Google Scholar] [CrossRef]
- Kummel, A.; Panke, S.; Heinemann, M. Putative regulatory sites unraveled by network-embedded thermodynamic analysis of metabolome data. Mol. Syst. Biol. 2006, 2, 2006.0034. [Google Scholar]
- Zamboni, N.; Kummel, A.; Heinemann, M. anNET: a tool for network-embedded thermodynamic analysis of quantitative metabolome data. BMC Bioinformatics 2008, 9, 199. [Google Scholar] [CrossRef]
- Karp, P.D.; Ouzounis, C.A.; Moore-Kochlacs, C.; Goldovsky, L.; Kaipa, P.; Ahrén, D.; Tsoka, S.; Darzentas, N.; Kunin, V.; López-Bigas, N. Expansion of the BioCyc collection of pathway/genome databases to 160 genomes. Nucleic Acids Res. 2005, 33, 6083–6089. [Google Scholar]
- Schomburg, I.; Chang, A.; Schomburg, D. BRENDA, enzyme data and metabolic information. Nucleic Acids Res. 2002, 30, 47–49. [Google Scholar] [CrossRef]
- de Matos, P.; Alcántara, R.; Dekker, A.; Ennis, M.; Hastings, J.; Haug, K.; Spiteri, I.; Turner, S.; Steinbeck, C. Chemical Entities of Biological Interest: an update. Nucleic Acids Res. 2010, 38, D249–D254. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Caspi, R.; Foerster, H.; Fulcher, C.A.; Kaipa, P.; Krummenacker, M.; Latendresse, M.; Paley, S.; Rhee, S.Y.; Shearer, A.G.; Tissier, C.; Walk, T.C.; Zhang, P.; Karp, P.D. The MetaCyc Database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res. 2008, 36, D623–D631. [Google Scholar]
- Bolton, E.E.; Wang, Y.; Thiessen, P.A.; Bryant, S.H. Chapter 12 PubChem: Integrated Platform of Small Molecules and Biological Activities. In Annu. Rep. Comput. Chem.; Ralph, A.W., David, C.S., Eds.; Elsevier, 2008; Volume 4, pp. 217–241. [Google Scholar]
- Canelas, A.B.; van Gulik, W.M.; Heijnen, J.J. Determination of the cytosolic free NAD/NADH ratio in Saccharomyces cerevisiae under steady-state and highly dynamic conditions. Biotechnol. Bioeng. 2008, 100, 734–743. [Google Scholar] [CrossRef]
- Jol, S.J.; Kümmel, A.; Terzer, M.; Stelling, J.; Heinemann, M. System-Level Insights into Yeast Metabolism by Thermodynamic Analysis of Elementary Flux Modes. PLoS Comput. Biol. 2012, 8, e1002415. [Google Scholar] [CrossRef] [Green Version]
- Henry, C.S.; Broadbelt, L.J.; Hatzimanikatis, V. Thermodynamics-Based Metabolic Flux Analysis. Biophys. J. 2007, 92, 1792–1805. [Google Scholar] [CrossRef]
- Price, N.D.; Famili, I.; Beard, D.A.; Palsson, B.Ø. Extreme Pathways and Kirchhoff's Second Law. Biophys. J. 2002, 83, 2879–2882. [Google Scholar] [CrossRef]
- Schellenberger, J.; Lewis, N.E.; Palsson, B.Ø. Elimination of Thermodynamically Infeasible Loops in Steady-State Metabolic Models. Biophys. J. 2011, 100, 544–553. [Google Scholar]
- Jankowski, M.D.; Henry, C.S.; Broadbelt, L.J.; Hatzimanikatis, V. Group Contribution Method for Thermodynamic Analysis of Complex Metabolic Networks. Biophys. J. 2008, 95, 1487–1499. [Google Scholar] [CrossRef]
- Finley, S.D.; Broadbelt, L.J.; Hatzimanikatis, V. Thermodynamic analysis of biodegradation pathways. Biotechnol. Bioeng. 2009, 103, 532–541. [Google Scholar] [CrossRef]
- Garg, S.; Yang, L.; Mahadevan, R. Thermodynamic analysis of regulation in metabolic networks using constraint-based modeling. BMC Research Notes 2010, 3, 125. [Google Scholar] [CrossRef]
- Bordel, S.; Nielsen, J. Identification of flux control in metabolic networks using non-equilibrium thermodynamics. Metab. Eng. 2010, 12, 369–377. [Google Scholar] [CrossRef]
- Hoppe, A.; Hoffmann, S.; Holzhutter, H.-G. Including metabolite concentrations into flux balance analysis: thermodynamic realizability as a constraint on flux distributions in metabolic networks. BMC Syst. Biol. 2007, 1, 23. [Google Scholar] [CrossRef]
- Heijnen, J.J. Approximative kinetic formats used in metabolic network modeling. Biotechnol. Bioeng. 2005, 91, 534–545. [Google Scholar] [CrossRef]
- Savageau, M.A. Biochemical systems analysis. I. Some mathematical properties of the rate law for the component enzymatic reactions. J. Theor. Biol. 1969, 25, 365–369. [Google Scholar] [CrossRef]
- Savageau, M.A. Mathematics of organizationally complex systems. Biomed. Biochim. Acta 1985, 44, 839–844. [Google Scholar]
- Voit, E.O.; Savageau, M.A. Accuracy of alternative representations for integrated biochemical systems. Biochemistry 1987, 26, 6869–6880. [Google Scholar] [CrossRef]
- Voit, E.O. Modelling metabolic networks using power-laws and S-systems. Essays Biochem. 2008, 45, 29–40. [Google Scholar] [CrossRef]
- Steuer, R.; Gross, T.; Selbig, J.; Blasius, B. Structural kinetic modeling of metabolic networks. Proc. Natl. Acad. Sci. USA 2006, 103, 11868–11873. [Google Scholar] [CrossRef]
- Nikerel, I.E.; van Winden, W.; van Gulik, W.; Heijnen, J. A method for estimation of elasticities in metabolic networks using steady state and dynamic metabolomics data and linlog kinetics. BMC Bioinformatics 2006, 7, 540. [Google Scholar] [CrossRef]
- Nikerel, I.E.; van Winden, W.A.; Verheijen, P.J.T.; Heijnen, J.J. Model reduction and a priori kinetic parameter identifiability analysis using metabolome time series for metabolic reaction networks with linlog kinetics. Metab. Eng. 2009, 11, 20–30. [Google Scholar] [CrossRef]
- Costa, R.S.; Machado, D.; Rocha, I.; Ferreira, E.C. Hybrid dynamic modeling of Escherichia coli central metabolic network combining Michaelis-Menten and approximate kinetic equations. Biosystems 2010, 100, 150–157. [Google Scholar] [CrossRef]
- Ralser, M.; Wamelink, M.; Kowald, A.; Gerisch, B.; Heeren, G.; Struys, E.; Klipp, E.; Jakobs, C.; Breitenbach, M.; Lehrach, H.; Krobitsch, S. Dynamic rerouting of the carbohydrate flux is key to counteracting oxidative stress. J. Biol. 2007, 6, 10. [Google Scholar] [CrossRef]
- Tran, L.M.; Rizk, M.L.; Liao, J.C. Ensemble Modeling of Metabolic Networks. Biophys. J. 2008, 95, 5606–5617. [Google Scholar] [CrossRef]
- Rizk, M.L.; Liao, J.C. Ensemble Modeling for Aromatic Production in Escherichia coli. PLoS ONE 2009, 4, e6903. [Google Scholar]
- Contador, C.A.; Rizk, M.L.; Asenjo, J.A.; Liao, J.C. Ensemble modeling for strain development of l-lysine-producing Escherichia coli. Metab. Eng. 2009, 11, 221–233. [Google Scholar] [CrossRef]
- Yizhak, K.; Benyamini, T.; Liebermeister, W.; Ruppin, E.; Shlomi, T. Integrating quantitative proteomics and metabolomics with a genome-scale metabolic network model. Bioinformatics 2010, 26, i255–i260. [Google Scholar] [CrossRef]
- Jamshidi, N.; Palsson, B.Ø. Mass Action Stoichiometric Simulation Models: Incorporating Kinetics and Regulation into Stoichiometric Models. Biophys. J. 2010, 98, 175–185. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Dromms, R.A.; Styczynski, M.P. Systematic Applications of Metabolomics in Metabolic Engineering. Metabolites 2012, 2, 1090-1122. https://doi.org/10.3390/metabo2041090
Dromms RA, Styczynski MP. Systematic Applications of Metabolomics in Metabolic Engineering. Metabolites. 2012; 2(4):1090-1122. https://doi.org/10.3390/metabo2041090
Chicago/Turabian StyleDromms, Robert A., and Mark P. Styczynski. 2012. "Systematic Applications of Metabolomics in Metabolic Engineering" Metabolites 2, no. 4: 1090-1122. https://doi.org/10.3390/metabo2041090