Evaluation of Extraction Protocols for Simultaneous Polar and Non-Polar Yeast Metabolite Analysis Using Multivariate Projection Methods



Abstract
:1. Introduction
2. Results and Discussion
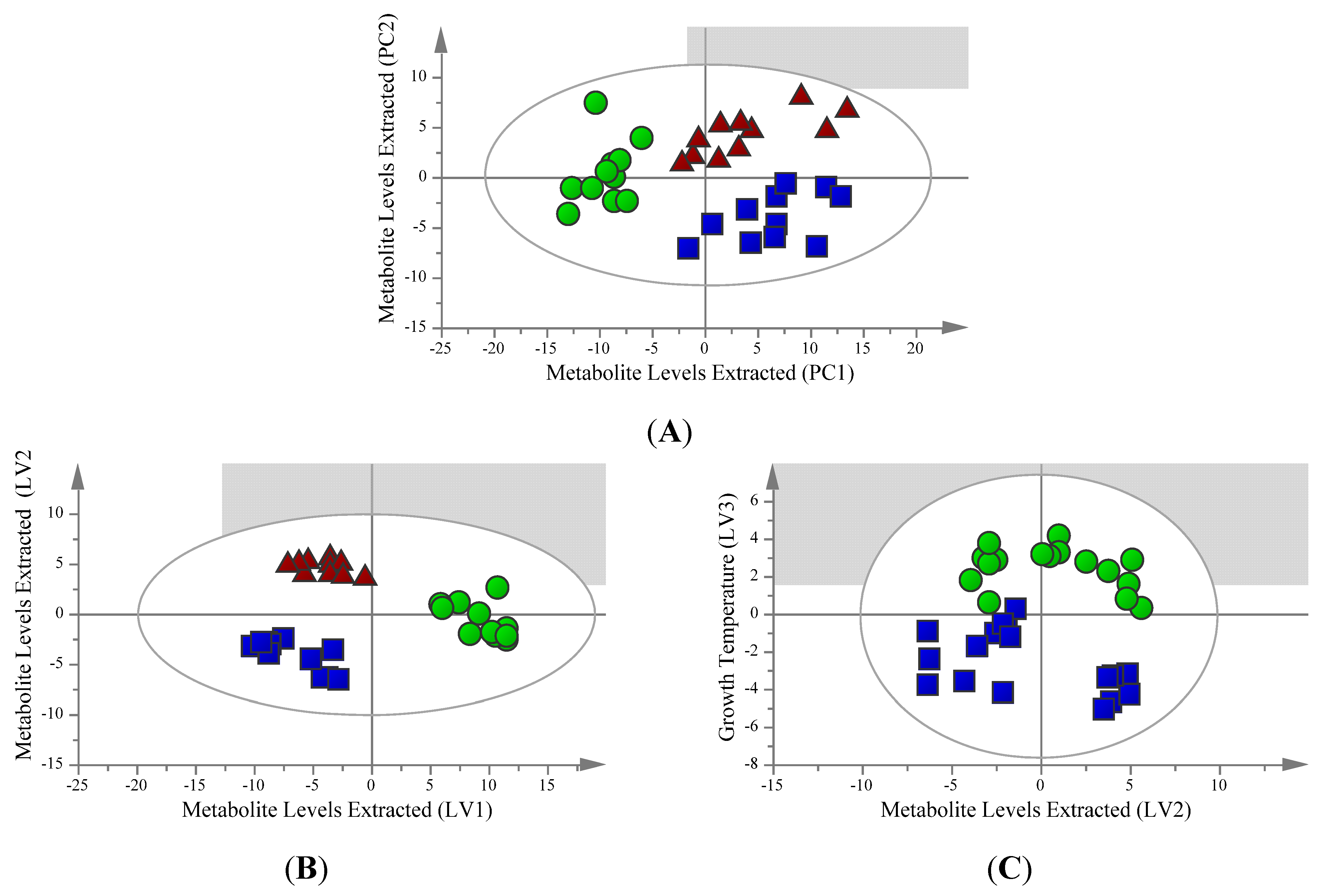
2.1. Unsupervised Analysis Clearly Differentiates Extraction Method
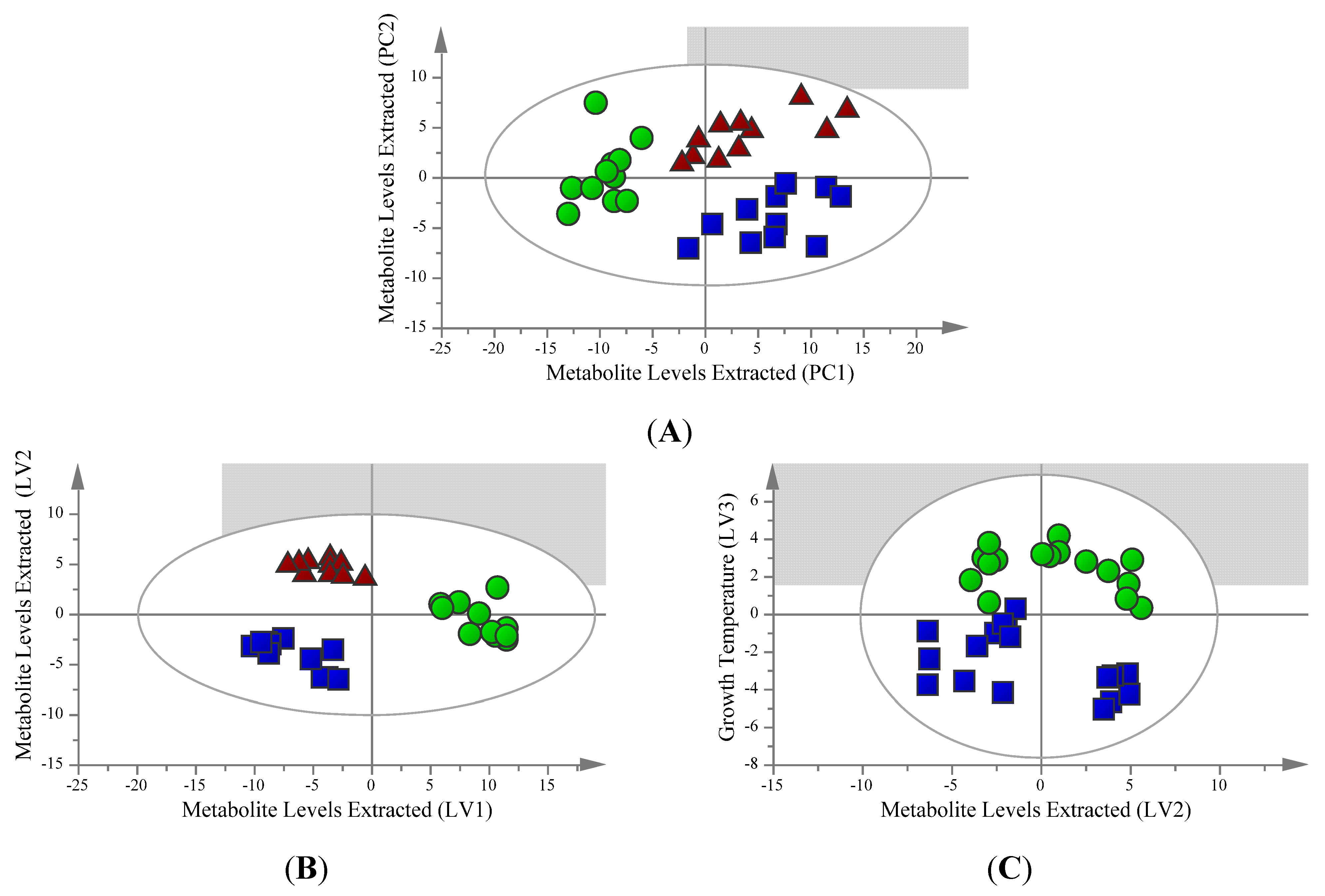
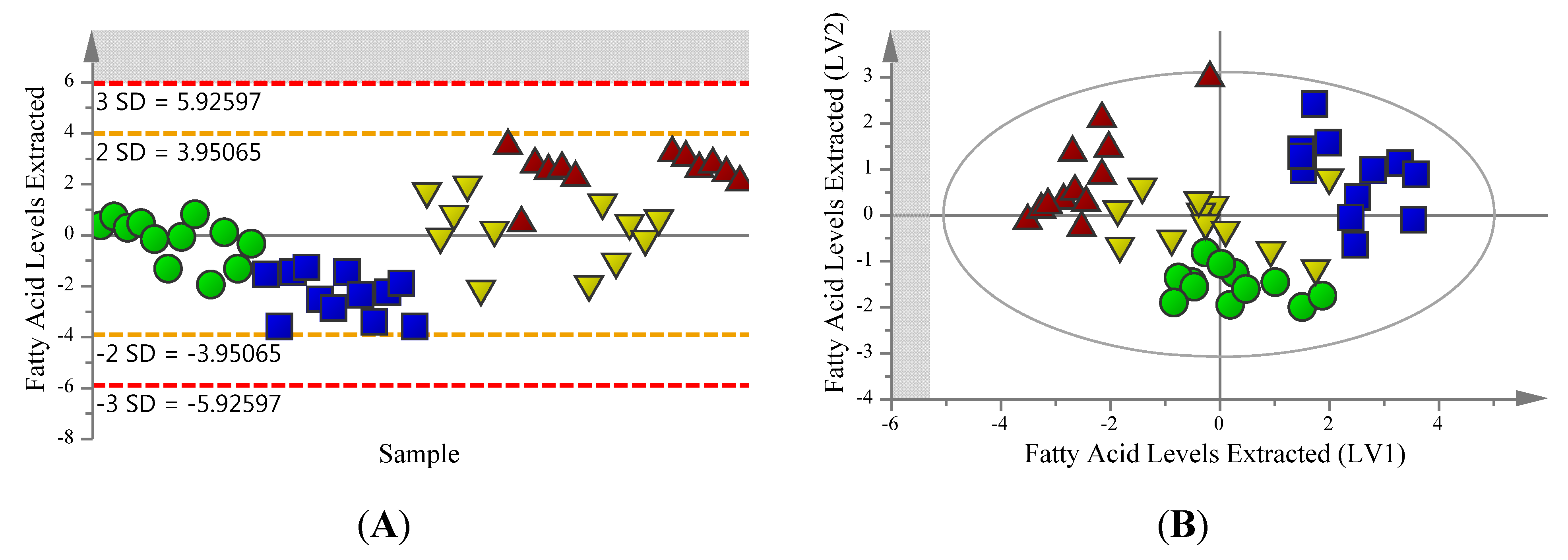
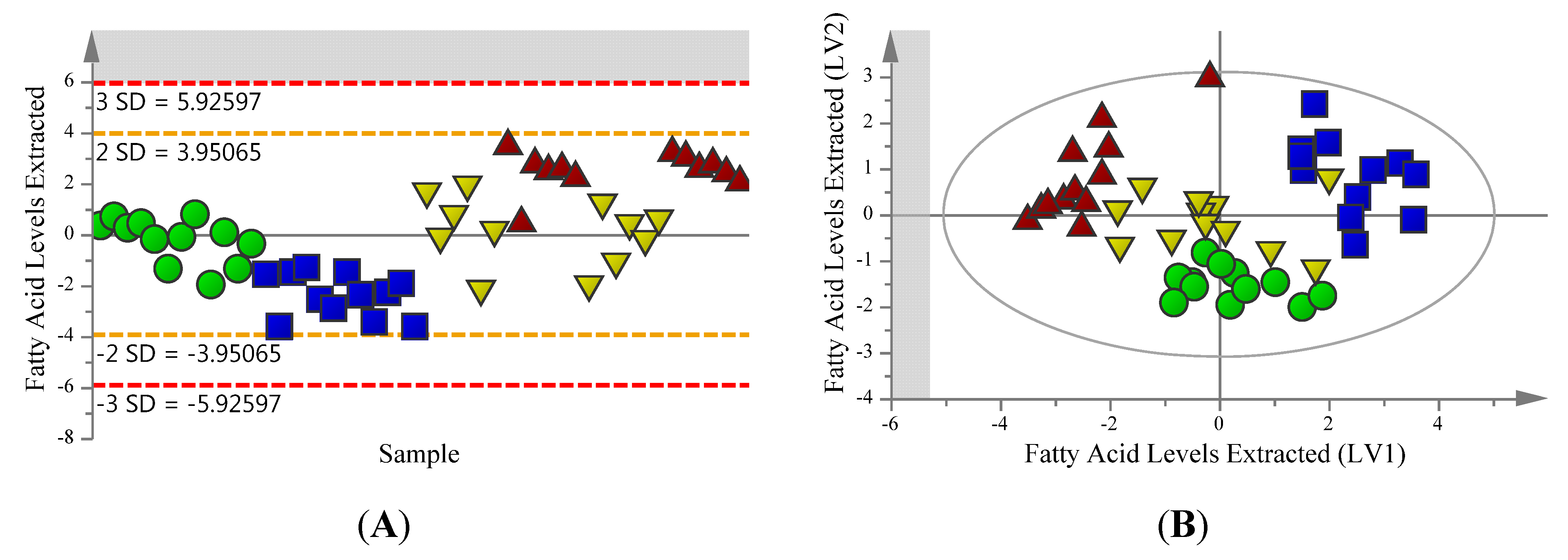
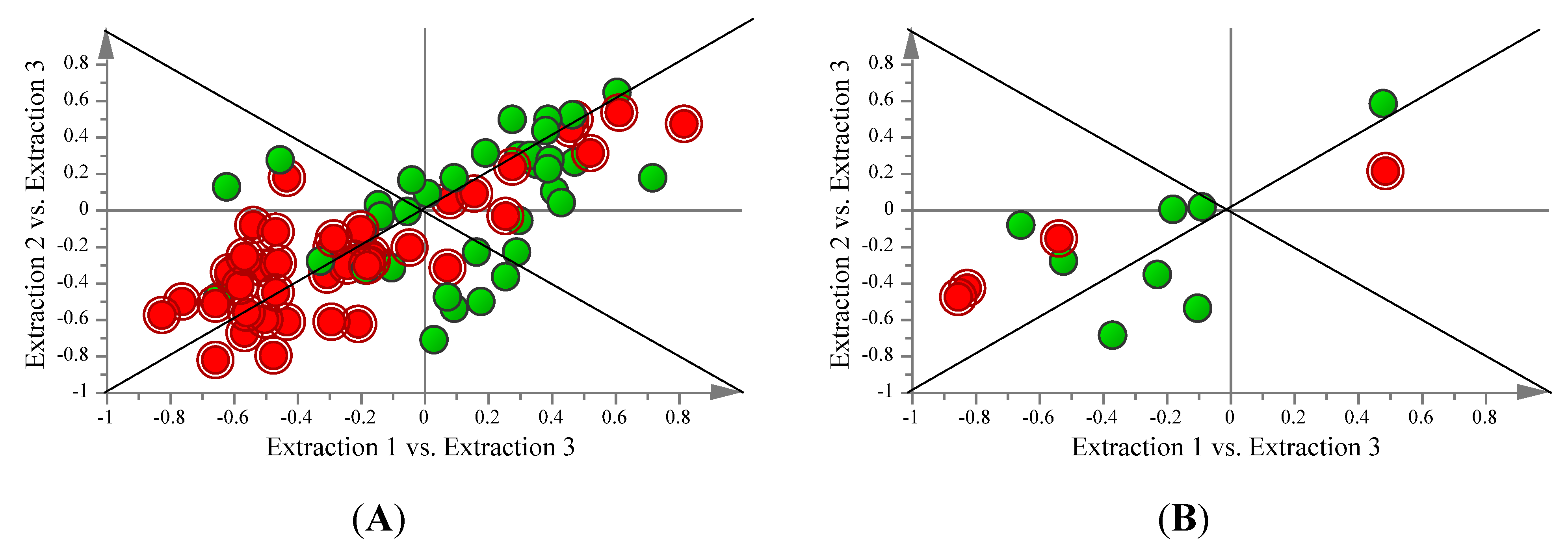
2.2. Supervised Analysis Identifies 36 Metabolites and Four Fatty Acids Differentiating the Extraction Methods

{kind=link}
{kind=link}
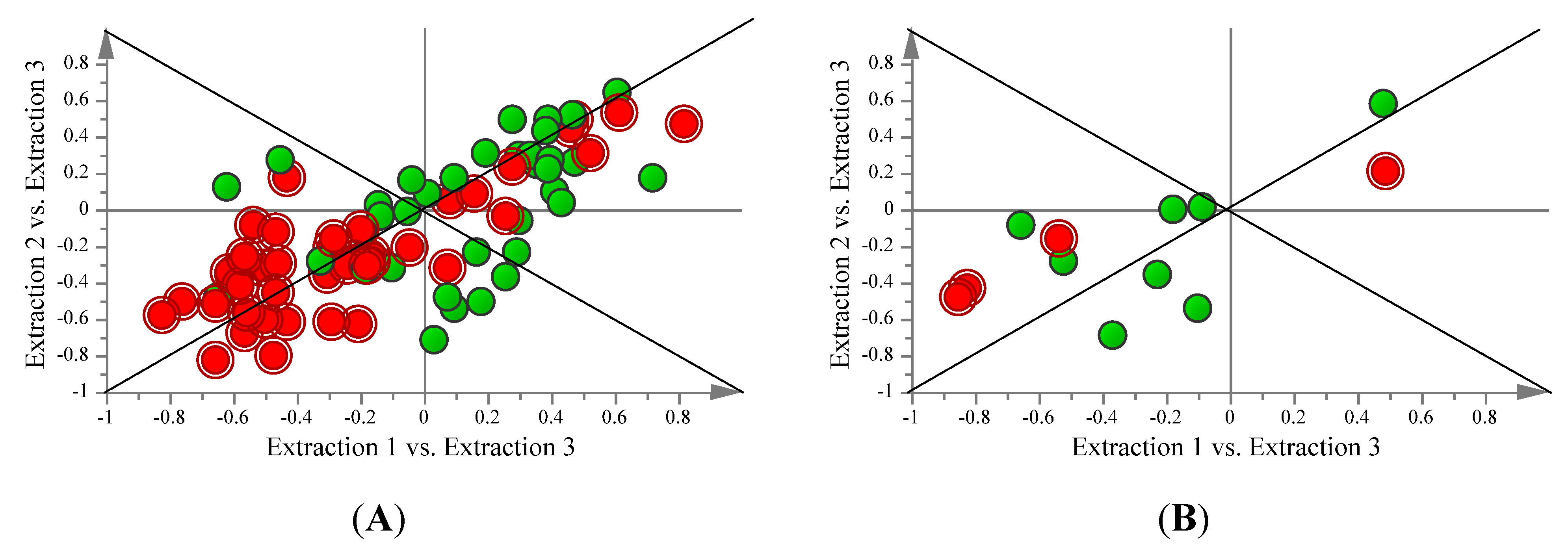{kind=link}
| Metabolite | VIP | Coefficient Extraction 1 | Coefficient Extraction 2 | Coefficient Extraction 3 |
|---|---|---|---|---|
| Threonine | 1.832 | 0.013 | 0.046 | −0.059 |
| Glycerol | 1.703 | −0.012 | −0.042 | 0.054 |
| Phenylalanine | 1.678 | 0.030 | 0.034 | −0.064 |
| Alanine-3cyano | 1.666 | 0.037 | 0.009 | −0.046 |
| Methionine | 1.649 | 0.014 | 0.041 | −0.055 |
| Proline | 1.630 | −0.001 | 0.029 | −0.028 |
| Sorbitol | 1.603 | 0.002 | 0.023 | −0.024 |
| Phosphoric Acid | 1.575 | −0.008 | 0.052 | −0.043 |
| Homoserine | 1.531 | −0.001 | 0.049 | −0.048 |
| Pyroglutamic Acid | 1.496 | 0.002 | 0.014 | −0.016 |
| Alanine | 1.476 | −0.023 | 0.025 | −0.002 |
| Ornithine | 1.461 | −0.011 | −0.046 | 0.057 |
| Serine-O acetyl | 1.382 | −0.029 | −0.035 | 0.064 |
| Fumaric Acid | 1.372 | 0.002 | 0.004 | −0.006 |
| Trehalose-alpha,alpha’-D | 1.340 | 0.013 | 0.002 | −0.015 |
| Alanine-beta | 1.327 | 0.007 | 0.006 | −0.013 |
| Succinic Acid | 1.310 | −0.006 | −0.011 | 0.017 |
| Malic acid, 2-isopropyl | 1.299 | 0.016 | 0.020 | −0.035 |
| Decan-1-ol, n- | 1.296 | 0.003 | −0.004 | 0.002 |
| Glycine | 1.271 | −0.027 | −0.014 | 0.041 |
| Valine | 1.267 | −0.010 | 0.033 | −0.023 |
| Aspartic Acid | 1.261 | 0.016 | 0.012 | −0.028 |
| Arginine [-NH3] | 1.259 | 0.030 | 0.003 | −0.033 |
| Glutamic Acid | 1.213 | 0.009 | 0.007 | −0.016 |
| Hexadecane, n- | 1.210 | −0.016 | 0.036 | −0.019 |
| Malic Acid | 1.190 | 0.005 | 0.002 | −0.006 |
| Uracil | 1.189 | −0.039 | −0.017 | 0.056 |
| Isoleucine | 1.162 | −0.009 | 0.019 | −0.011 |
| Glutamine, DL- | 1.156 | 0.029 | −0.023 | −0.007 |
| Octylamine | 1.131 | −0.005 | 0.005 | −0.001 |
| Tyramine | 1.119 | 0.014 | 0.012 | −0.026 |
| Butanoic Acid | 1.063 | −0.010 | 0.019 | −0.008 |
| Serine | 1.062 | −0.016 | 0.036 | −0.020 |
| Pentadecane, n- | 1.029 | −0.015 | 0.022 | −0.008 |
| Citric Acid | 1.024 | 0.018 | 0.005 | −0.023 |
| Heptadecan-1-ol | 1.010 | −0.011 | 0.011 | −0.001 |
| Palmitic Acid | 1.950 | 0.581 | −0.372 | −0.182 |
| Palmitoleic Acid | 1.273 | 0.173 | 0.082 | −0.221 |
| Oleic Acid | 1.239 | 0.123 | 0.066 | −0.164 |
| Stearic Acid | 1.139 | 0.058 | −0.197 | 0.121 |
2.3. Comparison of FAME and Aqueous Metabolite Profiles Obtained
2.4. Summary
3. Experimental Section
3.1. Yeast Growth and Harvesting
3.2. Metabolite Extraction
3.2.1. Extraction Protocol 1
3.2.2. Extraction Protocol 2
3.2.3. Extraction Protocol 3
3.3. Derivatization and Sample Preparation
3.4. GC-MS Data Acquisition
3.5. Data Processing and Interpretation
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Shearer, J.; Duggan, G.E.; Wejie, A.M.; Hittel, D.S.; Wasserman, D.H.; Vogel, H.J. Metabolomic profiling of dietary-induced insulin resistance in the high fat fed mouse. Diabetes Obes. Metab. 2008, 10, 950–958. [Google Scholar] [CrossRef]
- Booth, S.C.; Workentine, M.L.; Wen, J.; Shaykhutdinov, R.; Vogel, H.J.; Ceri, H.; Turner, R.J.; Weljie, A.M. Differences in metabolism between the biofilm and planktonic response to metal stress. J. Proteome Res. 2011, 10, 3190–3199. [Google Scholar] [CrossRef]
- Tremaroli, V.; Workentine, M.L.; Weljie, A.M.; Vogel, H.J.; Ceri, H.; Viti, C.; Tatti, E.; Zhang, P.; Hynes, A.P.; Turner, R.J.; et al. Metabolomics investigation of bacterial response to metal challenge. Appl. Environ. Microbiol. 2009, 75, 719–728. [Google Scholar] [CrossRef]
- Li, N.; Liu, J.; Qiu, H.; Harris, T.R.; Sirish, P.; Hammock, B.D.; Chiamvimovat, N. Use of metabolic profiling in the study of arachadonic acid metabolism in cardiovascular disease. Congestive Heart Fail. 2011, 17, 42–46. [Google Scholar] [CrossRef]
- Bogdanov, M.; Matson, W.R.; Wang, L.; Matson, T.; Saunder-Pullman, R.; Bressman, S.S.; Beal, M.F. Metabolomic profiling to develop blood biomarkers for Parkinson’s disease. Brain 2007, 131, 389–396. [Google Scholar]
- Le Belle, J.E.; Harris, N.G.; Williams, S.R.; Bhakoo, K.K. A comparison of cell and tissue extraction techniques using high-resolution 1H-NMR spectroscopy. NMR Biomed. 2002, 15, 37–44. [Google Scholar] [CrossRef]
- Lin, C.Y.; Wu, H.; Tjeerdema, R.S.; Viant, M.R. Evaluation of metabolite extraction strategies from tissue samples using NMR metabolomics. Metabolomics 2007, 3, 55–67. [Google Scholar]
- Aa, J.; Trygg, J.; Gullberg, J.; Johansson, A.I.; Jonsson, P.; Antti, H.; Marklund, S.L.; Moritz, T. Extraction and GC-MS analysis of the human blood plasma metabolome. Anal. Chem. 2005, 77, 8086–8094. [Google Scholar] [CrossRef]
- Winder, C.L.; Dunn, W.B.; Schuler, S.; Broadhurst, D.; Jarvis, R.; Stephens, G.M.; Goodacre, R. Global metabolic profiling of Escherichia coli cultures: An evaluation of methods for quenching and extraction of intracellular metabolites. Anal. Chem. 2008, 80, 2939–2948. [Google Scholar] [CrossRef]
- Sellick, C.A.; Knight, D.; Croxford, A.S.; Maqsood, A.R.; Stephens, G.M.; Goodacre, R.; Dickson, A.J. Evaluation of extraction processes for intracellular metabolite profiling of mammalian cells: Matching extraction approaches to cell type and metabolite targets. Metabolomics 2010, 6, 427–438. [Google Scholar] [CrossRef]
- Dettmer, K.; Nurnburger, N.; Kaspar, H.; Gruber, M.A.; Almstetter, M.F.; Oefner, P.J. Metabolite extraction form adherently growing mammalian cells for metabolomics studies: optimization of harvesting and extraction protocols. Anal. Bioanal. Chem. 2011, 399, 1127–1139. [Google Scholar] [CrossRef]
- Castrillo, J.I.; Hayes, A.; Mohammed, S.; Gaskell, S.J.; Oliver, S.G. An optimized protocol for metabolome analysis in yeast using direct infusion electrospray mass spectrometry. Phytochemistry 2003, 62, 929–937. [Google Scholar] [CrossRef]
- Want, E.J.; O’Maille, G.; Smith, C.A.; Brandon, T.R.; Uritboonthai, W.; Qin, C.; Trauger, S.A.; Siuzdak, G. Solvent-dependent metabolite distribution, clustering and protein extraction for serum profiling with mass spectrometry. Anal. Chem. 2006, 78, 743–752. [Google Scholar]
- Masson, P.; Alves, A.C.; Ebbels, T.M.D.; Nicholson, J.K.; Want, E.J. Optimization and evaluation of metabolite extraction protocols for untargeted metabolic profiling of liver samples by UPLC-MS. Anal. Chem. 2010, 82, 7779–7786. [Google Scholar] [CrossRef]
- Buscher, J.M.; Czernik, D.; Ewald, J.C.; Sauer, U.; Zamboni, N. Cross-platform comparison of methods for quantitative metabolomics of primary metabolism. Anal. Chem. 2009, 81, 2135–2143. [Google Scholar] [CrossRef]
- Geier, F.M.; Want, E.J.; Leroi, A.M.; Bundy, J.G. Cross-platform comparison of Caenorhabditis elegans tissue extraction strategies for comprehensive metabolome coverage. Anal. Chem. 2011, 83, 3730–3736. [Google Scholar] [CrossRef]
- Villas-Boas, S.G.; Hojer-Pedersen, J.; Akesson, M.; Smedsgaard, J.; Nielsen, J. Global metabolite analysis of yeast: evaluation of sample preparation methods. Yeast 2005, 22, 1155–1169. [Google Scholar] [CrossRef]
- Canelas, A.B.; ten Pierick, A.; Ras, C.; Seifar, R.M.; van Dam, J.C.; van Gulik, W.M.; Heijnen, J.J. Quantitative evaluation of intracellular metabolite extraction techniques for yeast metabolomics. Anal. Chem. 2009, 81, 7379–7389. [Google Scholar] [CrossRef]
- Cequier-Sanchez, E.; Rodriquez, C.; Ravelo, A.G.; Zarate, R. Dichloromethane as a solvent for lipid extraction and assessment of lipid classes and fatty acids from samples of different natures. J. Agric. Food Chem. 2008, 56, 4297–4303. [Google Scholar] [CrossRef]
- Matyash, V.; Liebisch, G.; Kurzchalia, T.V.; Shevchenko, A.; Schwudke, D. Lipid extraction by methyl-tert-butyl ether for high-throughput lipidomics. J. Lipid Res. 2008, 49, 1137–1145. [Google Scholar] [CrossRef]
- Gomez-Brandon, M.; Lores, M.; Dominguez, J. Comparison of extraction and derivitization methods for fatty acid analysis in solid environmental matrixes. Anal. Bioanal. Chem. 2008, 392, 505–514. [Google Scholar] [CrossRef]
- Guan, X.L.; Riezman, I.; Wenk, M.R.; Riezman, H. Yeast lipid analysis and quantification by mass spectrometry. Methods Enzymol. 2010, 480, 369–391. [Google Scholar]
- Nygren, H.; Seppanen-Laakso, T.; Castillo, S.; Hyotylainen, T.; Oresic, M. Liquid chromotography-mass spectrometry (LC-MS)-based lipidomics for studies of body fluids and tissues. Methods Mol. Biol. 2011, 708, 247–257. [Google Scholar] [CrossRef]
- Folch, J.; Lee, M.; Sloane, S. A simple method for the isolation and purification of total lipids from animal tissues. J. Biol. Chem. 1957, 266, 497–509. [Google Scholar]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef]
- Zaremberg, V.; Gajate, C.; Cacharro, L.M.; Mollinedo, F.; McMaster, C.R. Cytoxicity of and anti-cancer lysophospholipid through selective modification of lipid raft composition. J. Biol. Chem. 2005, 280, 38047–38058. [Google Scholar] [CrossRef]
- McCombie, G.; Medina-Gomez, G.; Lelliott, C.J.; Vidal-Piug, A.; Griffin, J.L. Metabolomic and lipidomic analysis of the heart of peroxisome proliferator-activated receptor-γ coactivator 1-β knock out mice on a high fat diet. Metabolites 2012, 2, 366–381. [Google Scholar] [CrossRef]
- Ejsing, C.S.; Sampaio, J.L.; Surendranath, V.; Duchoslav, E.; Ekroos, K.; Klemm, R.W.; Simons, K.; Shevchenko, A. Global analysis of the yeast lipidome by quantitative shotgun mass spectrometry. Proc. Natl. Acad. Sci. USA 2009, 106, 2136–2141. [Google Scholar] [CrossRef]
- Hiller, K.; Hangebrauk, J.; Jager, C.; Spura, J.; Schreiber, K.; Schomburg, D. MetaboliteDetector: Comprehensive analysis tool for targeted and non-targeted GC/MS based metabolome analysis. Anal. Chem. 2009, 81, 3429–3439. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tambellini, N.P.; Zaremberg, V.; Turner, R.J.; Weljie, A.M. Evaluation of Extraction Protocols for Simultaneous Polar and Non-Polar Yeast Metabolite Analysis Using Multivariate Projection Methods. Metabolites 2013, 3, 592-605. https://doi.org/10.3390/metabo3030592
Tambellini NP, Zaremberg V, Turner RJ, Weljie AM. Evaluation of Extraction Protocols for Simultaneous Polar and Non-Polar Yeast Metabolite Analysis Using Multivariate Projection Methods. Metabolites. 2013; 3(3):592-605. https://doi.org/10.3390/metabo3030592
Chicago/Turabian StyleTambellini, Nicolas P., Vanina Zaremberg, Raymond J. Turner, and Aalim M. Weljie. 2013. "Evaluation of Extraction Protocols for Simultaneous Polar and Non-Polar Yeast Metabolite Analysis Using Multivariate Projection Methods" Metabolites 3, no. 3: 592-605. https://doi.org/10.3390/metabo3030592