A Comprehensive Workflow of Mass Spectrometry-Based Untargeted Metabolomics in Cancer Metabolic Biomarker Discovery Using Human Plasma and Urine

Abstract
:
1. Overview
1.1. Hyphenated Separation and Detection Techniques
1.2. Untargeted Metabolic Profiling
1.3. Data Mining Techniques
1.4. Annotation and Identification
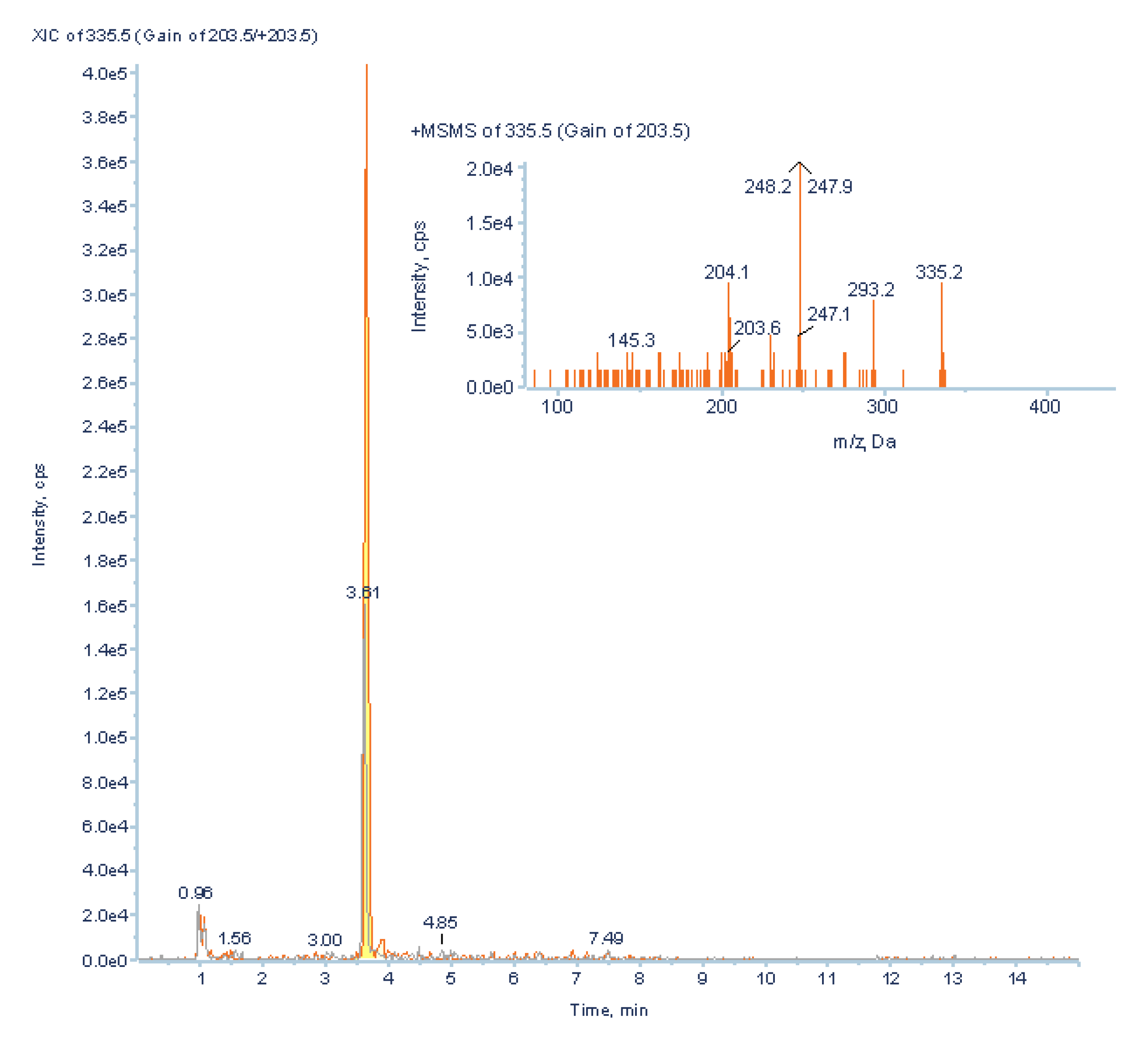
1.5. Predictive MRM Screening of Secondary Metabolites.
1.6. Pathway Analysis
2. The Application in Early Diagnosis of Pancreatic Cancer
2.1. Introduction
2.2. Procedure
2.2.1. Material
2.2.2. Sample Preparation
2.2.3. Untargeted Metabolic Profiling
2.2.3.1. LC-MS
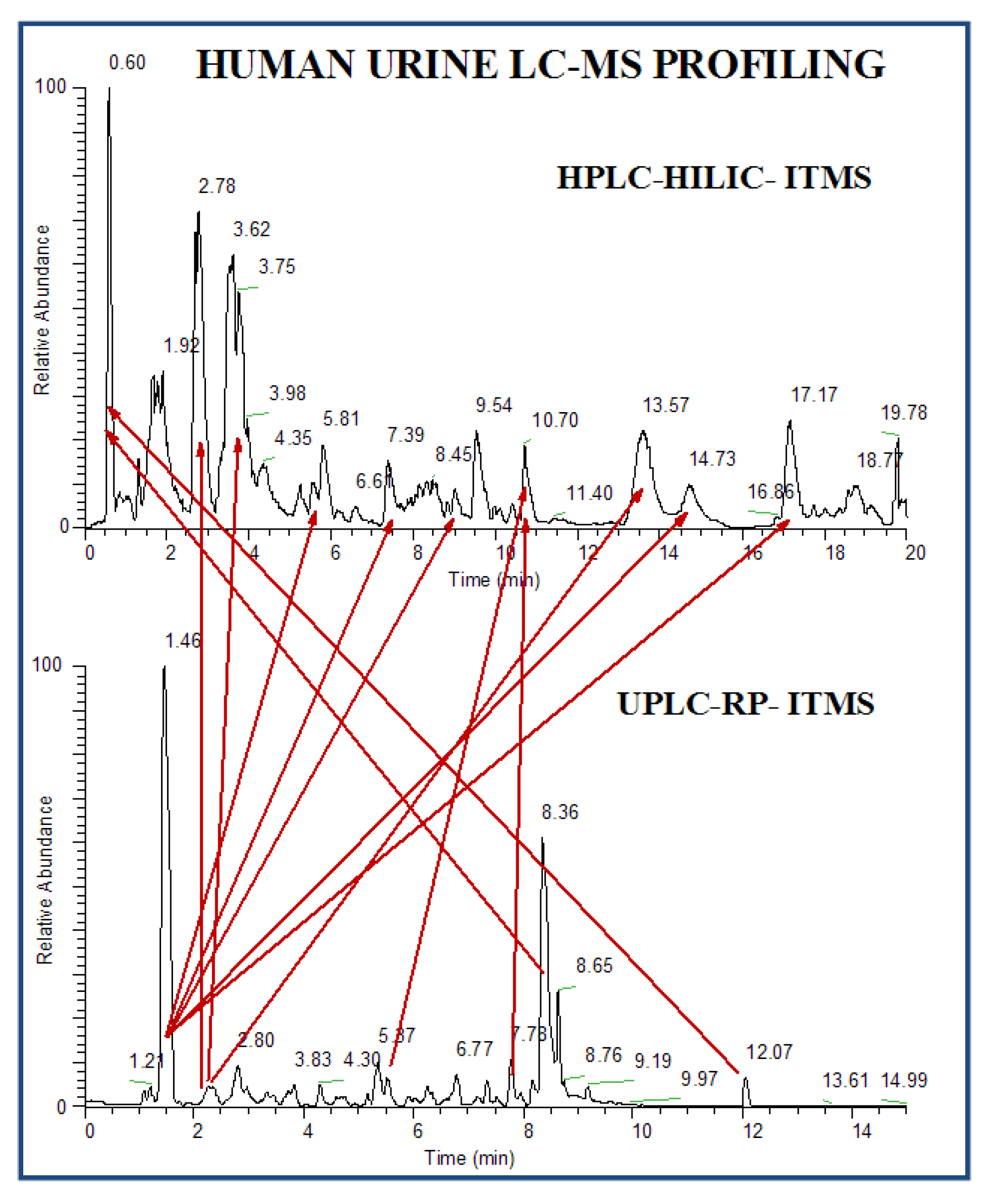
2.2.3.1.1. RP ESI-LC-MS
2.2.3.1.2. HILIC ESI-LC-MS
2.2.3.2. GC–MS
2.2.4. Data Mining
2.2.4.1. Free Open Source Software
2.2.4.2. Commercial Software
2.2.5. Annotation and Identification of Feature Components
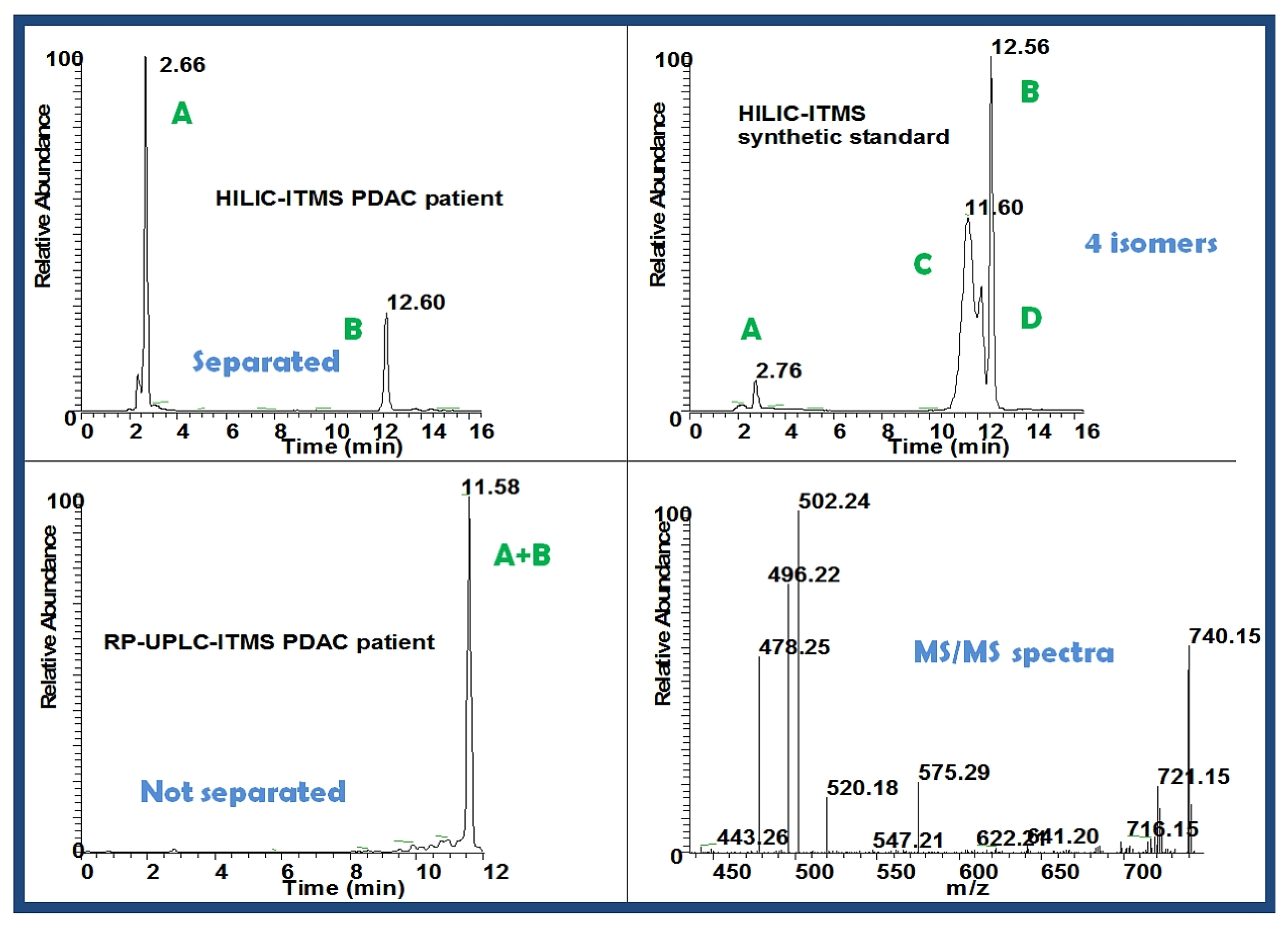
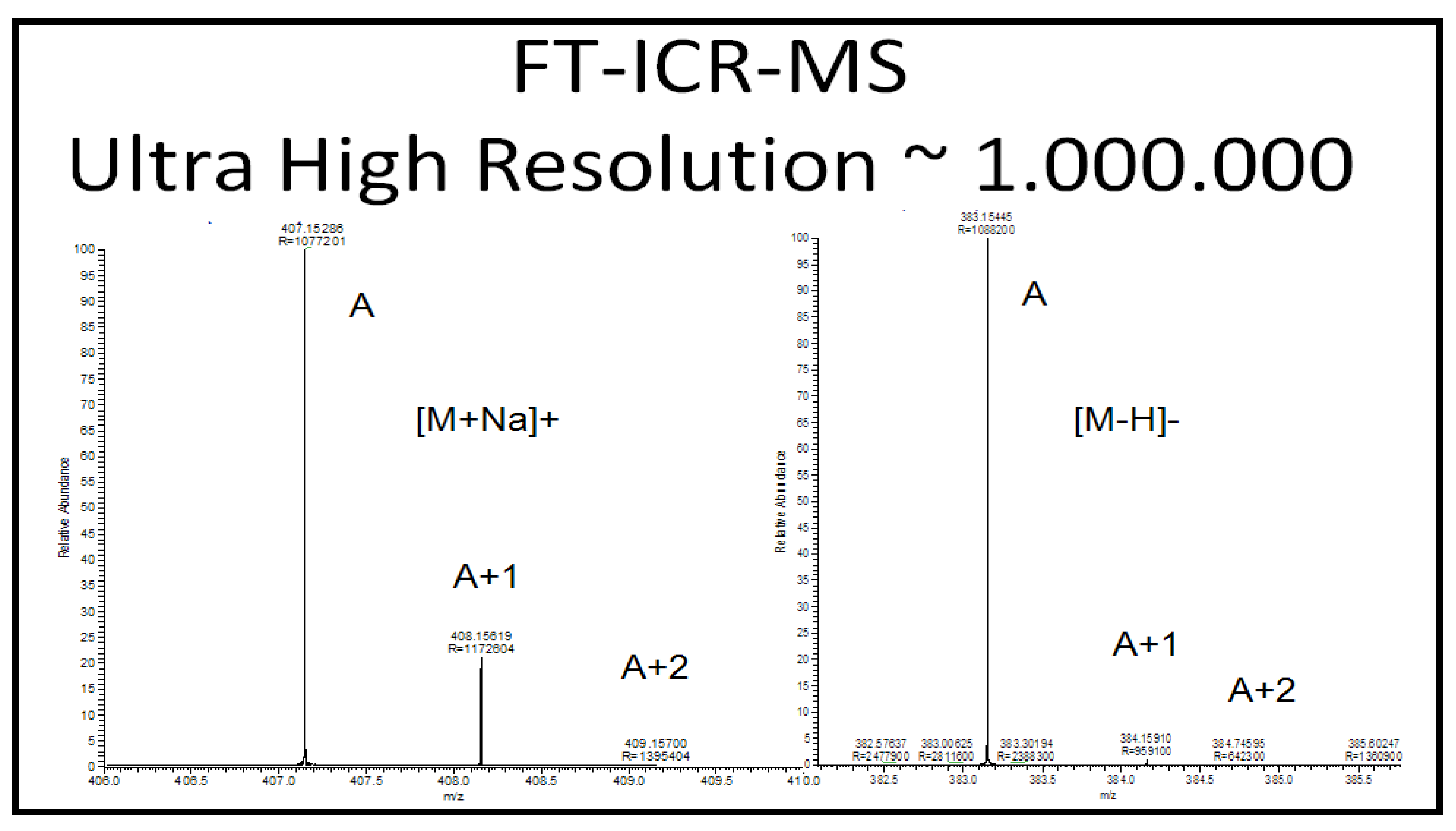
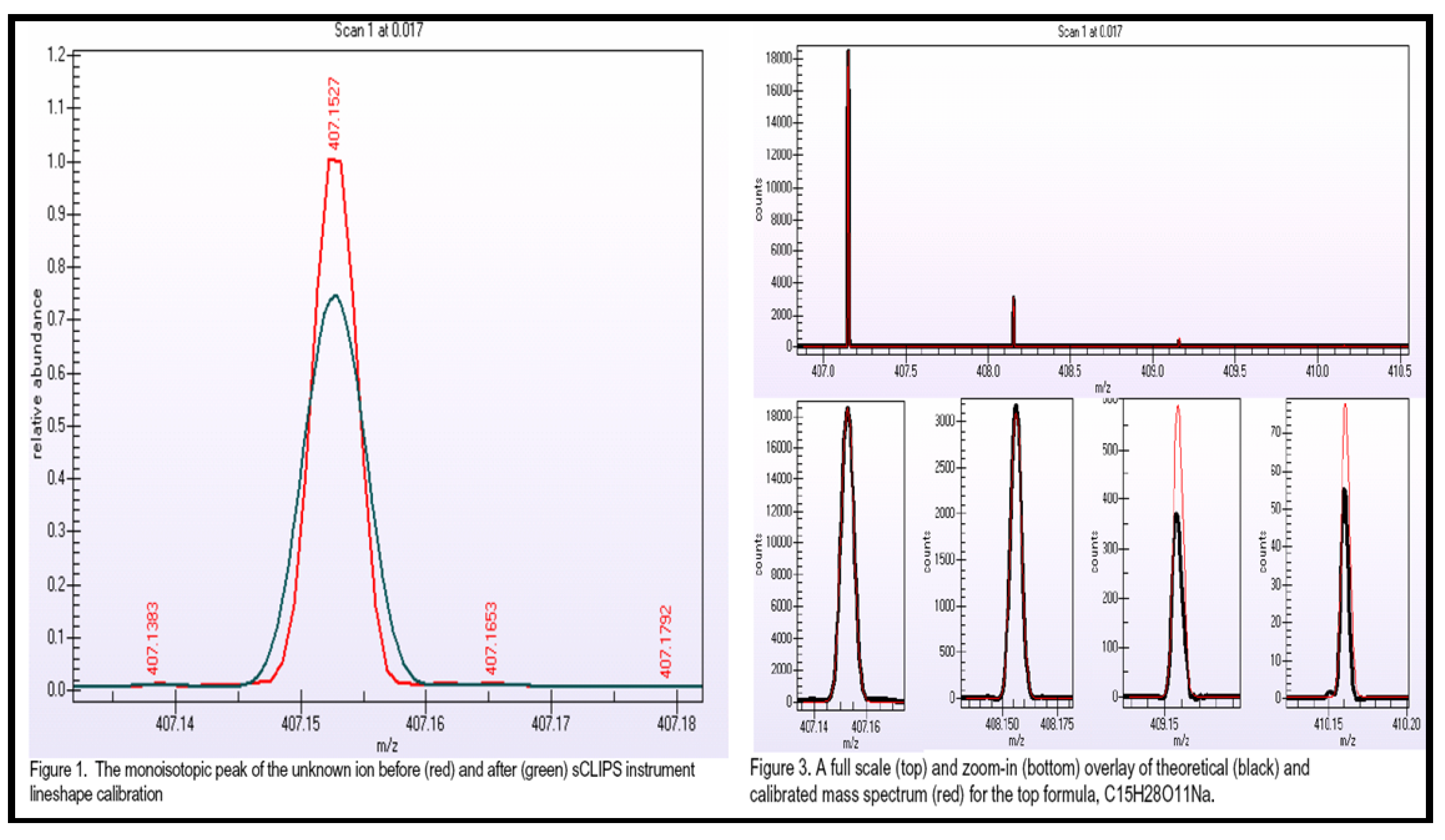
2.2.5.1. Accurate Mass, Isotope Pattern, and MS/MS
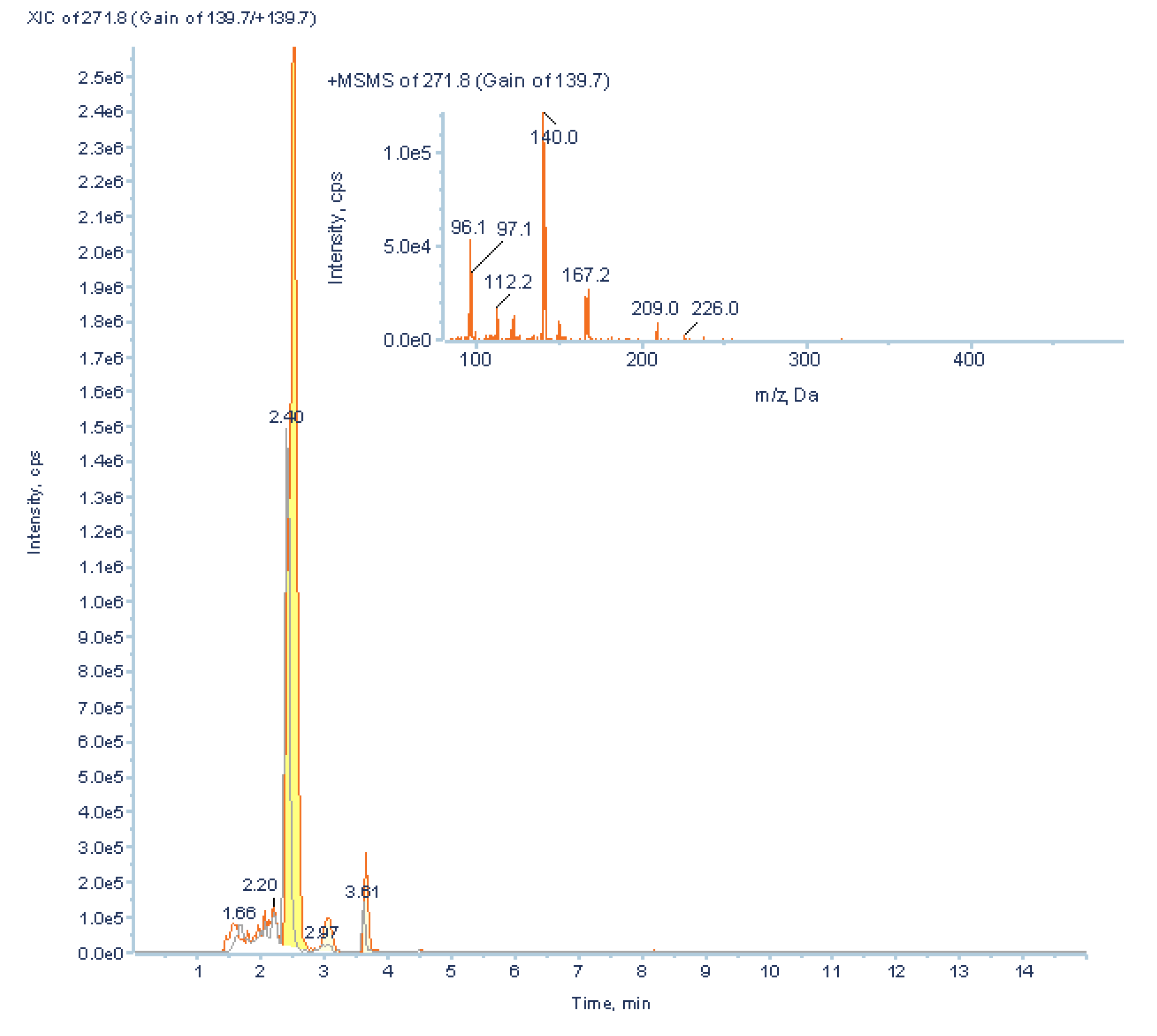
2.2.5.2. MSn Ion Tree
2.2.6. Pathway Analysis
2.3. Results and Discussion
2.3.1. Experimental Design
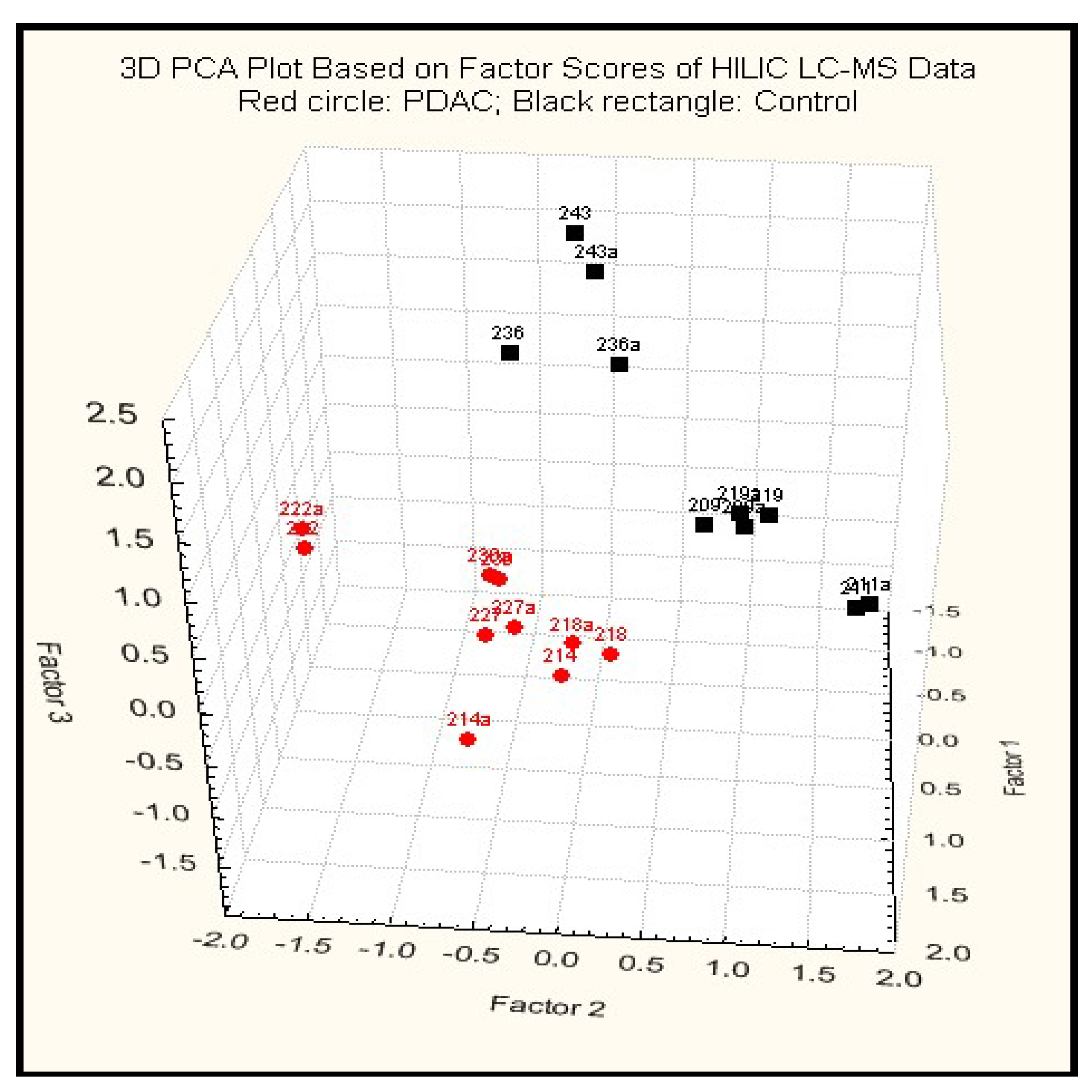
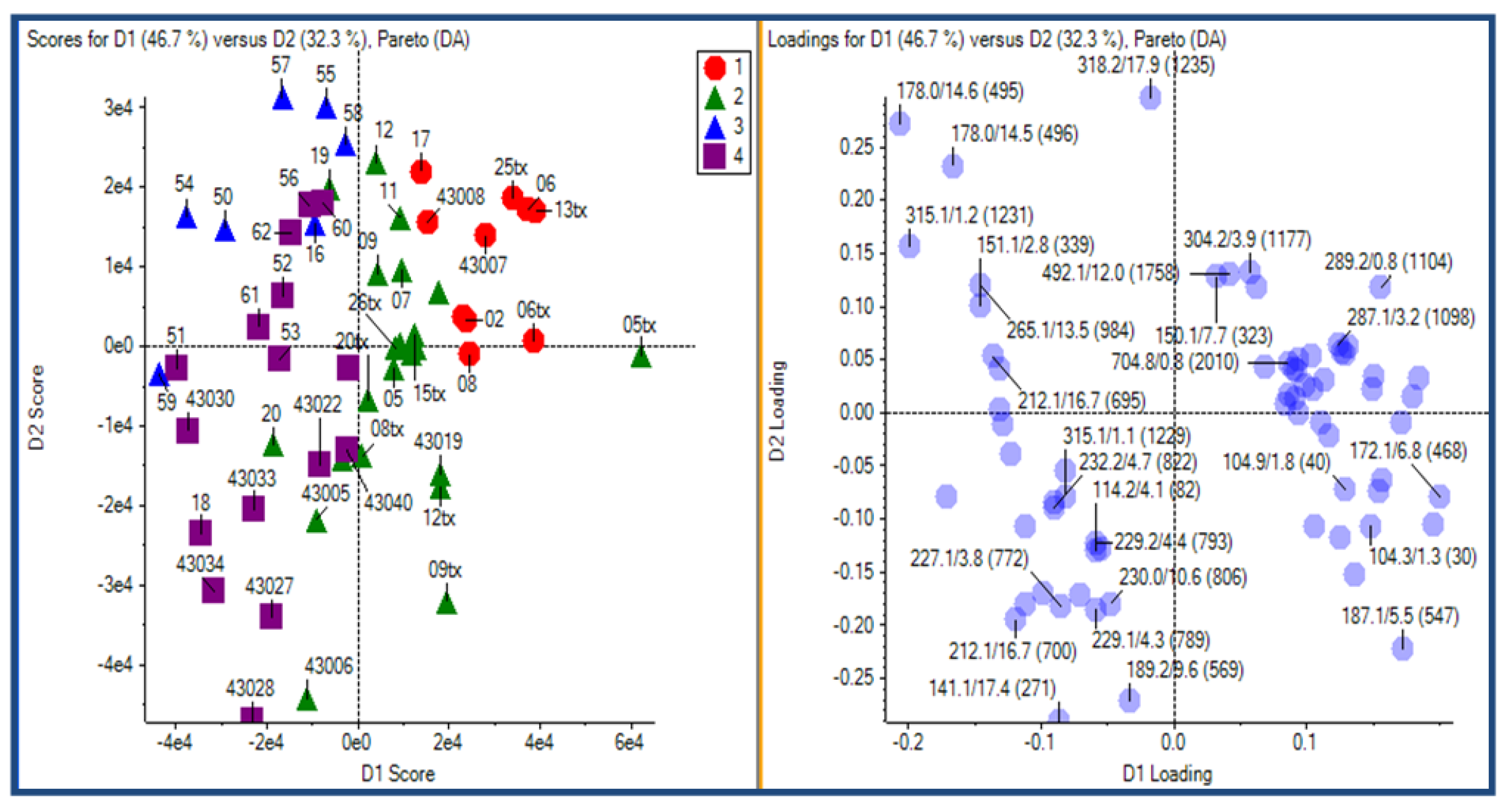
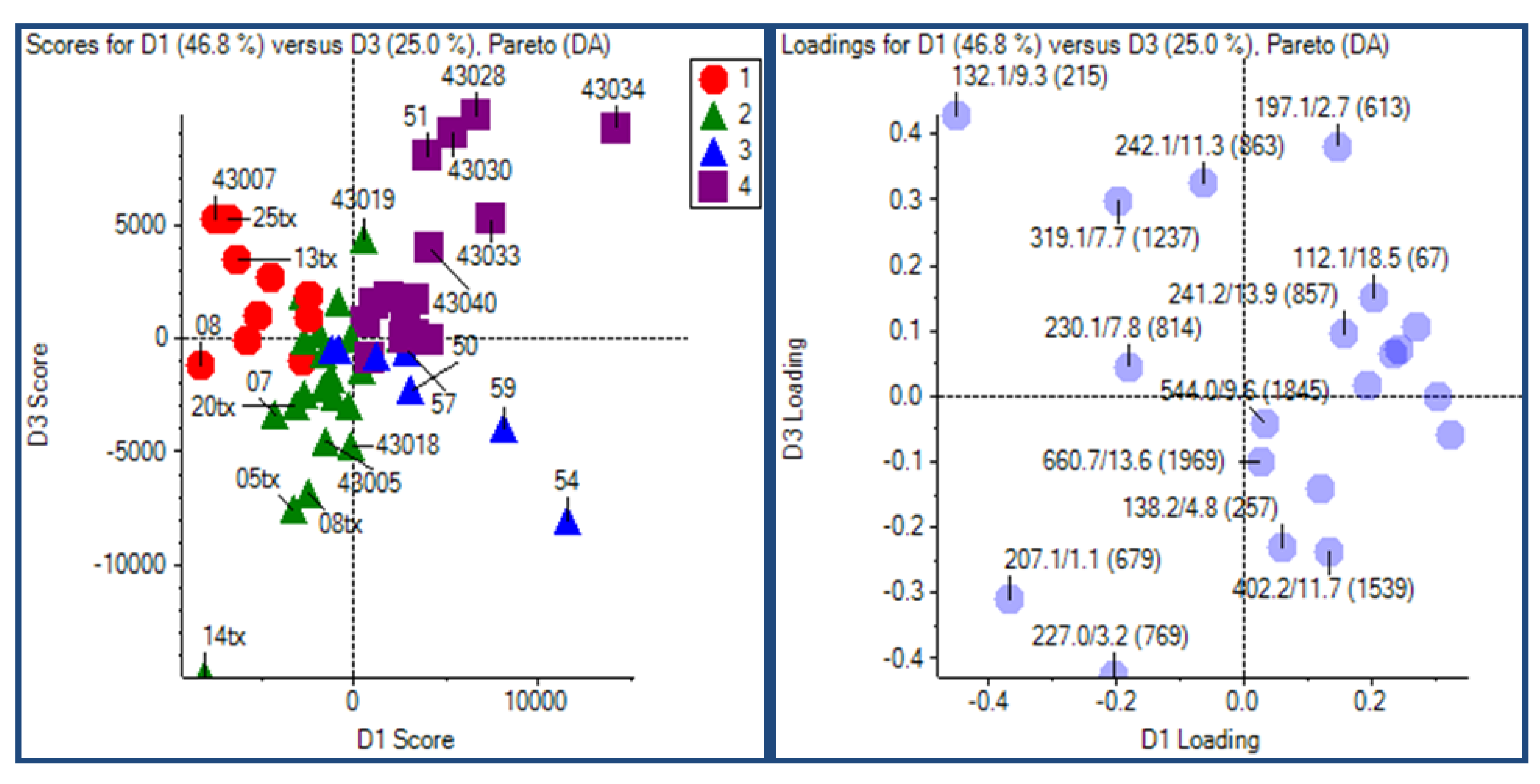
2.3.2. Principal Component Analysis

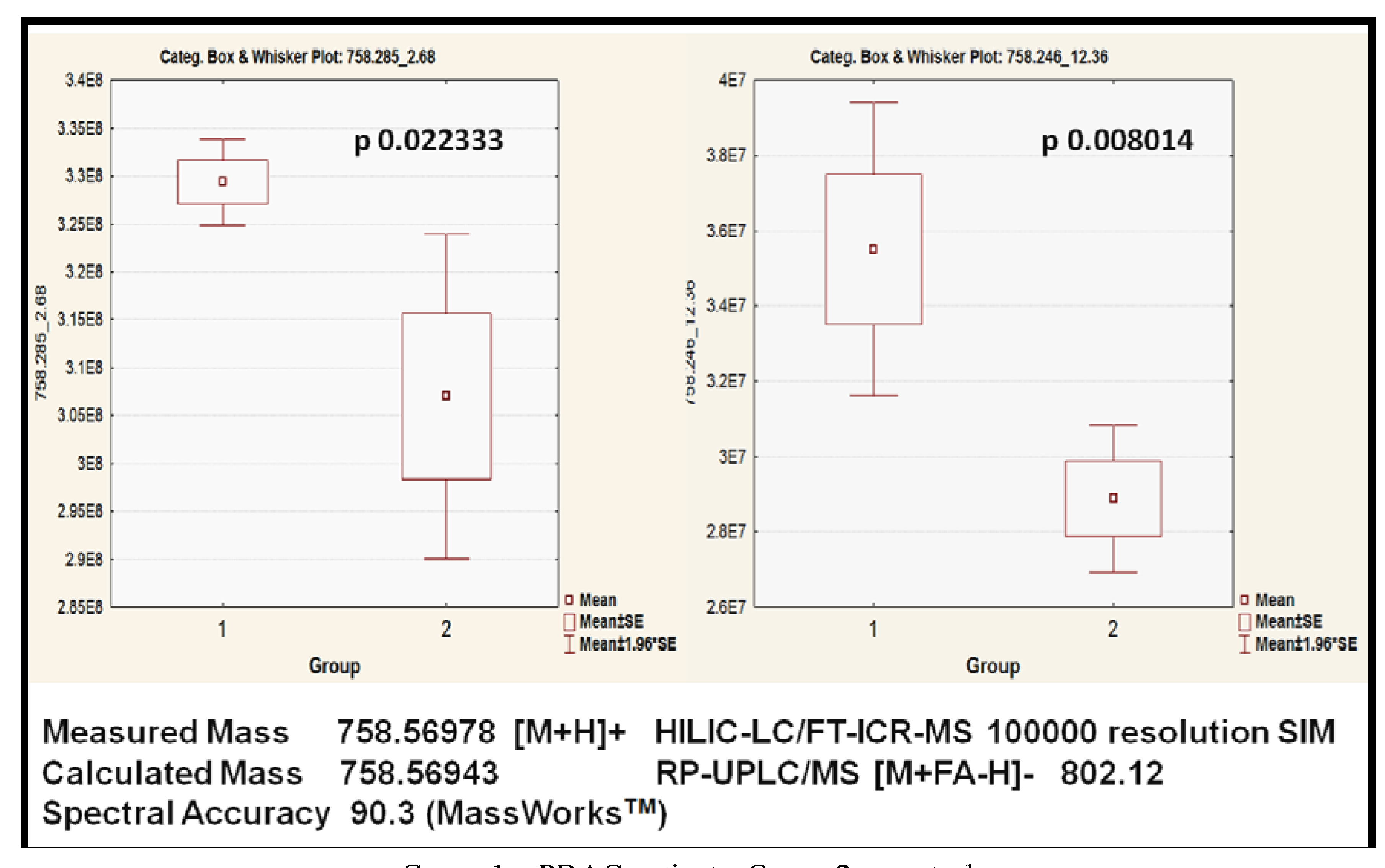
2.3.3. Putative Biomarkers

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substance Name | PubChem CID | m/z-Ion polarity | Profiling Method | p-Value | Fold Change |
|---|---|---|---|---|---|
| Increased in PDAC | |||||
| Arachidonic acid | 444899 | GC-TOF-MS | 0.040982 | 1.49 | |
| Erythritol | 8998 | GC-TOF-MS | 0.008525 | 1.53 | |
| Cholesterol | 5997 | GC-TOF-MS | 0.030047 | 1.85 | |
| N-Methylalanine | 5288725 | GC-TOF-MS | 0.024311 | 2.81 | |
| Lysine | 5962 | 147.1 pos | HILIC-LC/MS | 0.017356 | 1.03 |
| Deoxycholylglycine | 9675 | 448.53 neg | HILIC/RP-LC/MS | 0.000052 | 1.31 |
| Cholylglycine | 16219399 | 464.42 neg | HILIC/RP-LC/MS | 0.000001 | 2.61 |
| LysoPC (16:0) | 86554 | 496.2 pos | HILIC-LC/MS | 0.000645 | 1.33 |
| Tauroursodeoxycholic | 3034759 | 498.34 neg | RP-LC/MS | 0.004029 | 2.01 |
| Taurocholic acid | 6675 | 514.1 neg | HILIC/RP-LC/MS | 0.000312 | 1.75 |
| LysoPC(18:2) | 11988421 | 520.23 pos | RP-LC/MS | 0.013425 | 1.59 |
| PE(26:0) | 9546763 | 606.23 neg | RP-LC/MS | 0.012072 | 1.81 |
| PC (34:2) | 6021688 | 758.31 pos | HILIC-LC/MS | 0.008014 | 1.32 |
| Unknown | 753.12 pos | HILIC-LC/MS | 0.000002 | 1.27 | |
| Unknown | 265.07 pos | HILIC-LC/MS | 0.000005 | 1.17 | |
| Unknown | 332.07 pos | HILIC-LC/MS | 0.000031 | 1.37 | |
| Unknown | 633.19 pos | RP-LC/MS | 0.009372 | 1.8 | |
| Unknown | 414.15 pos | RP-LC/MS | 0.006497 | 1.69 | |
| Decreased in PDAC | |||||
| Glutamine | 5961 | 145.22 neg | HILIC/RP-LC/MS | 0.000021 | 1.2 |
| Hydrocinnamic acid | 107 | 149.12 neg | HILIC-LC/MS | 0.000252 | 1.38 |
| Phenylalanine | 6140 | 166.12 pos | RP-LC/MS | 0.036583 | 1.15 |
| Tryptamine | 1150 | 205.09 pos | RP-LC/MS | 0.016353 | 1.07 |
| Inosine | 6021 | 267.21 neg | RP-LC/MS | 0.000014 | 1.4 |
| Unknown | 187.12 neg | RP-LC/MS | 0.000246 | 1.11 |
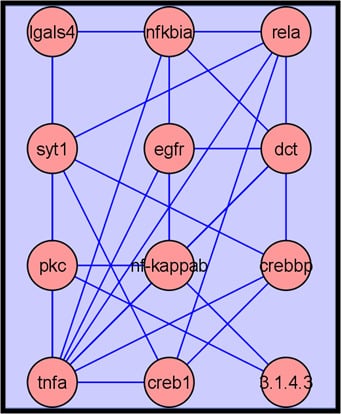
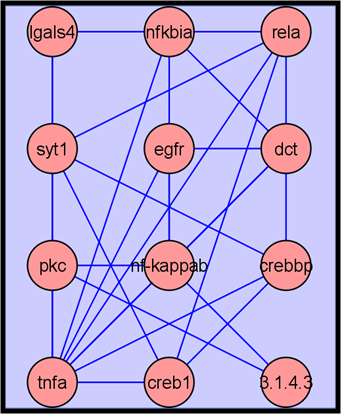
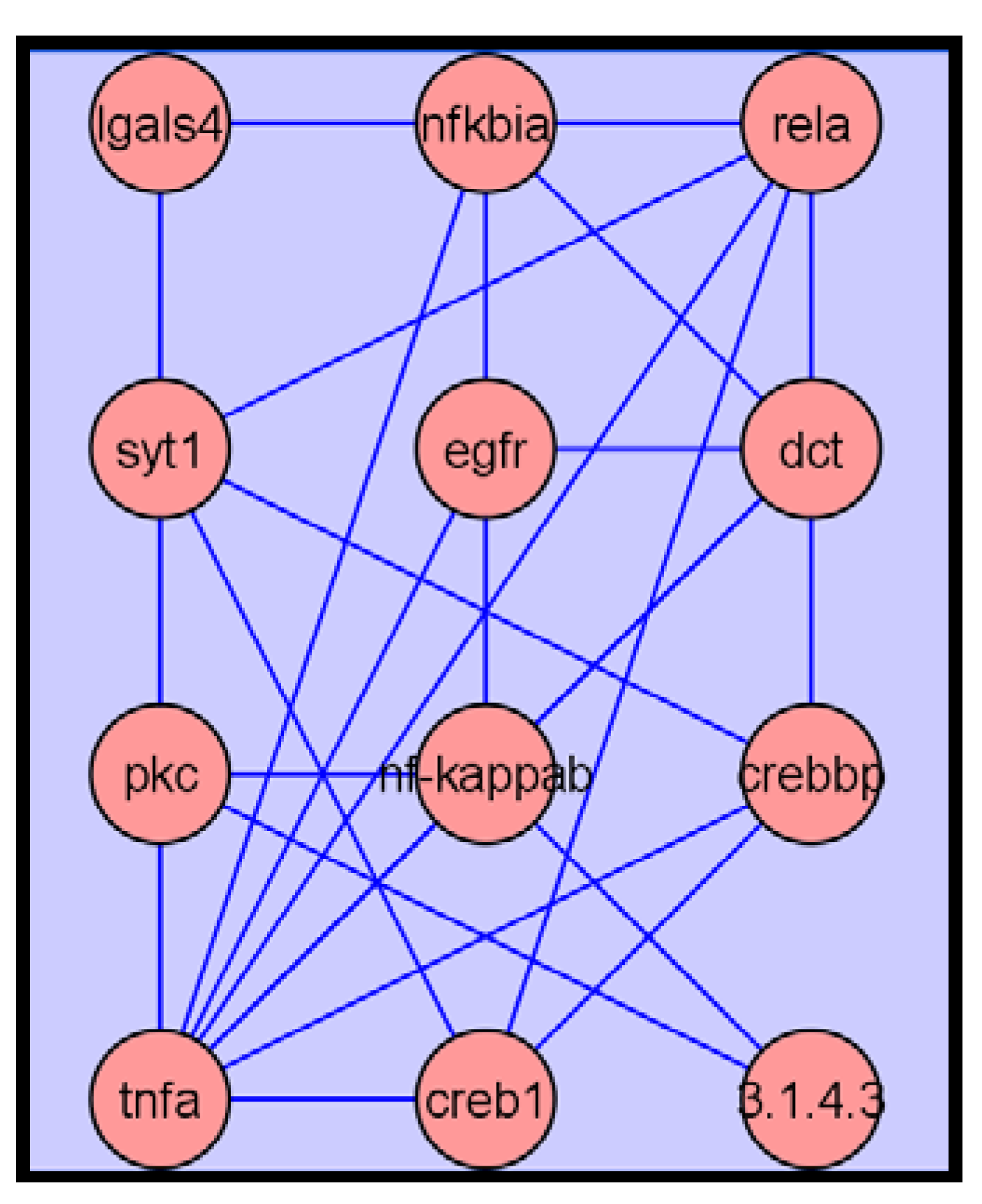
2.3.4. Metabolite Network Analysis




2.3.5. Validation
2.3.6. Summary
3. The Application in Early Diagnosis of Kidney Cancer
3.1. Introduction
3.2. Procedure
3.2.1. Sample Preparation
3.2.2. Untargeted Screening Using Full Scan Mode (See Section 2)
3.2.3. Subclass Screening Using Neutral Loss Scan, Precursor Ion Scan, and Predictive MRM
3.3. Results and Discussion
3.3.1. Untargeted Profiling





3.3.2. Low Abundant Subclass Screening
3.3.3. Summary


4. Conclusions
Acknowledgements
Conflicts of Interest
References
- Bentley, D.R. Genomic sequence information should be released immediately and freely in the public domain. Science 1996, 274, 533–534. [Google Scholar] [CrossRef]
- Bentley, D.R. Genomes for medicine. Nature 2004, 429, 440–445. [Google Scholar] [CrossRef]
- Kruglyak, L.; Nickerson, D.A. Variation is the spice of life. Nat. Genet. 2001, 27, 234–236. [Google Scholar] [CrossRef]
- Fiehn, O.; Kopka, J.; Trethewey, R.N.; Willmitzer, L. Identification of uncommon plant metabolites based on calculation of elemental compositions using gas chromatography and quadrupole mass spectrometry. Anal. Chem. 2000, 72, 3573–3580. [Google Scholar] [CrossRef]
- Tanaka, N.; Tolstikov, V.; Weckwerth, W.; Fiehn, O.; Fukusaki, H. Micro HPLC for Metabolomics. In Frontier of Metabolomic Research; Springer-Verlag: Tokyo, Japan, 2003; pp. 85–100. [Google Scholar]
- Ikegami, T.; Kobayashi, H.; Kimura, H.; Tolstikov, V.; Fiehn, O.; Tanaka, N. High-performance liquid chromatography for metabolomics: High-efficiency separations utilizing monolithic silica columns. In Metabolomics: The Frontier of Systems Biology; Springer-Verlag: Tokyo, 2005; pp. 107–126. [Google Scholar]
- Tanaka, N.; Kimura, H.; Tokuda, D.; Hosoya, K.; Ikegami, T.; Ishizuka, N.; Minakuchi, H.; Nakanishi, K.; Shintani, Y.; Furuno, M; et al. Simple and comprehensive two-dimensional reversed-phase HPLC using monolithic silica columns. Anal. Chem. 2004, 76, 1273–1281. [Google Scholar] [CrossRef]
- Tanaka, N.; Kobayashi, H. Monolithic columns for liquid chromatography. Anal. Bioanal. Chem. 2003, 376, 298–301. [Google Scholar]
- Tanaka, N.; Kobayashi, H.; Nakanishi, K.; Minakuchi, H.; Ishizuka, N. Monolithic LC columns. Anal. Chem. 2001, 73, 420A–429A. [Google Scholar]
- Tolstikov, V.V.; Fiehn, O.; Tanaka, N. Application of liquid chromatography-mass spectrometry analysis in metabolomics: reversed-phase monolithic capillary chromatography and hydrophilic chromatography coupled to electrospray ionization-mass spectrometry. In Metabolomics: Methods and Protocols (Methods in Molecular Biology); Weckwerth, W., Ed.; Humana Press: Totowa, NJ, USA, 2007; Volume 358, pp. 141–155. [Google Scholar]
- Tolstikov, V.V.; Lommen, A.; Nakanishi, K.; Tanaka, N.; Fiehn, O. Monolithic silica-based capillary reversed-phase liquid chromatography/electrospray mass spectrometry for plant metabolomics. Anal. Chem. 2003, 75, 6737–6740. [Google Scholar] [CrossRef]
- Plumb, R.S.; Granger, J.H.; Stumpf, C.L.; Johnson, K.A.; Smith, B.W.; Gaulitz, S.; Wilson, I.D.; Castro-Perez, J. A rapid screening approach to metabonomics using UPLC and q-TOF mass spectrometry: Application to age, gender and diurnal variation in normal/Zucker obese rats and black, white and nude mice. Analyst 2005, 130, 844–849. [Google Scholar] [CrossRef]
- Hemstrom, P.; Irgum, K. Hydrophilic interaction chromatography. J. Sep. Sci. 2006, 29, 1784–1821. [Google Scholar] [CrossRef]
- Takahashi, N. Three-dimensional mapping of N-linked oligosaccharides using anion-exchange, hydrophobic and hydrophilic interaction modes of high-performance liquid chromatography. J. Chromatogr. A 1996, 720, 217–225. [Google Scholar] [CrossRef]
- Tolstikov, V.V.; Fiehn, O. Analysis of highly polar compounds of plant origin: combination of hydrophilic interaction chromatography and electrospray ion trap mass spectrometry. Anal. Biochem. 2002, 301, 298–307. [Google Scholar] [CrossRef]
- Alpert, A.J. Electrostatic repulsion hydrophilic interaction chromatography for isocratic separation of charged solutes and selective isolation of phosphopeptides. Anal. Chem. 2008, 80, 62–76. [Google Scholar] [CrossRef]
- Mizzen, C.A.; Alpert, A.J.; Levesque, L.; Kruck, T.P.; McLachlan, D.R. Resolution of allelic and non-allelic variants of histone H1 by cation-exchange-hydrophilic-interaction chromatography. J. Chromatogr. B Biomed. Sci. Appl. 2000, 744, 33–46. [Google Scholar] [CrossRef]
- Alpert, A.J.; Shukla, M.; Shukla, A.K.; Zieske, L.R.; Yuen, S.W.; Ferguson, M.A.; Mehlert, A.; Pauly, M.; Orlando, R. Hydrophilic-interaction chromatography of complex carbohydrates. J. Chromatogr. A 1994, 676, 191–122. [Google Scholar] [CrossRef]
- Boutin, J.A.; Ernould, A.P.; Ferry, G.; Genton, A.; Alpert, A.J. Use of hydrophilic interaction chromatography for the study of tyrosine protein kinase specificity. J. Chromatogr. 1992, 583, 137–143. [Google Scholar]
- Alpert, A.J. Hydrophilic-interaction chromatography for the separation of peptides, nucleic acids and other polar compounds. J. Chromatogr. 1990, 499, 177–196. [Google Scholar] [CrossRef]
- Fiehn, O. Metabolite profiling in Arabidopsis. Arabidopsis Protoc. Methods Mol. Biol. 2006, 323, 439–447. [Google Scholar]
- Kind, T.; Tolstikov, V.V.; Fiehn, O.; Weiss, R.H. A comprehensive urinary metabolomic approach for identifying kidney cancer. Anal. Biochem. 2007, 363, 185–195. [Google Scholar] [CrossRef]
- Dietmair, S.; Timmins, N.E.; Gray, P.P.; Nielsen, L.K.; Krömer, J.O. Towards quantitative metabolomics of mammalian cells: Development of a metabolite extraction protocol. Anal. Biochem. 2010, 404, 155–164. [Google Scholar] [CrossRef]
- Zou, W.; Tolstikov, V.V. Probing genetic algorithms for feature selection in comprehensive metabolic profiling approach. Rapid Commun. Mass Spectrom. 2008, 22, 1312–1324. [Google Scholar] [CrossRef]
- Zou, W.; Tolstikov, V.V. Pattern recognition and pathway analysis with genetic algorithms in mass spectrometry based metabolomics. Algorithms 2009, 2, 638–666. [Google Scholar] [CrossRef]
- Scholz, M.; Fiehn, O. SetupX—A public study design database for metabolomic projects. Pac. Symp. Biocomput. 2007, 12, 169–180. [Google Scholar]
- Fiehn, O.; Wohlgemuth, G.; Scholz, M. Setup and Annotation of Metabolomic Experiments by Integrating Biological and Mass Spectrometric Metadata. Data Integr. Life Sci. 2005, 3615, 224–239. [Google Scholar]
- Fiehn, O.; Wohlgemuth, G.; Scholz, M.; Kind, T.; Lee, D.Y.; Lu, Y.; Moon, S.; Nikolau, B. Quality control for plant metabolomics: Reporting MSI-compliant studies. Plant J. 2008, 53, 691–704. [Google Scholar] [CrossRef]
- Shulaev, V. Metabolomics technology and bioinformatics. Brief. Bioinform. 2006, 7, 128–139. [Google Scholar] [CrossRef]
- Jain, A.K.; Duin, R.P.W.; Mao, J. Statistical pattern recognition: A review. Trans. Pattern Anal. Machine Intell. 2000, 22, 4–37. [Google Scholar] [CrossRef]
- MetaboAnalyst. Available online: http://www.metaboanalyst.ca/MetaboAnalyst/faces/Home.jsp/ (accessed on 9 June 2013).
- Scholz, M.; Gatzek, S.; Sterling, A.; Fiehn, O.; Selbig, J. Metabolite fingerprinting: detecting biological features by independent component analysis. Bioinformatics 2004, 20, 2447–2454. [Google Scholar] [CrossRef]
- Sansone, S.A.; Fan, T.; Goodacre, R.; Griffin, J.L.; Hardy, N.W.; Kaddurah-Daouk, R.; Kristal, B.S.; Lindon, J.; Mendes, P.; Morrison, N.; et al. The metabolomics standards initiative. Nat. Biotechnol. 2007, 25, 846–848. [Google Scholar]
- Johnson, H.E.; Broadhurst, D.; Goodacre, R.; Smith, A.R. Metabolic fingerprinting of salt-stressed tomatoes. Phytochemistry 2003, 62, 919–928. [Google Scholar] [CrossRef]
- Goodacre, R.; York, E.V.; Heald, J.K.; Scott, I.M. Chemometric discrimination of unfractionated plant extracts analyzed by electrospray mass spectrometry. Phytochemistry 2003, 62, 859–863. [Google Scholar] [CrossRef]
- Saeys, Y.; Inza, I.; Larranaga, P. A review of feature selection techniques in bioinformatics. Bioinformatics 2007, 23, 2507–2517. [Google Scholar] [CrossRef]
- Lee, J.W.; Lee, J.B.; Park, M.; Song, S.H. An extensive comparison of recent classification tools applied to microarray data. Comput. Stat. Data Anal. 2005, 48, 869–885. [Google Scholar] [CrossRef]
- Zhang, X.; Lu, X.; Shi, Q.; Xu, X.Q.; Leung, H.C.; Harris, L.; Iglehart, J.; Miron, A.; Liu, J.; Wong, W. Recursive SVM feature selection and sample classification for mass-spectrometry and microarray data. BMC Bioinformatics 2006, 7, 197. [Google Scholar] [CrossRef]
- Goodacre, R. Making sense of the metabolome using evolutionary computation: Seeing the wood with the trees. J. Exp. Bot. 2005, 56, 245–254. [Google Scholar] [CrossRef]
- Trevino, V.; Falciani, F. GALGO: An R package for multivariate variable selection using genetic algorithms. Bioinformatics 2006, 22, 1154–1156. [Google Scholar] [CrossRef]
- Jeffries, N.O. Performance of a genetic algorithm for mass spectrometry proteomics. BMC Bioinf. 2004. [Google Scholar] [CrossRef] [Green Version]
- Shulaev, V. Metabolic fingerprinting of breast cancer development. In Proceedings of Biomarker Discovery Summit-9th Annual Biomarkers, Philadelphia, PA, USA, 29 September–1 October, 2008.
- Zou, W.; Yasuor, H.; Fischer, A.J.; Tolstikov, V.V. Trace metabolic profiling and pathway analysis of clomazone using LC-MS-MS and high-resolution MS. LCGC 2011, 29, 760–769. [Google Scholar]
- Metlin. Available online: http://metlin.scripps.edu/ (accessed on 9 June 2013).
- MassBank. Available online: http://www.massbank.jp/ (accessed on 9 June 2013).
- Human Metabolome Database. Available online: http://www.hmdb.ca/ (accessed on 9 June 2013).
- Lipid Maps. Available online: http://www.lipidmaps.org/ (accessed on 9 June 2013).
- Binbase. Available online: http://fiehnlab.ucdavis.edu/projects/binbase_setupx/ (accessed on 9 June 2013).
- Kyoto Encyclopedia of Genes and Genomes. Available online: http://www.genome.jp/kegg/ (accessed on 9 June 2013).
- Kind, T.; Fiehn, O. Metabolomic database annotations via query of elemental compositions: mass accuracy is insufficient even at less than 1 ppm. BMC Bioinf. 2006, 7, 234. [Google Scholar] [CrossRef]
- Kind, T.; Fiehn, O. Seven Golden Rules for heuristic filtering of molecular formulas obtained by accurate mass spectrometry. BMC Bioinf. 2007, 8, 105. [Google Scholar] [CrossRef]
- Zou, W.; Wang, Y.D.; Gu, M.; Tolstikov, V.V. Optimization of mass accuracy, spectral accuracy, and resolution in metabolite identification using LTQ-FT Ultra hybrid mass spectrometer. In Proceedings of 57th ASMS Conference on Mass Spectrometry and Allied Topics, Philadelphia, PA, USA, June 2009.
- PubChem. Available online: http://pubchem.ncbi.nlm.nih.gov/ (accessed on 9 June 2013).
- Chemical Structure Lookup Service. Available online: http://cactus.nci.nih.gov/cgi-bin/lookup/search/ (accessed on 9 June 2013).
- CHEMnetBASE. Available online: http://dnp.chemnetbase.com/ (accessed on 9 June 2013).
- ChemSpider. Available online: http://chemspider.com/ (accessed on 9 June 2013).
- Serkova, N.J.; Brown, M.S. Quantitative analysis in magnetic resonance spectroscopy: From metabolic profiling to in vivo biomarkers. Bioanalysis 2012, 4, 321–341. [Google Scholar] [CrossRef]
- Serkova, N.J.; Glunde, K. Metabolomics of cancer. Methods Mol Biol. 2009, 520, 273–295. [Google Scholar] [CrossRef]
- Zou, W.; Tolstikov, V.V. Predictive multiple reactions monitoring (pMRM) in metabolomics. In Proceedings of 5th Annual Metabolomics Society International Conference, Edmonton, Alberta, Canada, August 2009.
- Yasuor, H.; Zou, W.; Tolstikov, V.V.; Tjeerdema, R.; Fischer, A. Differential oxidative metabolism and 5-ketoclomazone accumulation are involved in Echinochloa phyllopogon resistance to clomazone. Plant Physiol. 2010, 153, 319–326. [Google Scholar] [CrossRef]
- Tomco, P.; Holstege, D.; Zou, W.; Tjeerdema, R. Microbial degradation of clomazone under simulated california rice field conditions. J. Agric. Food Chem. 2010, 58, 3674–3680. [Google Scholar] [CrossRef]
- Duan, Y.Y.; Ma, X.C.; Zou, W.; Wang, C.; Saramipoor, I.; Ahuja, T.; Tolstikov, V.V.; Zern, M.A. Differentiation and characterization of metabolically functioning hepatocytes from human embryonic stem cells. Stem Cells 2010, 28, 674–686. [Google Scholar] [CrossRef]
- Ma, S.; Zhu, M. Recent advances in applications of liquid chromatography-tandem mass spectrometry to the analysis of reactive drug metabolites. Chem. Biol. Interact. 2009, 179, 25–37. [Google Scholar] [CrossRef]
- Ma, S.; Subramanian, R. Detecting and characterizing reactive metabolites by liquid chromatography/tandem mass spectrometry. J. Mass Spectrom. 2006, 41, 1121–1139. [Google Scholar] [CrossRef]
- Yao, M.; Ma, L.; Duchoslav, E.; Zhu, M. Rapid screening and characterization of drug metabolites using multiple ion monitoring dependent product ion scan and postacquisition data mining on a hybrid triple quadrupole-linear ion trap mass spectrometer. Rapid Commun. Mass Spectrom. 2009, 23, 1683–1693. [Google Scholar] [CrossRef]
- Li, A.C.; Gohdes, M.A.; Shou, W.Z. “N-in-one” strategy for metabolite identification using a liquid chromatography/hybrid triple quadrupole linear ion trap instrument using multiple dependent product ion scans triggered with full mass scan. Rapid Commun. Mass Spectrom. 2007, 21, 1421–1430. [Google Scholar] [CrossRef]
- Gao, H.; Materne, O.L.; Howe, D.L.; Brummel, C.L. Method for rapid metabolite profiling of drug candidates in fresh hepatocytes using liquid chromatography coupled with a hybrid quadrupole linear ion trap. Rapid Commun. Mass Spectrom. 2007, 21, 3683–3693. [Google Scholar] [CrossRef]
- Holcapek, M.; Kolarova, L.; Nobilis, M. High-performance liquid chromatography-tandem mass spectrometry in the identification and determination of phase I and phase II drug metabolites. Anal. Bioanal. Chem. 2008, 391, 59–78. [Google Scholar] [CrossRef]
- Fiehn, O. Combining genomics, metabolome analysis, and biochemical modelling to understand metabolic networks. Comp.Funct.Genomics 2001, 2, 155–168. [Google Scholar] [CrossRef]
- MetPA. Available online: http://metpa.metabolomics.ca/MetPA/ (accessed on 9 June 2013).
- Ingenuity Pathway Analysis. Available online: http://www.ingenuity.com/ (accessed on 9 June 2013).
- Cline, M.S.; Smoot, M.; Cerami, E.; Kuchinsky, A.; Landys, N.; Workman, C.; Christmas, R.; Avila-Campilo, I.; Creech, B; Gross, B.; et al. Integration of biological networks and gene expression data using cytoscape. Nat. Protocols. 2007, 2, 2366–2382. [Google Scholar] [CrossRef]
- Urayama, S.; Zou, W.; Brooks, K.; Tolstikov, V.V. Comprehensive mass spectrometry based metabolic profiling of blood plasma reveals potent discriminatory classifiers of pancreatic cancer. Rapid Commun. Mass Spectrom. 2010, 24, 613–620. [Google Scholar] [CrossRef]
- Hruban, R.H.; Klein, A.P.; Eshleman, J.R.; Axilbund, J.E.; Goggins, M. Familial pancreatic cancer: from genes to improved patient care. Exp. Rev. Gastroenterol. Hepatol. 2007, 1, 81–88. [Google Scholar] [CrossRef]
- Abbruzzese, J.L. The challenge of pancreatic cancer. Int. J. Gastrointest Cancer 2003, 33, 1–2. [Google Scholar] [CrossRef]
- Li, D.; Xie, K.; Wolff, R.; Abbruzzese, J.L. Pancreatic Cancer. Lancet 2004, 363, 1049–1057.
- Bardeesy, N.; DePinho, R.A. Pancreatic cancer biology and genetics. Nat. Rev. Cancer 2002, 2, 897–909. [Google Scholar] [CrossRef]
- Griffin, J.F.; Smalley, S.R.; Jewell, W.; Paradelo, J.C.; Reymond, R.D.; Hassanein, R.E.; Evans, R.G. Patterns of failure after curative resection of pancreatic carcinoma. Cancer 1990, 66, 56–61. [Google Scholar] [CrossRef]
- MSConvert. Available online: http://proteowizard.sourceforge.net/tools/msconvert.html/ (accessed on 9 June 2013).
- Statistica. Available online: http://www.statsoft.com/ (accessed on 9 June 2013).
- Tolstikov, V.V. Metabolic biomarkers discovery project. In Proceedings of ACS National Meeting & Exposition, Anaheim, CA, USA; 2011. [Google Scholar]
- Tolstikov, V.V. Metabolic biomarkers discovery project (MBDP). In Proceedings of Pancreatic Cancer Diagnostic Test Development, Molecular Diagnostics World Congress, South San Francisco, CA, USA; 2011. [Google Scholar]
- Tolstikov, V.V. Mass spectrometry-derived metabolic biomarkers and signatures in diagnostic development. In Proceedings of Biomarker Discovery Summit, Philadelphia, PA, USA, 29 September–1 October 2008.
- Kemsley, E.K.; Le Gall, G.; Dainty, J.R.; Watson, A.D.; Harvey, L.J.; Tapp, H.S.; Colquhoun, I.J. Multivariate techniques and their application in nutrition: A metabolomics case study. Br. J. Nutr. 2007, 98, 1–14. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zou, W.; She, J.; Tolstikov, V.V. A Comprehensive Workflow of Mass Spectrometry-Based Untargeted Metabolomics in Cancer Metabolic Biomarker Discovery Using Human Plasma and Urine. Metabolites 2013, 3, 787-819. https://doi.org/10.3390/metabo3030787
Zou W, She J, Tolstikov VV. A Comprehensive Workflow of Mass Spectrometry-Based Untargeted Metabolomics in Cancer Metabolic Biomarker Discovery Using Human Plasma and Urine. Metabolites. 2013; 3(3):787-819. https://doi.org/10.3390/metabo3030787
Chicago/Turabian StyleZou, Wei, Jianwen She, and Vladimir V. Tolstikov. 2013. "A Comprehensive Workflow of Mass Spectrometry-Based Untargeted Metabolomics in Cancer Metabolic Biomarker Discovery Using Human Plasma and Urine" Metabolites 3, no. 3: 787-819. https://doi.org/10.3390/metabo3030787