Generation and Evaluation of a Genome-Scale Metabolic Network Model of Synechococcus elongatus PCC7942


 and
and
Abstract
:1. Introduction
2. Results and Discussion

{kind=link}
{kind=link}
{kind=link}
| Chromosome | Plasmid pANL | Plasmid pANS | |
|---|---|---|---|
| Length of DNA (base pairs) | 2,695,903 | 46,366 | 7835 |
| G+C (%) | ~55.47 | 52.9 | ~59 |
| RNA genes | 54 | - | - |
| rRNA genes | 6 | - | - |
| tRNA genes | 45 | - | - |
| Other RNA genes | 3 | - | - |
| Protein genes | 2856 | 50 | 8 |
| With predicted function | 1682 | 17 | - |
| Without predicted function | 1174 | 33 | - |
| Total genes | 2906 | 50 | 8 |

| General Overview | iSyf715 |
|---|---|
| Genes | 715 |
| Metabolic reactions | 851 |
| Metabolites | 838 |
| Enzymes | 530 |
| Multimeric enzymes and enzymatic complexes | 79 |
| Reactions overview | |
| Reversible reactions | 326 |
| Irreversible reactions | 525 |
| Reactions with assigned genes | 735 |
| Enzymatic conversion | 710 |
| Protein-mediated transport (active and passive-mediated transports) | 25 |
| Reactions with no cognate genes | 116 |
| Non-enzymatic conversion (spontaneous) | 13 |
| Passive transport reactions (simple diffusion) | 16 |
| EC reactions not annotated | 76 |
| Unassigned reactions | 11 |
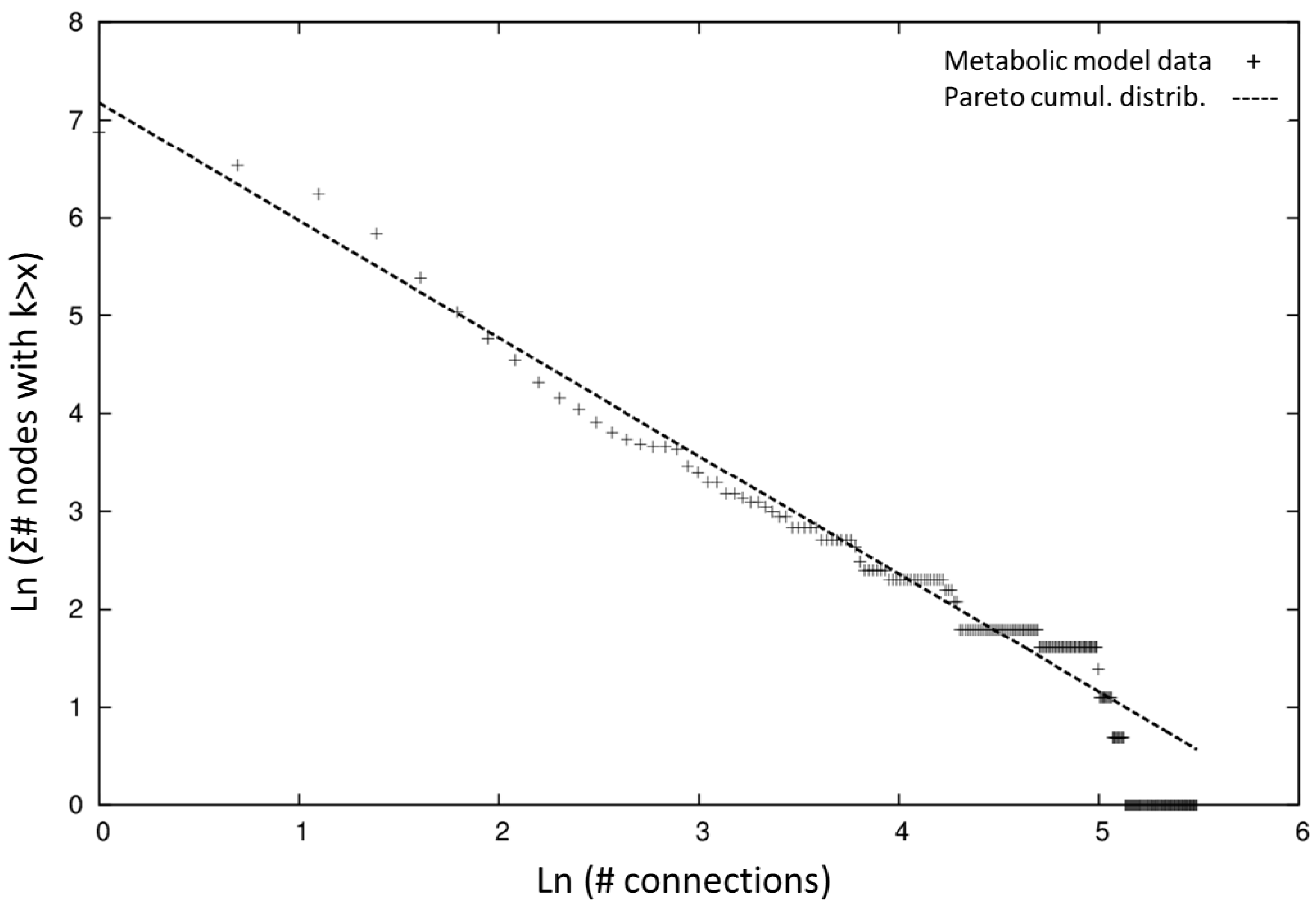
2.1. Network Topology and Connectivity Analysis
| Metabolic Hubs | Neighbors in iSyf715 | Neighbors in iSyn811 [12] | Neighbors in E. coli [45] | Neighbors in Yeast [19] |
|---|---|---|---|---|
| H2O | 243 | 219 | 697 | - |
| Phosphate | 169 | 112 | 81 | 113 |
| ADP | 159 | 111 | 253 | 131 |
| ATP | 148 | 136 | 338 | 166 |
| H+ | 149 | 153 | 923 | 188 |
| Diphosphate | 110 | 84 | 28 | - |
| CO2 | 69 | 72 | 53 | 66 |
| AMP | 74 | 21 | 86 | 48 |
| NADPH | 74 | 68 | 66 | 57 |
| NADP+ | 72 | 68 | 39 | 61 |
| L-glutamate | 52 | 44 | 52 | 56 |
| NAD+ | 46 | 52 | 79 | 58 |
| NADH | 45 | 48 | 75 | 52 |
| oxygen O2 | 45 | 40 | 40 | 31 |
| S-adenosyl-L-methionine | 37 | 28 | 18 | 19 |
| Ammonia | 44 | 28 | 22 | - |
| coenzyme A | 29 | 23 | 71 | 39 |
| Pyruvate | 32 | 20 | 61 | 20 |
| L-glutamine | 30 | 21 | 18 | 23 |
| Glutathione | 32 | 26 | 17 | 10 |
| S-adenosyl-L-homocysteine | 25 | 24 | 12 | 14 |

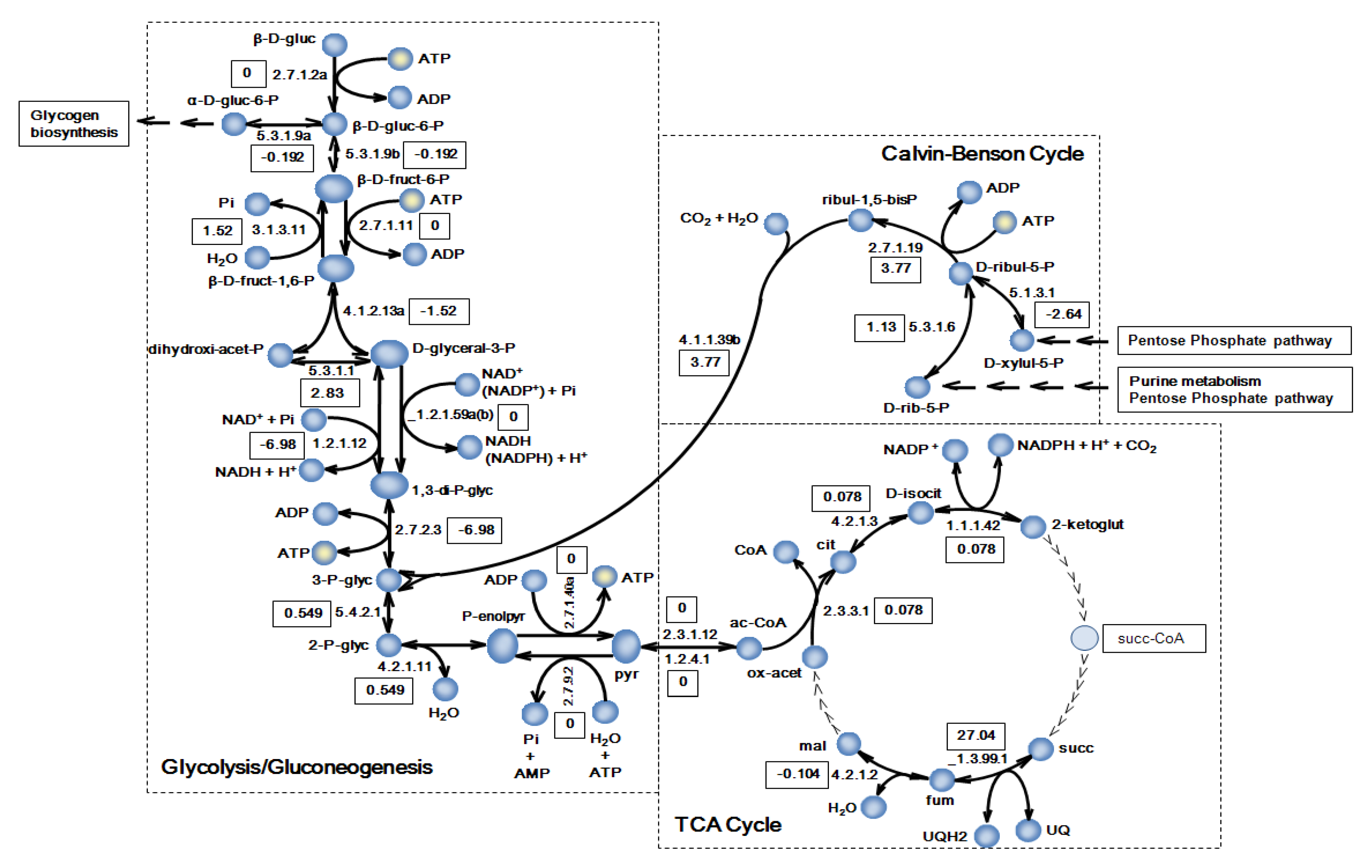
2.2. Simulation of the Model
| Metabolites | mmol/gDW | Metabolites | mmol/gDW |
|---|---|---|---|
| Proteins | 0.000459 | Ribonucleotides | |
| Carbohydrates | AMP | 0.140389293 | |
| Glycogen | 0.53439 | UMP | 0.140389293 |
| Antenna chromophores | GMP | 0.123745851 | |
| Zeaxanthin | 0.00079 | CMP | 0.123745851 |
| Beta-carotene | 0.000875 | Lipids | |
| Trans-lycopene | 0.00820225 | 14C-lipid | 0.028 |
| Chlorophyll a | 0.0057 | 16C-lipid | 0.0042 |
| Phycocyanobiline | 0.0285 | 18C-lipid | 0.00448 |
| Deoxyribonucleotides | (9Z)16C-lipid | 0.0066 | |
| dATP | 0.0201156 | (9Z)18C-lipid | 0.00625 |
| dTTP | 0.0201156 | ||
| dGTP | 0.02538445 | ||
| dCTP | 0.02538445 |

3. Methods
3.1. Genome-Scale Metabolic Network Reconstruction
3.2. Linear Programming for Flux Balance Analysis
3.3. Biomass Composition
4. Conclusions
Acknowledgments
Supplementary Information
- Supplementary File S1: Table S1, Table S2 and detailed description of biomass reaction.
- Supplementary File S2: iSyf715 model with constraints ready to be simulated in OptGene/BioOpt.
- Supplementary File S3: iSyf715 model with gene/reaction association in Excel.
- Supplementary File S4: iSyf715 model in SBML.
- Supplementary File S5: iSyf715 metabolic flux values.
Supplementary Files
Supplementary File 1Author Contributions
Conflicts of Interest
References
- Shestakov, S.V.; Khyen, N.T. Evidence for genetic transformation in blue-green alga Anacystis nidulans R2. Mol. Gen. Genet. 1970, 107, 372–375. [Google Scholar] [CrossRef] [PubMed]
- Andersson, C.R.; Tsinoremas, N.F.; Shelton, J.; Lebedeva, N.V.; Yarrow, J.; Min, H.; Golden, S.S. Application of bioluminescence to the study of circadian rhythms in cyanobacteria. Method. Enzymol. 2000, 305, 527–542. [Google Scholar]
- Waterbury, J.B.; Watson, S.W.; Valois, F.W.; Franks, D.G. Biological and ecological characterization of the marine unicellular cyanobacterium Synechococcus. Can. Bull. Fish Aquat. Sci. 1986, 214, 71–120. [Google Scholar]
- Rippka, R.; Deruelles, J.; Waterbury, J.B.; Herdman, M.; Stanier, R.Y. Generic assignments, strain histories and properties of pure cultures of cyanobacteria. J. Gen. Microbiol. 1979, 111, 1–61. [Google Scholar] [CrossRef]
- Scanlan, D.J.; Nyree, J.W. Molecular ecology of the marine cyanobacterial genera Prochlorococcus and Synechococcus. FEMS Microbiol. Ecol. 2000, 40, 1–12. [Google Scholar]
- Ducat, D.C.; Way, J.C.; Silver, P.A. Engineering cyanobacteria to generate high-value products. Trends Biotechnol. 2011, 29, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Montagud, A.; Gamermann, D.; Fernández de Córdoba, P.; Urchueguía, J.F. Synechocystis sp. PCC6803 metabolic models for the enhanced production of biofuels. Crit. Rev. Biotechnol. 2013, 8551, 1–15. [Google Scholar]
- Snoep, J.L.; Bruggeman, F.; Olivier, B.G.; Westerhoff, H.V. Towards building the silicon cell: A modular approach. BioSystems 2006, 83, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Papin, J.A.; Price, N.D.; Wiback, S.J.; Fell, D.A.; Palsson, B.Ø. Metabolic pathways in the post-genome era. Trends Biochem. Sci. 2003, 28, 250–258. [Google Scholar]
- Szallasi, Z.; Periwal, V.; Stelling, J. Modeling in Cellular System Biology: From Concepts to Nuts and Bolts; The MIT Press: Cambridge, MA, USA, 2006. [Google Scholar]
- Montagud, A.; Navarro, E.; Fernández de Córdoba, P.; Urchueguía, J.F.; Patil, K.R. Reconstruction and analysis of genome-scale metabolic model of a photosynthetic bacterium. BMC Syst. Biol. 2010, 4, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Montagud, A.; Zelezniak, A.; Navarro, E.; de Córdoba, P.F.; Urchueguía, J.F.; Patil, K.R. Flux coupling and transcriptional regulation within the metabolic network of the photosynthetic bacterium Synechocystis sp. PCC6803. Biotechnol. J. 2011, 6, 330–342. [Google Scholar] [CrossRef]
- Park, J.M.; Kim, T.Y.; Lee, S.Y. Genome-scale reconstruction and in silico analysis of the Ralstonia eutropha H16 for polyhydroxyalkanoate synthesis, lithoautotrophic growth, and 2-methyl citric acid production. BMC Syst. Biol. 2011, 5, 101–112. [Google Scholar] [PubMed]
- Milne, C.B.; Eddy, J.A.; Raju, R.; Ardekani, S.; Kim, P.-J.; Senger, R.S.; Jin, Y.-S.; Blaschek, H.P.; Price, N.D. Metabolic network reconstruction and genome-scale model of butanol-producing strain Clostridium beijerinckii NCIMB 8052. BMC Syst. Biol. 2011, 5, 130–145. [Google Scholar] [CrossRef] [PubMed]
- Van den Hondel, C.A.; Verbeek, S.; Van der Ende, A.; Weisbeek, P.J.; Borrias, W.E.; van Arkel, G.A. Introduction of transposon Tn901 into a plasmid of Anacystis nidulans: Preparation for cloning in cyanobacteria. Proc. Natl. Acad. Sci. 1980, 77, 1570–1574. [Google Scholar]
- Van der Plas, J. Identification of replication and stability functions in the complete nucleotide sequence of plasmid pUH24 from the cyanobacterium Synechococcus sp. PCC7942. Mol. Microbiol. 1992, 6, 653–664. [Google Scholar]
- Chen, Y.C.; Holtman, K.C.; Magnuson, R.D.; Youderian, P.A.; Golden, S.S. The complete sequence and functional analysis of pANL, the large plasmid of the unicellular freshwater cyanobacterium Synechococcus elongatus PCC7942. Plasmid 2008, 59, 176–192. [Google Scholar] [CrossRef] [PubMed]
- Weise, S.; Grosse, I.; Klukas, C.; Koschützki, D.; Scholz, U.; Schreiber, F.; Junker, B.H. Meta-All: A system for managing metabolic pathway information. BMC Bioinform. 2006, 7, 465. [Google Scholar] [CrossRef]
- Förster, J.; Famili, I.; Fu, P.; Palsson, B.Ø.; Nielsen, J. Genome-scale reconstruction of the Saccharomyces cerevisiae metabolic network. Genome Res. 2003, 13, 244–253. [Google Scholar]
- Stephanopoulos, G.; Aristidou, A.A.; Nielsen, J.H. Metabolic engineering: principles and methodologies; Academic Press: San Diego, CA, USA, 1998. [Google Scholar]
- Price, N.D.; Papin, J.A.; Schilling, C.H.; Palsson, B.Ø. Genome-scale microbial in silico models: The constraints-based approach. Trends Biotechnol. 2003, 21, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Durot, M.; Bourguignon, P.Y.; Schachter, V. Genome-scale models of bacterial metabolism: Reconstruction and applications. FEMS Microbiol. Rev. 2009, 33, 164–190. [Google Scholar] [CrossRef] [PubMed]
- Price, N.D.; Reed, J.L.; Palsson, B.Ø. Genome-scale models of microbial cells: Evaluating the consequences of constraints. Nat. Rev. Microbiol. 2004, 2, 886–897. [Google Scholar] [CrossRef] [PubMed]
- Orth, J.D.; Thiele, I.; Palsson, B.Ø. What is flux balance analysis? Nat. Biotechnol. 2010, 28, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Thiele, I.; Palsson, B.Ø. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protoc. 2010, 5, 93–121. [Google Scholar]
- Feist, A.M.; Herrgard, M.J.; Thiele, I.; Reed, J.L.; Palsson, B.Ø. Reconstruction of biochemical networks in microorganisms. Nat. Rev. Microbiol. 2009, 7, 129–143. [Google Scholar]
- Notebaart, R.A.; van Enckevort, F.H.; Francke, C.; Siezen, R.J.; Teusink, B. Accelerating the reconstruction of genome-scale metabolic networks. BMC Bioinform. 2006, 7, 296. [Google Scholar] [CrossRef] [Green Version]
- NCBI. Entrez Genome for Synechococcus elongatus PCC7942. Available online: http://www.ncbi.nlm.nih.gov/genome/430?project_id=58045 (accessed on 24 February 2011).
- Karp, P.D.; Paley, S.; Romero, P. The Pathway Tools software. Bioinformatics 2002, 18 (Suppl 1), S225–S232. [Google Scholar]
- Reyes, R.; Gamermann, D.; Montagud, A.; Fuente, D.; Triana, J.; Urchueguía, J.F.; de Córdoba, P.F. Automation on the generation of genome-scale metabolic models. J. Comput. Biol. 2012, 7, 1295–1306. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Furumichi, M.; Tanabe, M.; Hirakawa, M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucl. Acids Res. 2010, 38, 355–360. [Google Scholar] [CrossRef]
- Caspi, R.; Foerster, H.; Fulcher, C.A.; Karp, P.D. MetaCyc: A multiorganism database of metabolic pathways and enzymes. Nucl. Acids Res. 2006, 34, 511–516. [Google Scholar] [CrossRef]
- Voet, D.; Voet, J.G. Biochemistry; John Wiley & Sons: Hoboken, NJ, USA, 2012. [Google Scholar]
- Nelson, D.L.; Cox, M.M. Lehninger Principles of Biochemistry; Freeman, W.H., Ed.; W. H. Freeman: New York, NY, USA, 2008. [Google Scholar]
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. Biochemistry; W.H. Freeman& Palgrave MacMillan: Basingstoke, Hampshire, UK, 2011. [Google Scholar]
- Mathews, C.K.; van Holde, K.E.; Appling, D.R.; Anthony-Cahill, S.J. Biochemistry; Prentice Hall: New York, NY, USA, 2012. [Google Scholar]
- Chang, A.; Scheer, M.; Grote, A.; Schomburg, I.; Schomburg, D. BRENDA, AMENDA and FRENDA the enzyme information system: new content and tools in 2009. Nucl. Acids Res. 2008, 36, 190–195. [Google Scholar] [CrossRef]
- Schilling, C.H.; Letscher, D.; Palsson, B.Ø. Theory for the systemic definition of metabolic pathways and their use in interpreting metabolic function from a pathway-oriented perspective. J. Theor. Biol. 2000, 203, 229–248. [Google Scholar]
- Pearce, J.; Carr, N.G. The metabolism of acetate by the blue-green algae, Anabaena variabilis and Anacystis nidulans. J. Gen. Microbiol. 1967, 49, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Donizelli, M.; Rodriguez, N.; Dharuri, H.; Endler, L.; Chelliah, V.; Li, L.; He, E.; Henry, A.; Stefan, M.I.; et al. BioModels Database: An enhanced, curated and annotated resource for published quantitative kinetic models. BMC Syst. Biol. 2010, 4, 92. [Google Scholar] [CrossRef] [PubMed]
- Barabási, A.L.; Albert, R. Emergence of scaling in random networks. Science 1999, 286, 509–512. [Google Scholar]
- Albert, R.; Jeong, H.; Barabási, A.L. Error and attack tolerance of complex networks. Nature 2000, 406, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Barabási, A.L.; Bonabeau, E. Scale-free networks. Sci. Am. 2003, 288, 50–59. [Google Scholar]
- Barabási, A.L.; Oltvai, Z.N. Network biology understanding the cell’s functional organization. Nat. Rev. Genet. 2004 2004, 5, 101–113. [Google Scholar]
- Feist, A.M.; Henry, C.S.; Reed, J.L.; Krummenacker, M.; Joyce, A.R.; Karp, P.D.; Broadbelt, L.J.; Hatzimanikatis, V.; Palsson, B.Ø. A genome-scale metabolic reconstruction for Escherichia coli K-12 MG1655 that accounts for 1260 ORFs and thermodynamic information. Mol. Syst. Biol. 2007, 3, 121. [Google Scholar] [PubMed]
- Csete, M.; Doyle, J. Bow ties, metabolism and disease. Trends Biotechnol. 2004, 22, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Hardy, M. “Pareto’s Law”. Math. Intell. 2010, 32, 38–43. [Google Scholar] [CrossRef]
- Newman, M.E.J. “Power laws, Pareto distributions and Zipf's law”. Contemp. Phys. 2005, 46, 323–351. [Google Scholar]
- Adamic, L.A.; Huberman, B.A. “Zipf’s law and the Internet”. Glottometrics 2002, 3, 143–150. [Google Scholar]
- Wagner, A.; Fell, D.A. The small world inside large metabolic networks. Proc. R. Soc. B. 2001, 268, 1803–1810. [Google Scholar] [CrossRef] [PubMed]
- Rosales-Loaiza, N.; Ortega, J.; Mora, R. Influencia de la salinidad sobre crecimiento y composición bioquímica de la cianobacteria Synechococcus sp. Cienc. Mar. 2005, 31, 349–355. [Google Scholar]
- Kajiwara, S.; Yamada, H.; Ohkuni, N.; Ohtaguchi, K. Design of the bioreactor for carbon dioxide fixation by Synechococcus PCC7492. Energy Convers. Manag. 1997, 38, 529–532. [Google Scholar] [CrossRef]
- Shastri, A.A.; Morgan, J.A. Flux balance analysis of photoautotrophic metabolism. Biotechnol. Prog. 2005, 21, 1617–1626. [Google Scholar] [CrossRef] [PubMed]
- Kuan, D. Growth optimization of Synechococcus elongatus PCC7942 in lab flask and 2D photobioreactor. Master Thesis; The University of British Columbia: Vancouver, BC, Canada, 2013. Available online: https://circle.ubc.ca/bitstream/handle/2429/45010/ubc_2013_fall_kuan_david.pdf?sequence=1 (accessed on 14/08/14).
- Lewis, N.E.; Hixson, K.K.; Conrad, T.M.; Lerman, J.A.; Charusanti, P.; Polpitiya, A.D.; Adkins, J.N.; Schramm, G.; Purvine, S.O.; Lopez-Ferrer, D.; et al. Omic data from evolved E. coli are consistent with computed optimal growth from genome-scale models. Mol. Syst. Biol. 2010, 6, 390. [Google Scholar]
- Imam, S.; Yilmaz, S.; Sohmen, U.; Gorzalski, A.S.; Reed, J.L.; Noguera, D.R.; Donohue, T.J. iRsp1095: A genome-scale reconstruction of the Rhodobacter sphaeroides metabolic network. BMC Syst. Biol. 2011, 5, 116. [Google Scholar] [CrossRef] [PubMed]
- Munekaga, Y.; Hashimoto, M.; Miyaka, C.; Tomizawa, K.-I.; Endo, T.; Tasaka, M.; Shikanai, T. Cyclic electron flow around photosystem I is essential for photosynthesis. Nature 2004, 429, 579–582. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Daviet, L.; Schalk, M.; Siewers, V.; Nielsen, J. Establishing a platform cell factory through engineering of yeast acetyl-CoA metabolism. Met. Eng. 2013, 15, 48–54. [Google Scholar] [CrossRef]
- Shi, S.; Chen, Y.; Siewers, V.; Nielsen, J. Improving production of malonyl coenzyme A-derived metabolites by abolishing Snf1-dependent regulation of Acc1. mBio 2014, 5, e01130–e01140. [Google Scholar] [PubMed]
- Krivoruchko, A.; Serrano-Amatriain, C.; Chen, Y.; Siewer, V.; Nielsen, J. Improving biobutanol production in engineered Saccharomyces cerevisiae by manipulation of acetyl-CoA metabolism. J. Ind. Microbiol. Biotechnol. 2013, 40, 1051–1056. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, B.B.; Gruissem, W.; Jones, R.L. Biochemistry and Molecular Biology of Plants; American Society of Plants Physiologists: Poole, UK, 2001. [Google Scholar]
- Robertson, B.R.; Tezuka, N.; Watanabe, M.M. Phylogenetic analyses of Synechococcus strains (cyanobacteria) using sequences of 16S rDNA and part of the phycocyanin operon reveal multiple evolutionary lines and reflect phycobilin content. Int. J. Syst. Evol. Microbiol. 2001, 51, 861–871. [Google Scholar] [CrossRef] [PubMed]
- Patil, K.R.; Rocha, I.; Förster, J.; Nielsen, J. Evolutionary programming as a platform for in silico metabolic engineering. BMC Bioinform. 2005, 6, 308. [Google Scholar] [CrossRef] [Green Version]
- Cvijovic, M.; Olivares-Hernández, R.; Agren, R.; Dahr, N.; Vongsangnak, W.; Nookaew, I.; Patil, K.R.; Nielsen, J. BioMet Toolbox: genome-wide analysis of metabolism. Nucleic Acids Res. 2010, 38 Suppl, W144–W149. [Google Scholar] [PubMed]
- BioOpt software. Available online: http://biomet-toolbox.org/index.php?page=downtools-bioOpt (accessed on 14 August 2014).
- Gamermann, D.; Montagud, A.; Conejero, J.A.; Urchueguía, J.F.; de Córdoba, P.F. New approach for phylogeneic tree recovery based on genome-scale metabolic networks. J. Comput. Biol. 2014, 21, 508–519. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.T.; Stolyar, S.M.; Pinchuk, G.E.; Hill, E.A.; Kucek, L.A.; Brown, R.N.; Lipton, M.S.; Osterman, A.; Fredrickson, J.K.; Konopka, A.E.; et al. Genome-scale modeling of light-driven reductant partitioning and carbon fluxes in diazotrophic unicellular cyanobacterium Cyanothece sp. ATCC 51142. PLoS Comput. Biol. 2012, 8, e1002460. [Google Scholar] [CrossRef]
- Hamilton, J.J.; Reed, J.L. Identification of functional differences in metabolic networks using comparative genomics and constraint-based models. PLoS One 2012, 7, e34670. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Triana, J.; Montagud, A.; Siurana, M.; Fuente, D.; Urchueguía, A.; Gamermann, D.; Torres, J.; Tena, J.; De Córdoba, P.F.; Urchueguía, J.F. Generation and Evaluation of a Genome-Scale Metabolic Network Model of Synechococcus elongatus PCC7942. Metabolites 2014, 4, 680-698. https://doi.org/10.3390/metabo4030680
Triana J, Montagud A, Siurana M, Fuente D, Urchueguía A, Gamermann D, Torres J, Tena J, De Córdoba PF, Urchueguía JF. Generation and Evaluation of a Genome-Scale Metabolic Network Model of Synechococcus elongatus PCC7942. Metabolites. 2014; 4(3):680-698. https://doi.org/10.3390/metabo4030680
Chicago/Turabian StyleTriana, Julián, Arnau Montagud, Maria Siurana, David Fuente, Arantxa Urchueguía, Daniel Gamermann, Javier Torres, Jose Tena, Pedro Fernández De Córdoba, and Javier F. Urchueguía. 2014. "Generation and Evaluation of a Genome-Scale Metabolic Network Model of Synechococcus elongatus PCC7942" Metabolites 4, no. 3: 680-698. https://doi.org/10.3390/metabo4030680