
Using Molecular Networking for Microbial Secondary Metabolite Bioprospecting


Abstract
:1. Introduction
2. Results and Discussion
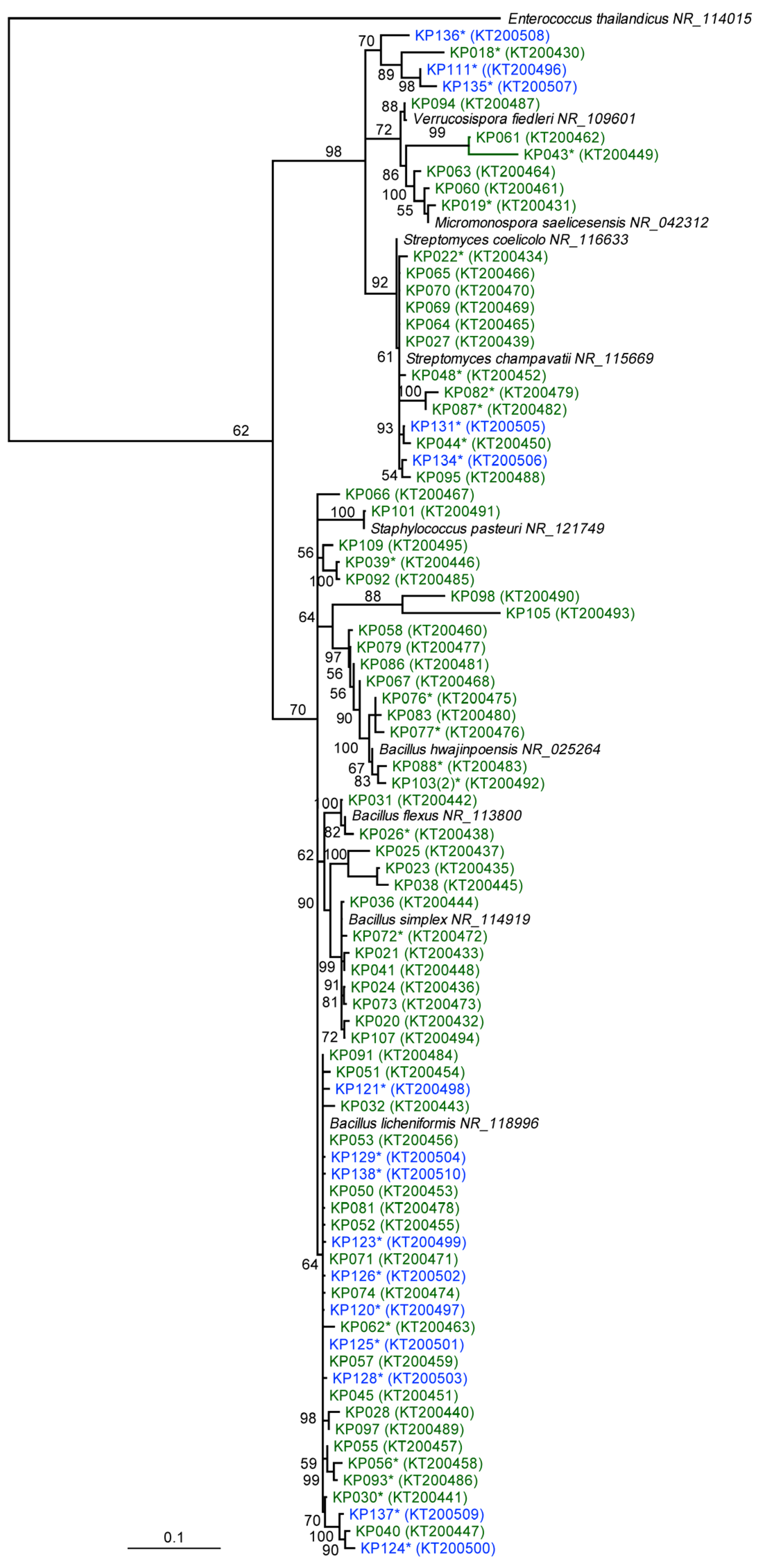
2.1. Phylogenetic and Taxonomic Identification of Antarctic and Scottish Bacterial Isolates

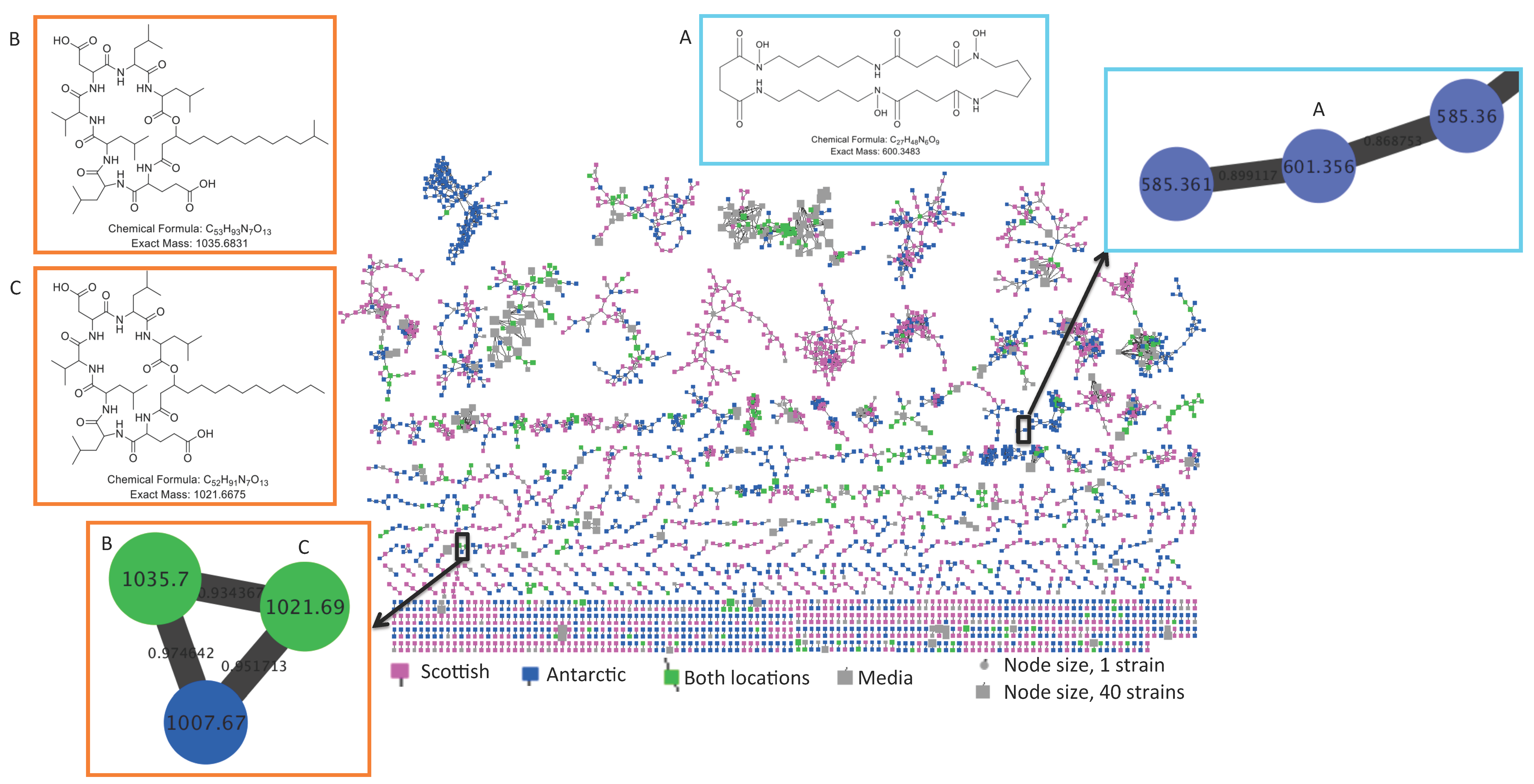
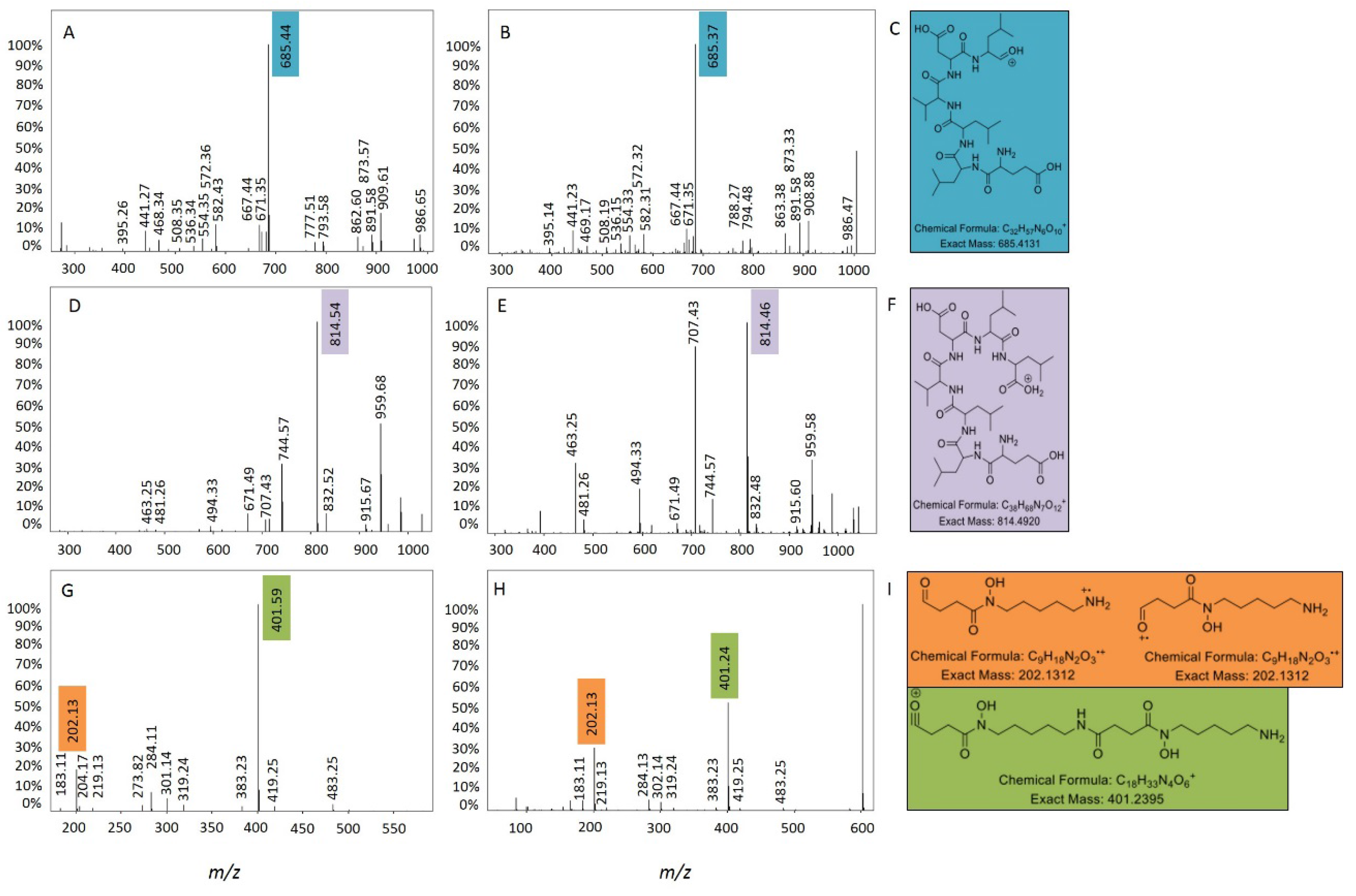
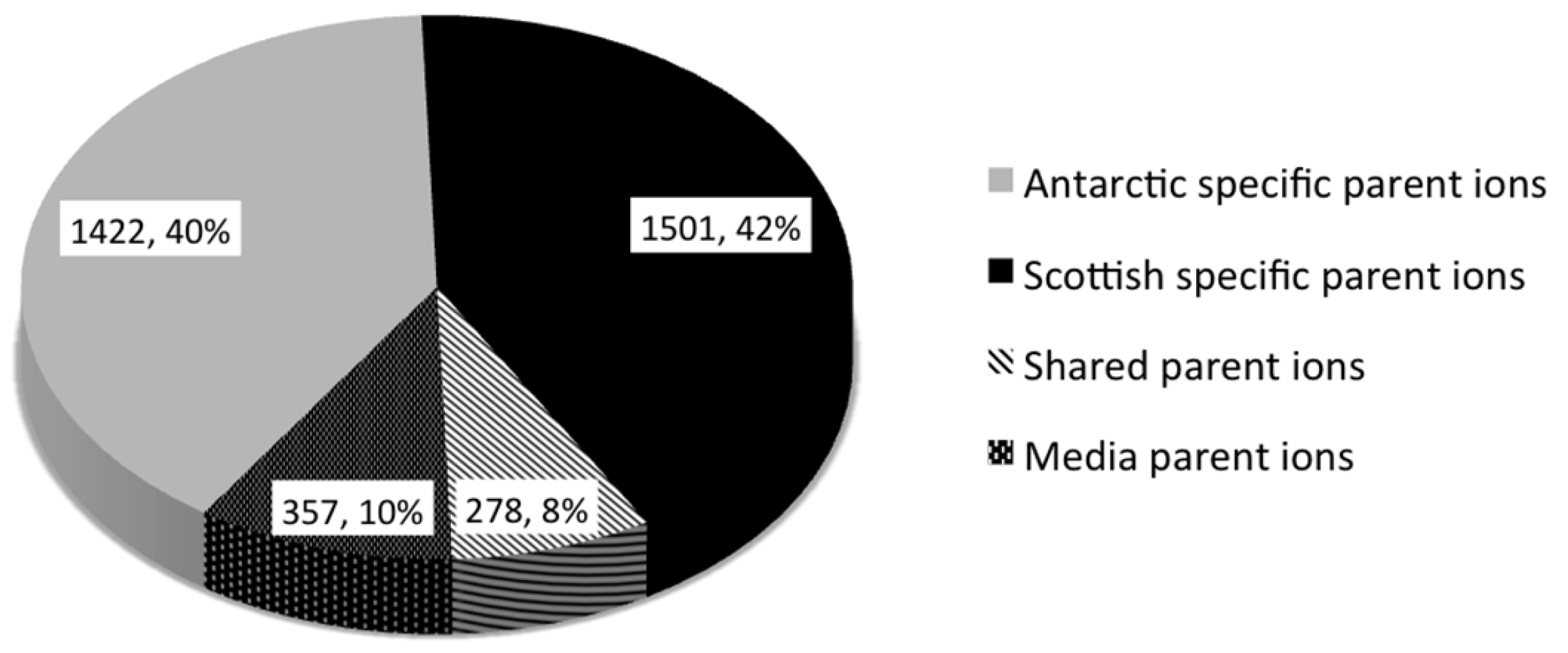
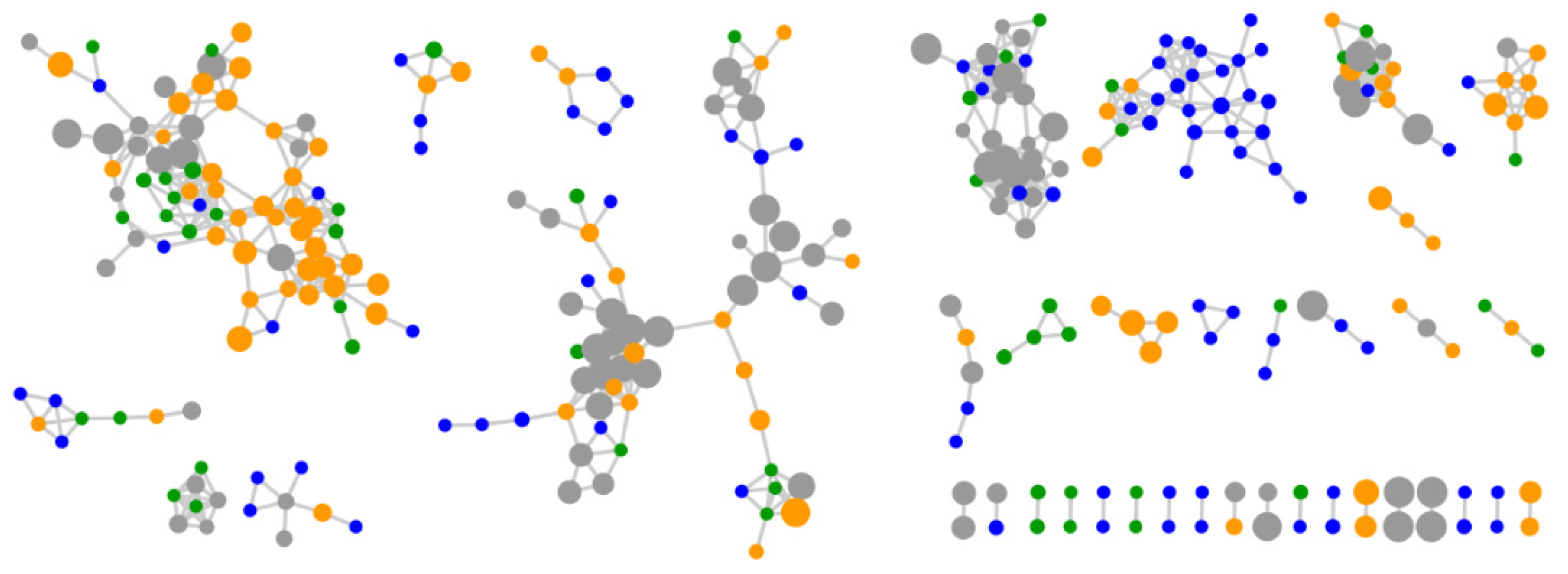
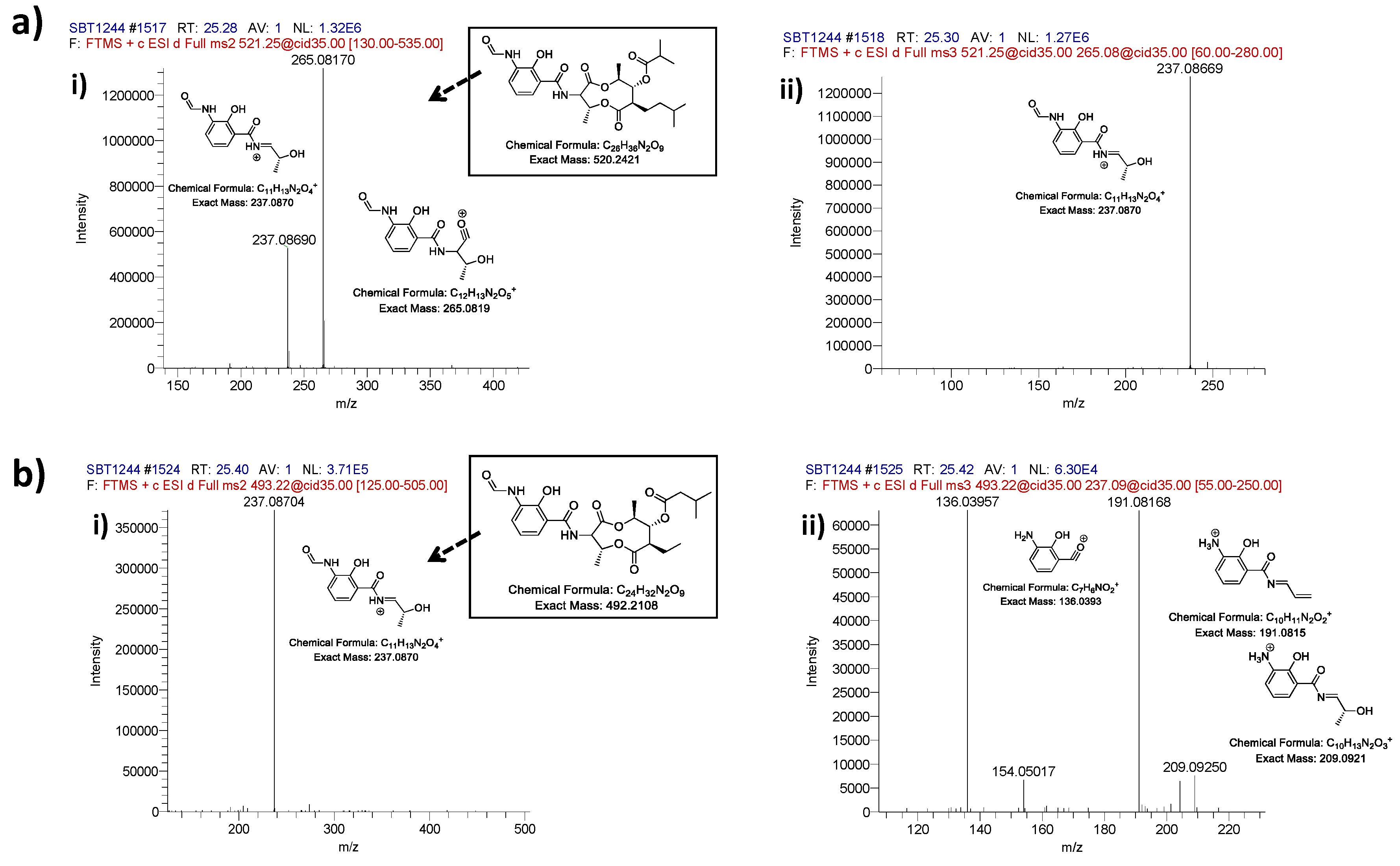
2.2. Comparative Metabolomics





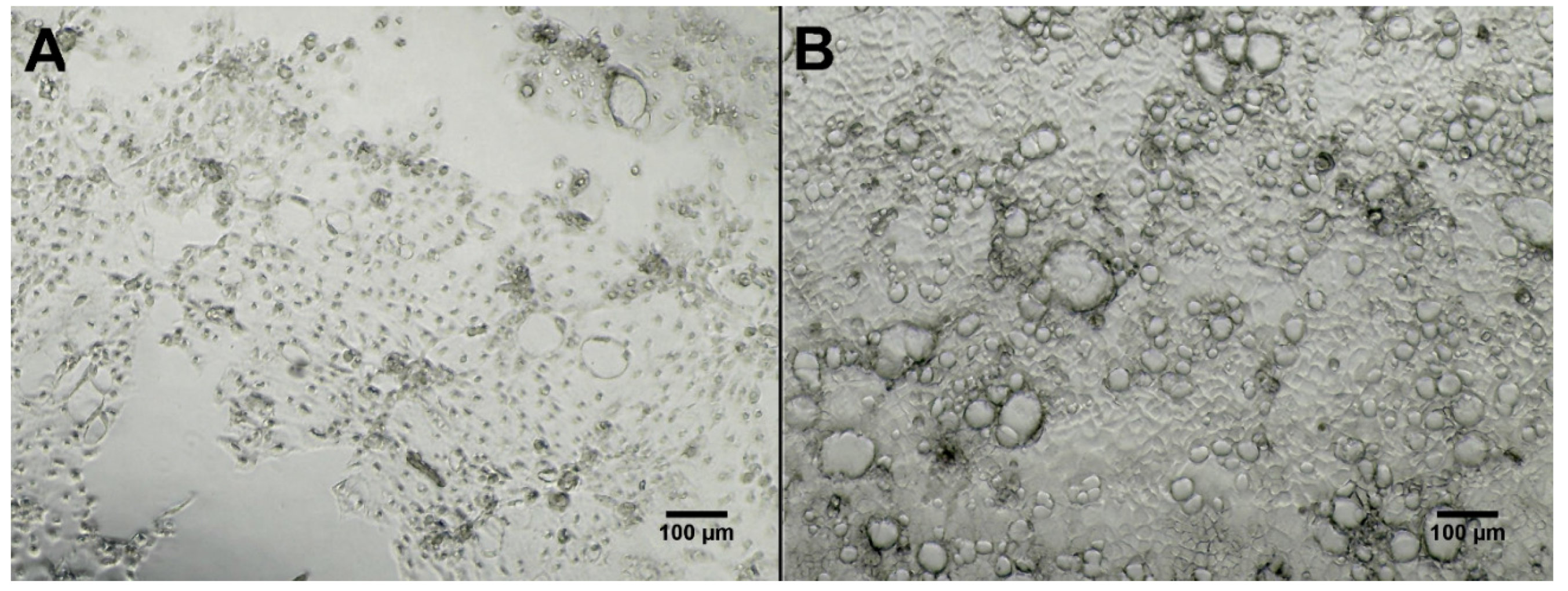
2.3. Bioactivity Testing

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | IC50 (µg/mL) |
|---|---|
| KP022 | 6.2 |
| KP039 | 55.5 |
| KP044 | 0.1 |
| KP077 | 1.6 |
| KP082 | 30.0 |
| KP087 | 6.1 |
| KP131 | 31.4 |

3. Experimental Section
3.1. Sample Collection and Processing
3.2. Cultivable Bacterial Diversity
3.3. Bacterial Fermentation and Metabolite Extraction
3.4. Molecular Networking
3.5. Mass Spectrometry Chemical Dereplication
3.6. DNA Extraction, PCR Amplification, 16S rRNA Gene Sequencing of Bacterial Isolates
3.7. Taxonomic Identification and Phylogeny of Bacterial Isolates
3.8. Bioactivity Testing of Fermentation Extracts Against Three Cell Lines
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- David, A.H. Streptomyces in Nature and Medicine: the Antibiotic Makers; Oxford University Press: Oxford, UK, 2007. [Google Scholar]
- Kayser, O.; Kiderlen, A.F.; Croft, S.L. Natural products as antiparasitic drugs. Parasitol. Res. 2003, 90 (Suppl. 2), S55–S62. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.D.; Konig, G.M.; Angerhofer, C.K.; Greenidges, P.; Linden, A.; Desqueyroux-Faundez, R. Antimalarial Activity: The Search for Marine-Derived Natural Products with Selective Antimalarial Activity. J. Nat. Prod. 1996, 59, 710–716. [Google Scholar] [CrossRef] [PubMed]
- Clark, B.R.; Engene, N.; Teasdale, M.E.; Rowley, D.C.; Matainaho, T.; Valeriote, F.A.; Gerwick, W.H. Natural products chemistry and taxonomy of the marine cyanobacterium Blennothrix cantharidosmum. J. Nat. Prod. 2008, 71, 1530–1537. [Google Scholar] [CrossRef] [PubMed]
- Mann, J. Natural products as immunosuppressive agents. Nat. Prod. Rep. 2001, 18, 417–430. [Google Scholar] [CrossRef] [PubMed]
- Miyanaga, A.; Janso, J.E.; McDonald, L.; He, M.; Liu, H.; Barbieri, L.; Eustáquio, A.S.; Fielding, E.N.; Carter, G.T.; Jensen, P.R.; et al. Discovery and assembly-line biosynthesis of the lymphostin pyrroloquinoline alkaloid family of mTOR inhibitors in Salinispora bacteria. J. Am. Chem. Soc. 2011, 133, 13311–13313. [Google Scholar] [CrossRef] [PubMed]
- Simmons, T.L.; Andrianasolo, E.; McPhail, K.; Flatt, P.; Gerwick, W.H. Marine natural products as anticancer drugs. Mol. Cancer Ther. 2005, 4, 333–342. [Google Scholar] [PubMed]
- Eustáquio, A.S.; McGlinchey, R.P.; Liu, Y.; Hazzard, C.; Beer, L.L.; Florova, G.; Alhamadsheh, M.M.; Lechner, A.; Kale, A.J.; Kobayashi, Y.; et al. Biosynthesis of the salinosporamide A polyketide synthase substrate chloroethylmalonyl-coenzyme A from S-adenosyl-L-methionine. Proc. Natl. Acad. Sci. USA. 2009, 106, 12295–12300. [Google Scholar]
- Wyche, T.P.; Hou, Y.; Braun, D.; Cohen, H.C.; Xiong, M.P.; Bugni, T.S. First natural analogs of the cytotoxic thiodepsipeptide thiocoraline A from a marine Verrucosispora sp. J. Org. Chem. 2011, 76, 6542–6547. [Google Scholar] [CrossRef] [PubMed]
- Kohanski, M.A.; Dwyer, D.J.; Collins, J.J. How antibiotics kill bacteria: From targets to networks. Nat. Rev. Microbiol. 2010, 8, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K. Platforms for antibiotic discovery. Nat. Rev. Drug Discov. 2013, 12, 371–387. [Google Scholar] [CrossRef] [PubMed]
- Chait, R.; Vetsigian, K.; Kishony, R. What counters antibiotic resistance in nature? Nat. Chem. Biol. 2012, 8, 2–5. [Google Scholar] [CrossRef] [PubMed]
- Torella, J.P.; Chait, R.; Kishony, R. Optimal drug synergy in antimicrobial treatments. PLoS Comput. Biol. 2010. [Google Scholar] [CrossRef]
- Sogin, M.L.; Morrison, H.G.; Huber, J.A.; Mark Welch, D.; Huse, S.M.; Neal, P.R.; Arrieta, J.M.; Herndl, G.J. Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proc. Natl. Acad. Sci. USA. 2006, 103, 12115–12120. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; Fierer, N.; Lauber, C.L.; Caporaso, J.G.; Knight, R.; Grogan, P. Soil bacterial diversity in the Arctic is not fundamentally different from that found in other biomes. Environ. Microbiol. 2010, 12, 2998–3006. [Google Scholar] [CrossRef] [PubMed]
- Ganzert, L.; Bajerski, F.; Wagner, D. Bacterial community composition and diversity of five different permafrost-affected soils of Northeast Greenland. FEMS Microbiol. Ecol. 2014, 89, 426–441. [Google Scholar] [CrossRef] [PubMed]
- Steven, B.; Pollard, W.H.; Greer, C.W.; Whyte, L.G. Microbial diversity and activity through a permafrost/ground ice core profile from the Canadian high Arctic. Environ. Microbiol. 2008, 10, 3388–3403. [Google Scholar] [CrossRef] [PubMed]
- Sunagawa, S.; Woodley, C.M.; Medina, M. Threatened corals provide underexplored microbial habitats. PLoS ONE 2010. [Google Scholar] [CrossRef] [PubMed]
- Hentschel, U.; Piel, J.; Degnan, S.M.; Taylor, M.W. Genomic insights into the marine sponge microbiome. Nat. Rev. Microbiol. 2012, 10, 641–654. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Sun, W.; Tang, C.; Jin, L.; Zhang, F.; Li, Z. Phylogenetic diversity of actinobacteria associated with soft coral Alcyonium gracllimum and stony coral Tubastraea coccinea in the East China Sea. Microb. Ecol. 2013, 66, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Hochmuth, T.; Piel, J. Polyketide synthases of bacterial symbionts in sponges—Evolution-based applications in natural products research. Phytochemistry 2009, 70, 1841–1849. [Google Scholar] [CrossRef] [PubMed]
- Montalvo, N.F.; Mohamed, N.M.; Enticknap, J.J.; Hill, R.T. Novel actinobacteria from marine sponges. Antonie Van Leeuwenhoek 2005, 87, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Duncan, K.R.; Haltli, B.; Gill, K.A.; Correa, H.; Berrué, F.; Kerr, R.G. Exploring the diversity and metabolic potential of actinomycetes from temperate marine sediments from Newfoundland, Canada. J. Ind. Microbiol. Biotechnol. 2015, 42, 57–72. [Google Scholar] [CrossRef] [PubMed]
- Bredholt, H.; Fjaervik, E.; Johnsen, G.; Zotchev, S.B. Actinomycetes from sediments in the Trondheim fjord, Norway: Diversity and biological activity. Mar. Drugs 2008, 6, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Riedlinger, J.; Reicke, A.; Zähner, H.; Krismer, B.; Bull, A.T.; Maldonado, L.A.; Ward, A.C.; Goodfellow, M.; Bister, B.; Bischoff, D.; et al. Abyssomicins, inhibitors of the para-aminobenzoic acid pathway produced by the marine Verrucosispora strain AB-18–032. J. Antibiot. 2004, 57, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.G.; Buchanan, G.O.; Feling, R.H.; Kauffman, C.A.; Jensen, P.R.; Fenical, W. New cytotoxic salinosporamides from the marine Actinomycete Salinispora tropica. J. Org. Chem. 2005, 70, 6196–6203. [Google Scholar] [CrossRef] [PubMed]
- Bérdy, J. Bioactive microbial metabolites. J. Antibiot. 2005, 58, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Nithya, C.; Devi, M.G.; Karutha Pandian, S. A novel compound from the marine bacterium Bacillus pumilus S6–15 inhibits biofilm formation in gram-positive and gram-negative species. Biofouling 2011, 27, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.-C.; Lin, C.-F.; Chang, W.-Y.; Kuo, J.; Huang, Y.-T.; Chung, P.-J.; Hwang, T.-L. Bioactive secondary metabolites of a marine Bacillus sp. inhibit superoxide generation and elastase release in human neutrophils by blocking formyl peptide receptor 1. Molecules 2013, 18, 6455–6468. [Google Scholar] [CrossRef] [PubMed]
- Mondol, M.A.M.; Shin, H.J.; Islam, M.T. Diversity of secondary metabolites from marine Bacillus species: Chemistry and biological activity. Mar. Drugs 2013, 11, 2846–2872. [Google Scholar] [CrossRef] [PubMed]
- Blunt, J.W.; Copp, B.R.; Hu, W.-P.; Munro, M.H.G.; Northcote, P.T.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2007, 24, 31–86. [Google Scholar] [CrossRef] [PubMed]
- Pathom-aree, W.; Stach, J.E.M.; Ward, A.C.; Horikoshi, K.; Bull, A.T.; Goodfellow, M. Diversity of actinomycetes isolated from Challenger Deep sediment (10,898 m) from the Mariana Trench. Extremophiles 2006, 10, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Bazire, A.; Diab, F.; Jebbar, M.; Haras, D. Influence of high salinity on biofilm formation and benzoate assimilation by Pseudomonas aeruginosa. J. Ind. Microbiol. Biotechnol. 2007, 34, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Ballav, S.; Kerkar, S.; Thomas, S.; Augustine, N. Halophilic and halotolerant actinomycetes from a marine saltern of Goa, India producing anti-bacterial metabolites. J. Biosci. Bioeng. 2015, 119, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Starič, N.; Danevčič, T.; Stopar, D. Vibrio sp. DSM 14379 pigment production—A competitive advantage in the environment? Microb. Ecol. 2010, 60, 592–598. [Google Scholar]
- McDougald, D.; Rice, S.A.; Barraud, N.; Steinberg, P.D.; Kjelleberg, S. Should we stay or should we go: Mechanisms and ecological consequences for biofilm dispersal. Nat. Rev. Microbiol. 2012, 10, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Penn, K.; Jenkins, C.; Nett, M.; Udwary, D.W.; Gontang, E.A.; McGlinchey, R.P.; Foster, B.; Lapidus, A.; Podell, S.; Allen, E.E.; et al. Genomic islands link secondary metabolism to functional adaptation in marine Actinobacteria. ISME J. 2009, 3, 1193–1203. [Google Scholar] [CrossRef] [PubMed]
- Ziemert, N.; Lechner, A.; Wietz, M.; Millán-Aguiñaga, N.; Chavarria, K.; Jensen, P.R. Diversity and Evolution of Secondary Metabolism in the Marine Actinomycete Genus Salinispora. Proc. Natl. Acad. Sci. USA. 2014, 111, E1130–E1139. [Google Scholar] [CrossRef] [PubMed]
- Bull, A.T. Clean technology: Industry and environment, a viable partnership? Biologist 2000, 47, 61–64. [Google Scholar] [PubMed]
- Andrianasolo, E.H.; Haramaty, L.; Rosario-Passapera, R.; Vetriani, C.; Falkowski, P.; White, E.; Lutz, R. Ammonificins C and D, hydroxyethylamine chromene derivatives from a cultured marine hydrothermal vent bacterium, Thermovibrio ammonificans. Mar. Drugs 2012, 10, 2300–2311. [Google Scholar] [CrossRef] [PubMed]
- Watrous, J.; Roach, P.; Alexandrov, T.; Heath, B.S.; Yang, J.Y.; Kersten, R.D.; van der Voort, M.; Pogliano, K.; Gross, H.; Raaijmakers, J.M.; et al. Mass spectral molecular networking of living microbial colonies. Proc. Natl. Acad. Sci. USA. 2012, 109, E1743–E1752. [Google Scholar] [CrossRef] [PubMed]
- Frank, A.M.; Bandeira, N.; Shen, Z.; Tanner, S.; Briggs, S.P.; Smith, R.D.; Pevzner, P.A. Clustering millions of tandem mass spectra. J. Proteome Res. 2008, 7, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.D.; Wu, C.-H.; Moree, W.J.; Lamsa, A.; Medema, M.H.; Zhao, X.; Gavilan, R.G.; Aparicio, M.; Atencio, L.; Jackson, C.; et al. MS/MS networking guided analysis of molecule and gene cluster families. Proc. Natl. Acad. Sci. USA. 2013, 110, E2611–E2620. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.Y.; Sanchez, L.M.; Rath, C.M.; Liu, X.; Boudreau, P.D.; Bruns, N.; Glukhov, E.; Wodtke, A.; de Felicio, R.; Fenner, A.; et al. Molecular Networking as a Dereplication Strategy. J. Nat. Prod. 2013, 76, 1686–1699. [Google Scholar] [CrossRef] [PubMed]
- Duncan, K.R.; Crüsemann, M.; Lechner, A.; Sarkar, A.; Li, J.; Ziemert, N.; Wang, M.; Bandeira, N.; Moore, B.S.; Dorrestein, P.C.; et al. Molecular Networking and Pattern-Based Genome Mining Improves Discovery of Biosynthetic Gene Clusters and their Products from Salinispora Species. Chem. Biol. 2015, 22, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Kersten, R.D.; Ziemert, N.; Gonzalez, D.J.; Duggan, B.M.; Nizet, V.; Dorrestein, P.C.; Moore, B.S. Glycogenomics as a mass spectrometry-guided genome-mining method for microbial glycosylated molecules. Proc. Natl. Acad. Sci. USA. 2013, 110, E4407–E4416. [Google Scholar] [CrossRef] [PubMed]
- Kersten, R.D.; Yang, Y.-L.; Xu, Y.; Cimermancic, P.; Nam, S.-J.; Fenical, W.; Fischbach, M.A.; Moore, B.S.; Dorrestein, P.C. A mass spectrometry-guided genome mining approach for natural product peptidogenomics. Nat. Chem. Biol. 2011, 7, 794–802. [Google Scholar] [CrossRef] [PubMed]
- Briand, E.; Bormans, M.; Gugger, M.; Dorrestein, P.C.; Gerwick, W.H. Changes in secondary metabolic profiles of Microcystis aeruginosa strains in response to intraspecific interactions. Environ. Microbiol. 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winnikoff, J.R.; Glukhov, E.; Watrous, J.; Dorrestein, P.C.; Gerwick, W.H. Quantitative molecular networking to profile marine cyanobacterial metabolomes. J. Antibiot. 2014, 67, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Dorrestein, P.C. Emerging mass spectrometry techniques for the direct analysis of microbial colonies. Curr. Opin. Microbiol. 2014, 19, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Klitgaard, A.; Nielsen, J.B.; Frandsen, R.J.N.; Andersen, M.R.; Nielsen, K.F. Combining Stable Isotope Labeling and Molecular Networking for Biosynthetic Pathway Characterization. Anal. Chem. 2015, 87, 6520–6526. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, L.A.; Stach, J.E.M.; Pathom-Aree, W.; Ward, A.C.; Bull, A.T.; Goodfellow, M. Diversity of cultivable actinobacteria in geographically widespread marine sediments. Antonie Van Leeuwenhoek 2005, 87, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Duncan, K.; Haltli, B.; Gill, K.A.; Kerr, R.G. Bioprospecting from marine sediments of New Brunswick, Canada: Exploring the relationship between total bacterial diversity and actinobacteria diversity. Mar. Drugs 2014, 12, 899–925. [Google Scholar] [CrossRef] [PubMed]
- Murray, A.E.; Grzymski, J.J. Diversity and genomics of Antarctic marine micro-organisms. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2007, 362, 2259–2271. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, W.L.; Munakata, N.; Horneck, G.; Melosh, H.J.; Setlow, P. Resistance of Bacillus endospores to extreme terrestrial and extraterrestrial environments. Microbiol. Mol. Biol. Rev. 2000, 64, 548–572. [Google Scholar] [CrossRef] [PubMed]
- Sneath, P. Longevity of microorganisms. Nature 1962, 195, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Haselwandter, K.; Ebner, M.R. Microorganisms surviving for 5300 years. FEMS Microbiol. Lett. 1994, 116, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.R.; Gontang, E.; Mafnas, C.; Mincer, T.J.; Fenical, W. Culturable marine actinomycete diversity from tropical Pacific Ocean sediments. Environ. Microbiol. 2005, 7, 1039–1048. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, W.; Jin, Y.; Meifang, J.; Yu, X. A comparative study on the phylogenetic diversity of culturable actinobacteria isolated from five marine sponge species. Antonie Van Leeuwenhoek 2008, 93, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, L.A.; Fragoso-Yáñez, D.; Pérez-García, A.; Rosellón-Druker, J.; Quintana, E.T. Actinobacterial diversity from marine sediments collected in Mexico. Antonie Van Leeuwenhoek 2009, 95, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Mincer, T.J.; Jensen, P.R.; Kauffman, C.A.; Fenical, W. Widespread and Persistent Populations of a Major New Marine Actinomycete Taxon in Ocean Sediments. Appl. Environ. Microbiol. 2002, 68, 5005–5011. [Google Scholar] [CrossRef] [PubMed]
- Weyland, H. Actinomycetes of the bottom sediments of various seas. In Proceedings of the Colloque International de Bacteriologie Marine, Brest, France, 1–5 October 1986; pp. 73–79.
- Sass, A.M.; McKew, B.A; Sass, H.; Fichtel, J.; Timmis, K.N.; McGenity, T.J. Diversity of Bacillus-like organisms isolated from deep-sea hypersaline anoxic sediments. Saline Syst. 2008. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, M.A.C.; Cavalett, A.; Spinner, A.; Rosa, D.C.; Jasper, R.B.; Quecine, M.C.; Bonatelli, M.L.; Pizzirani-Kleiner, A.; Corção, G.; Lima, A.O.D.S. Phylogenetic identification of marine bacteria isolated from deep-sea sediments of the eastern South Atlantic Ocean. Springerplus 2013, 2, 127. [Google Scholar] [CrossRef] [PubMed]
- Palomo, S.; González, I.; de La Cruz, M.; Martín, J.; Tormo, J.R.; Anderson, M.; Hill, R.T.; Vicente, F.; Reyes, F.; Genilloud, O. Sponge-derived Kocuria and Micrococcus spp. as sources of the new thiazolyl peptide antibiotic kocurin. Mar. Drugs 2013, 11, 1071–1086. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.R.; Williams, P.G.; Oh, D.-C.; Zeigler, L.; Fenical, W. Species-specific secondary metabolite production in marine actinomycetes of the genus Salinispora. Appl. Environ. Microbiol. 2007, 73, 1146–1152. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.R.; Mafnas, C. Biogeography of the marine actinomycete Salinispora. Environ. Microbiol. 2006, 8, 1881–1888. [Google Scholar] [CrossRef] [PubMed]
- Charlop-Powers, Z.; Owen, J.G.; Reddy, B.V.B.; Ternei, M.A; Brady, S.F. Chemical-biogeographic survey of secondary metabolism in soil. Proc. Natl. Acad. Sci. USA. 2014, 111, 3757–3762. [Google Scholar] [CrossRef] [PubMed]
- Dunshee, B.R.; Leben, C.; Keitt, G.W.; Strong, F.M. The Isolation and Properties of Antimycin A. J. Am. Chem. Soc. 1949, 71, 2436–2437. [Google Scholar] [CrossRef]
- Viegelmann, C.; Margassery, L.M.; Kennedy, J.; Zhang, T.; O’Brien, C.; O’Gara, F.; Morrissey, J.P.; Dobson, A.D.W.; Edrada-Ebel, R. Metabolomic profiling and genomic study of a marine sponge-associated Streptomyces sp. Mar. Drugs 2014, 12, 3323–3351. [Google Scholar] [CrossRef] [PubMed]
- Liaw, C.-C.; Chen, P.-C.; Shih, C.-J.; Tseng, S.-P.; Lai, Y.-M.; Hsu, C.-H.; Dorrestein, P.C.; Yang, Y.-L. Vitroprocines, new antibiotics against Acinetobacter baumannii, discovered from marine Vibrio sp. QWI-06 using mass-spectrometry-based metabolomics approach. Sci. Rep. 2015. [Google Scholar] [CrossRef] [PubMed]
- Vijayabharathi, R.; Bruheim, P.; Andreassen, T.; Raja, D.S.; Devi, P.B.; Sathyabama, S.; Priyadarisini, V.B. Assessment of resistomycin, as an anticancer compound isolated and characterized from Streptomyces aurantiacus AAA5. J. Microbiol. 2011, 49, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Schorn, M.; Zettler, J.; Noel, J.P.; Dorrestein, P.C.; Moore, B.S.; Kaysser, L. Genetic basis for the biosynthesis of the pharmaceutically important class of epoxyketone proteasome inhibitors. ACS Chem. Biol. 2014, 9, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Ohba, M.; Mizuki, E.; Uemori, A. Parasporin, a new anticancer protein group from Bacillus thuringiensis. Anticancer Res. 2009, 29, 427–433. [Google Scholar] [PubMed]
- Ma, E.L.; Choi, Y.J.; Choi, J.; Pothoulakis, C.; Rhee, S.H.; Im, E. The anticancer effect of probiotic Bacillus polyfermenticus on human colon cancer cells is mediated through ErbB2 and ErbB3 inhibition. Int. J. Cancer 2010, 127, 780–790. [Google Scholar] [PubMed]
- Howe, J.A.; Shimmield, T.M.; Diaz, R. Deep-water sedimentary environments of the northwestern Weddell Sea and South Sandwich Islands, Antarctica. Deep Sea Res. 2004, 51, 1489–1514. [Google Scholar] [CrossRef]
- Sea, N.W.; Howe, J.A.; Wilson, C.R.; Shimmield, T.M.; Diaz, R.J.; Carpenter, L.W. Recent deep-water sedimentation, trace metal and radioisotope geochemistry across the Southern Ocean and Northern Weddell Sea, Antarctica. Deep Sea Res. 2007, 54, 1652–1681. [Google Scholar]
- Shiring, E.B.; Gottlieb, D. Methods for the Characterization of Streptomyces species. Int. J. Syst. Evol. Microbiol. 1966, 16, 313–340. [Google Scholar]
- Macintyre, L.; Zhang, T.; Viegelmann, C.; Martinez, I.J.; Cheng, C.; Dowdells, C.; Abdelmohsen, U.R.; Gernert, C.; Hentschel, U.; Edrada-Ebel, R. Metabolomic tools for secondary metabolite discovery from marine microbial symbionts. Mar. Drugs 2014, 12, 3416–3448. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, E.W.; Mendoza, L.; Shteynberg, D.; Farrah, T.; Lam, H.; Sun, Z.; Nilsson, E.; Pratt, B.; Prazen, B.; Eng, J.K.; et al. A Guided Tour of the Trans-Proteomic Pipeline. Proteomics 2011, 10, 1150–1159. [Google Scholar] [CrossRef] [PubMed]
- GNPS. The Future of Natural Products Research and Mass Spectrometry. Available online: http://gnps.ucsd.edu/ProteoSAFe/static/gnps-splash.jsp (accessed on 7 January 2016).
- Guthals, A.; Watrous, J.D.; Dorrestein, P.C.; Bandeira, N. The spectral networks paradigm in high throughput mass spectrometry. Mol. Biosyst. 2012, 8, 2535–2544. [Google Scholar] [CrossRef] [PubMed]
- Keller, A.; Eng, J.; Zhang, N.; Li, X.; Aebersold, R. A uniform proteomics MS/MS analysis platform utilizing open XML file formats. Mol. Syst. Biol. 2005. [Google Scholar] [CrossRef] [PubMed]
- Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.-L.; Ideker, T. Cytoscape 2.8: New features for data integration and network visualization. Bioinformatics 2011, 27, 431–432. [Google Scholar] [CrossRef] [PubMed]
- Cline, M.S.; Smoot, M.; Cerami, E.; Kuchinsky, A.; Landys, N.; Workman, C.; Christmas, R.; Avila-Campilo, I.; Creech, M.; Gross, B.; et al. Integration of biological networks and gene expression data using Cytoscape. Nat. Protoc. 2007, 2, 2366–2382. [Google Scholar] [CrossRef] [PubMed]
- Chambers, M.C.; Maclean, B.; Burke, R.; Amodei, D.; Ruderman, D.L.; Neumann, S.; Gatto, L.; Fischer, B.; Pratt, B.; Egertson, J.; et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 2012, 30, 918–920. [Google Scholar] [CrossRef] [PubMed]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Oresic, M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinform. 2010. [Google Scholar] [CrossRef] [PubMed]
- Hopwood, D.A.; Bibb, M.J.; Chater, K.F.; Kieser, T.; Bruton, C.J.; Kieser, H.M.; Lydiate, D.J.; Smith, C.P.; Ward, J.M.; Schrempf, H. Genetic Manipulation of Streptomyces: A laboratory manual. Biochem. Educ. 1986. [Google Scholar] [CrossRef]
- Weisburg, W.G.; Barns, S.M.; Pelletie, D.A; Lane, D.J. 16S ribosomal DNA amplification for phylogenetic study. J. Bacteriol. 1991, 173, 697–703. [Google Scholar] [PubMed]
- Sambrook, J.; Russel, D.W. Molecular Cloning: A Laboratory Manual; Cold pring Harbor Laboratory Press: New York, NY, USA, 2001. [Google Scholar]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [PubMed]
- Jukes, T.H.; Cantor, C.R. Evolution of Protein Models; Academic Press: New York, NY, USA, 1969. [Google Scholar]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Purves, K.; Macintyre, L.; Brennan, D.; Hreggviðsson, G.Ó.; Kuttner, E.; Ásgeirsdóttir, M.E.; Young, L.C.; Green, D.H.; Edrada-Ebel, R.; Duncan, K.R. Using Molecular Networking for Microbial Secondary Metabolite Bioprospecting. Metabolites 2016, 6, 2. https://doi.org/10.3390/metabo6010002
Purves K, Macintyre L, Brennan D, Hreggviðsson GÓ, Kuttner E, Ásgeirsdóttir ME, Young LC, Green DH, Edrada-Ebel R, Duncan KR. Using Molecular Networking for Microbial Secondary Metabolite Bioprospecting. Metabolites. 2016; 6(1):2. https://doi.org/10.3390/metabo6010002
Chicago/Turabian StylePurves, Kevin, Lynsey Macintyre, Debra Brennan, Guðmundur Ó. Hreggviðsson, Eva Kuttner, Margrét E. Ásgeirsdóttir, Louise C. Young, David H. Green, Ruangelie Edrada-Ebel, and Katherine R. Duncan. 2016. "Using Molecular Networking for Microbial Secondary Metabolite Bioprospecting" Metabolites 6, no. 1: 2. https://doi.org/10.3390/metabo6010002