
Metabolomic Profiling of the Effects of Melittin on Cisplatin Resistant and Cisplatin Sensitive Ovarian Cancer Cells Using Mass Spectrometry and Biolog Microarray Technology


Abstract
:1. Introduction
2. Results
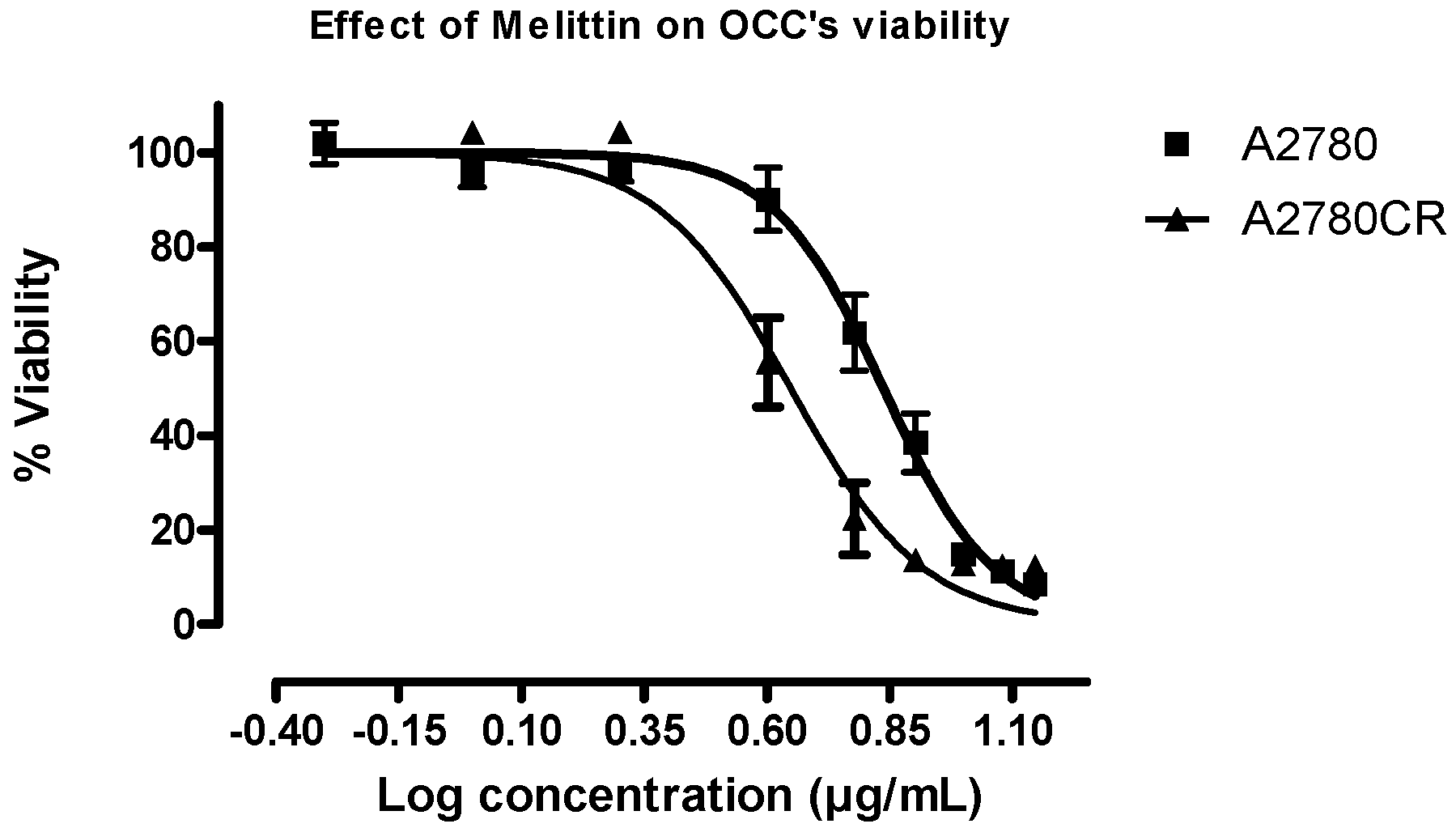
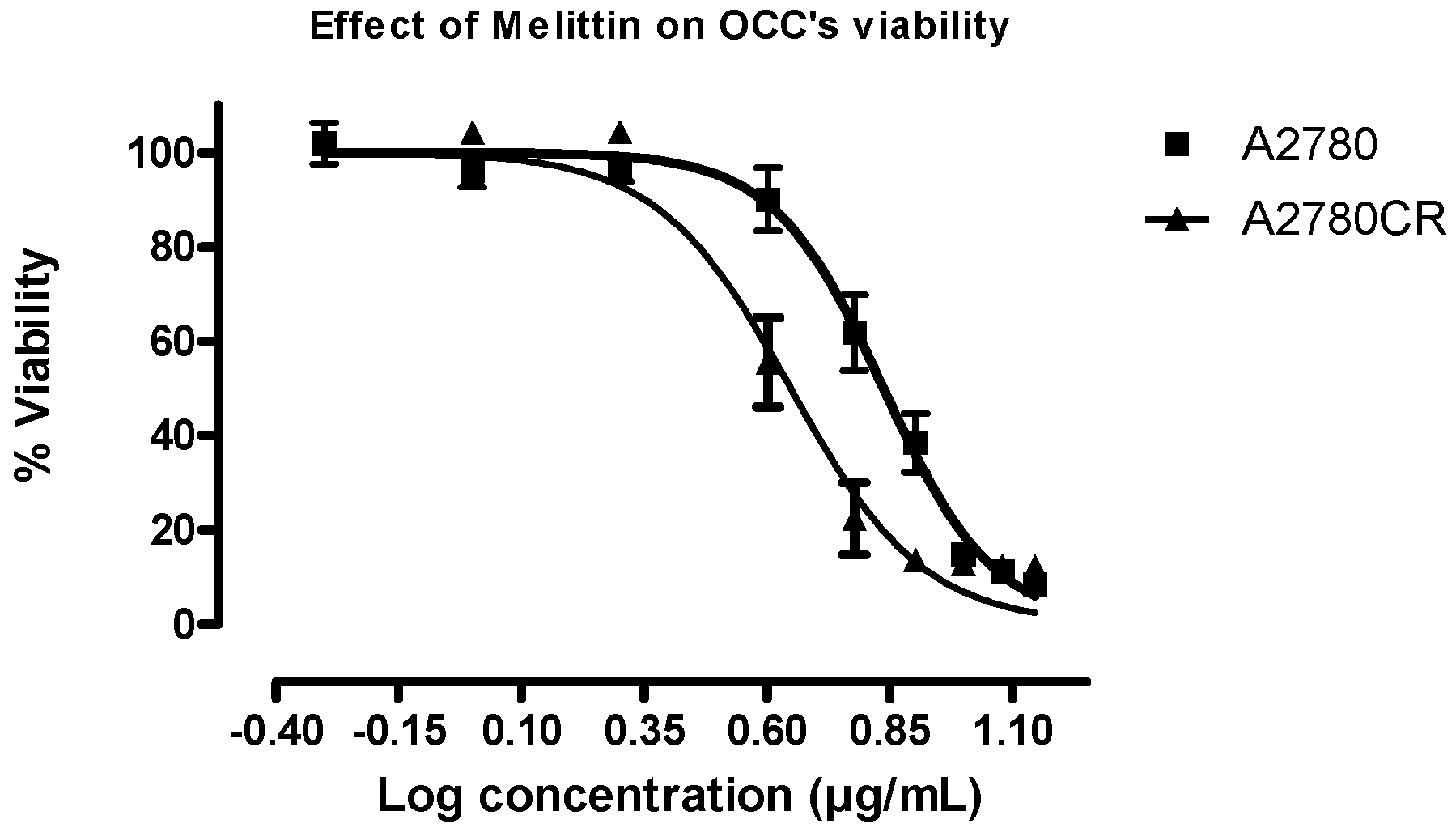
2.1. Melittin Sensitivity of the Ovarian Cancer Cells
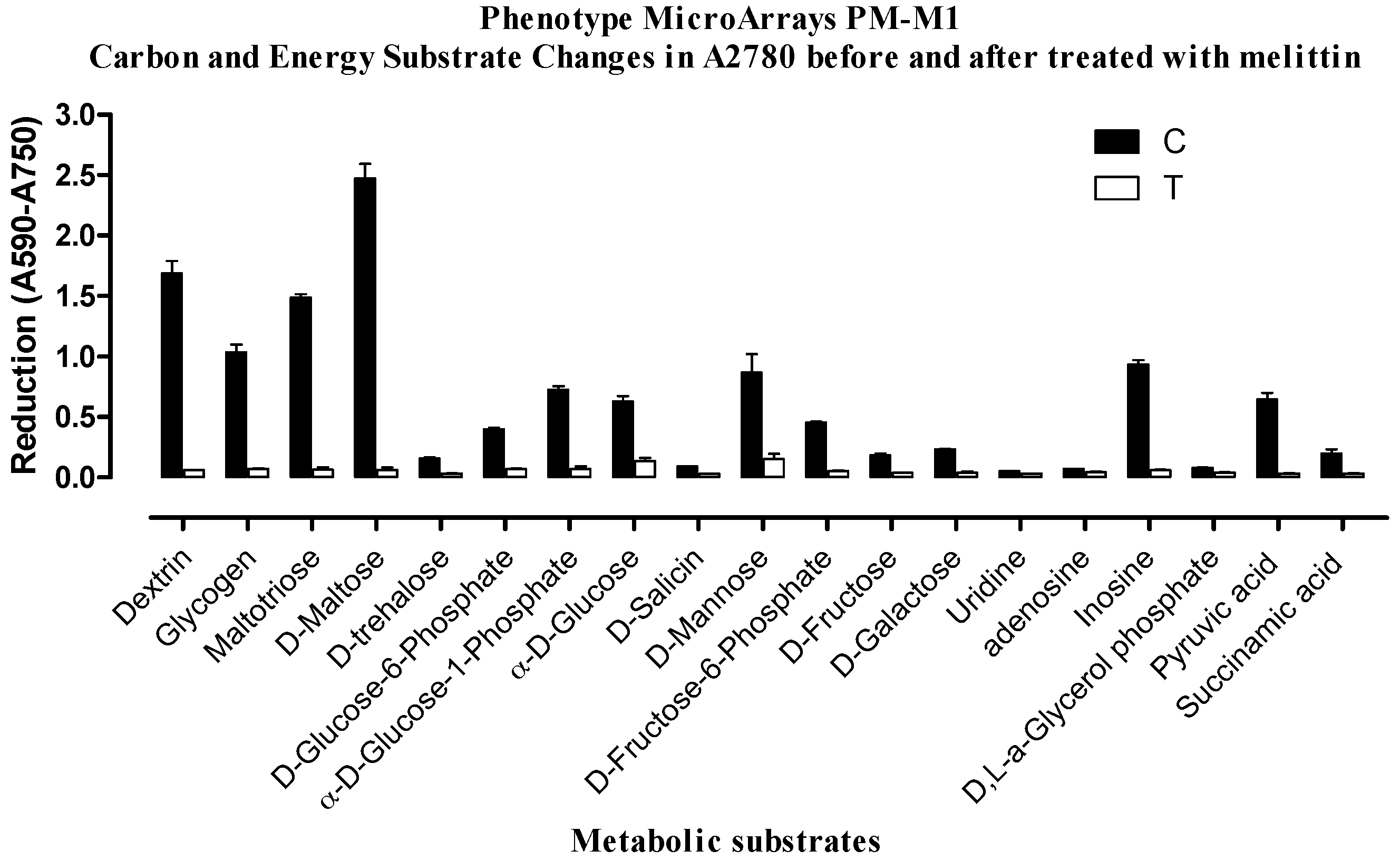
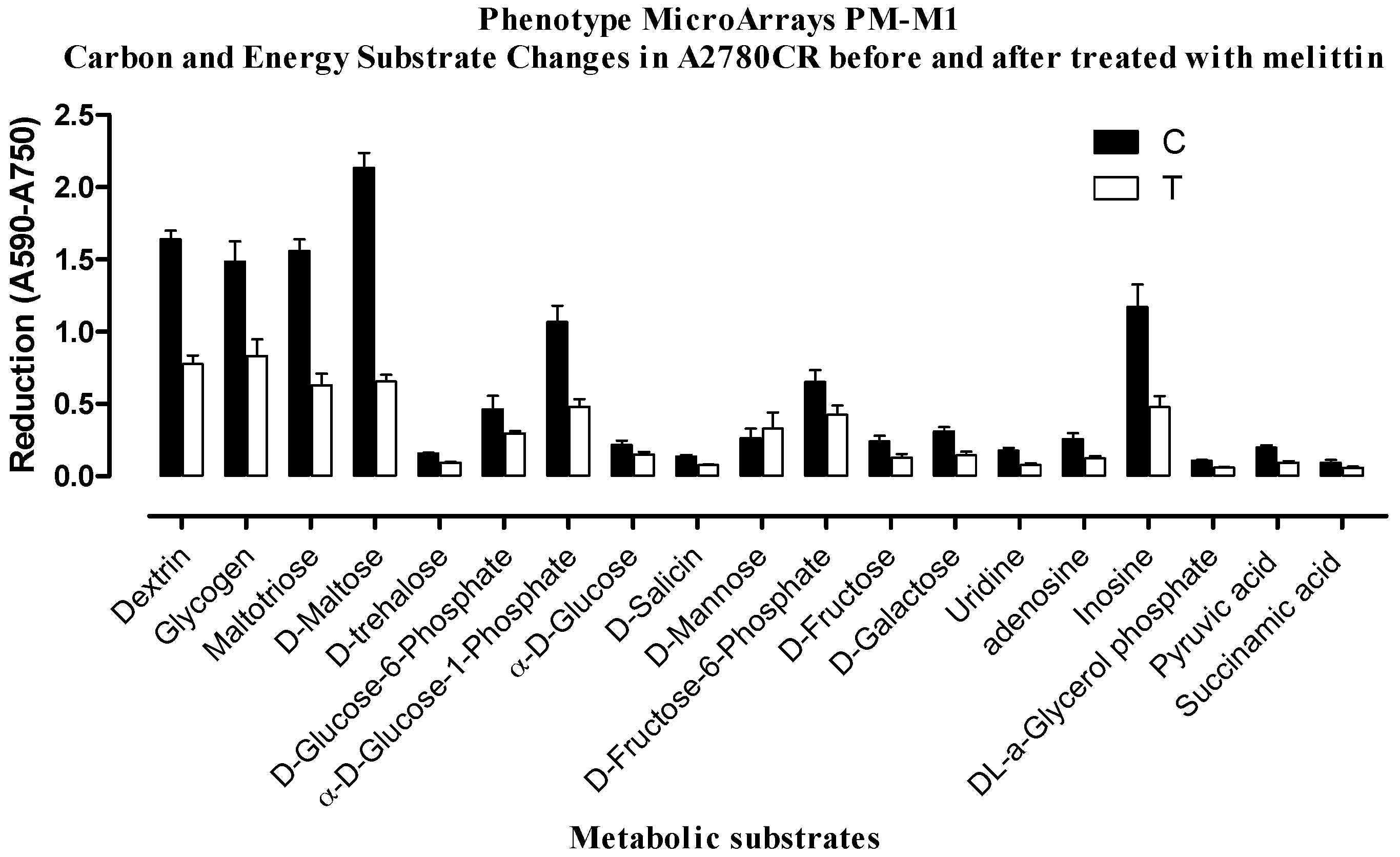
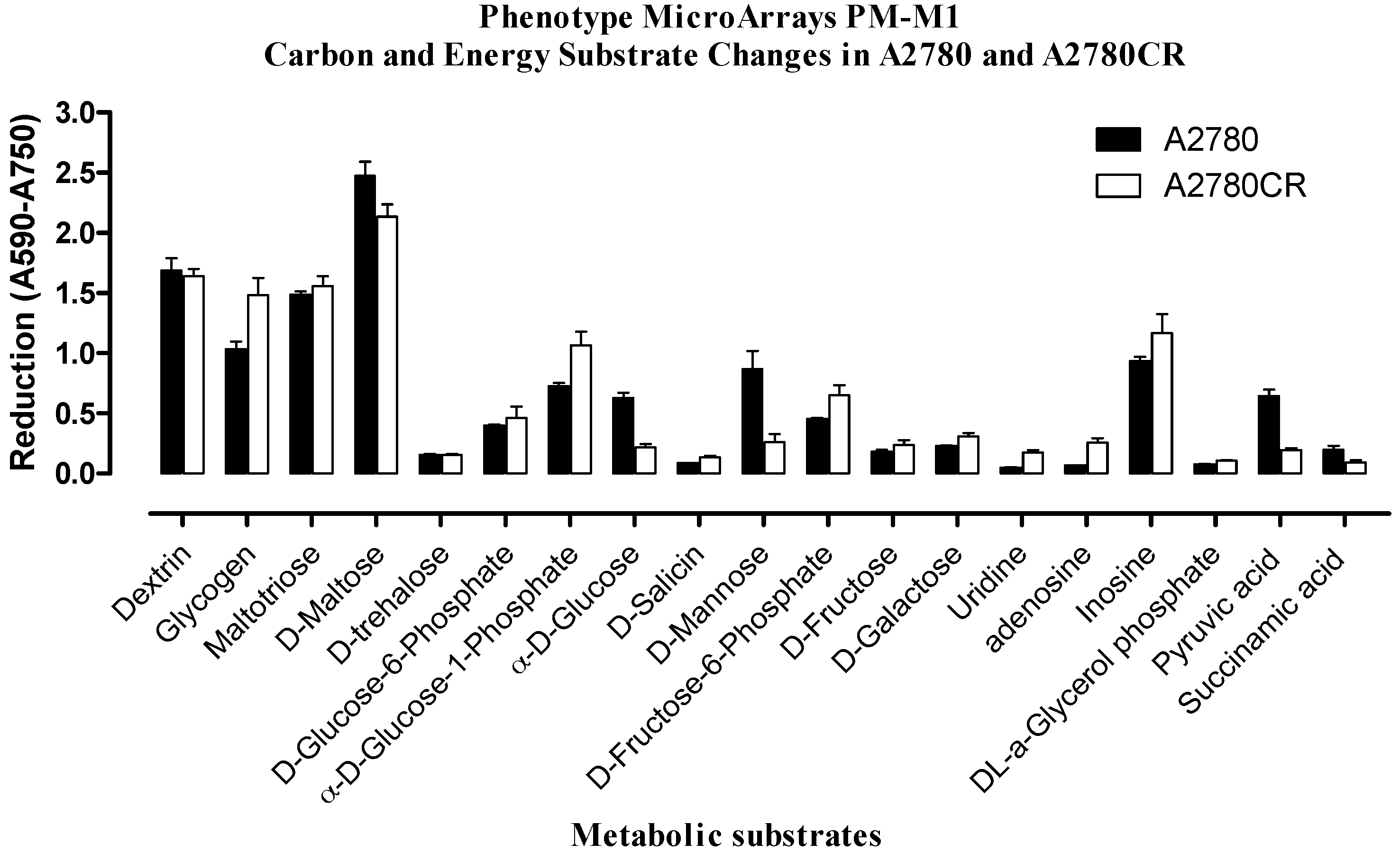
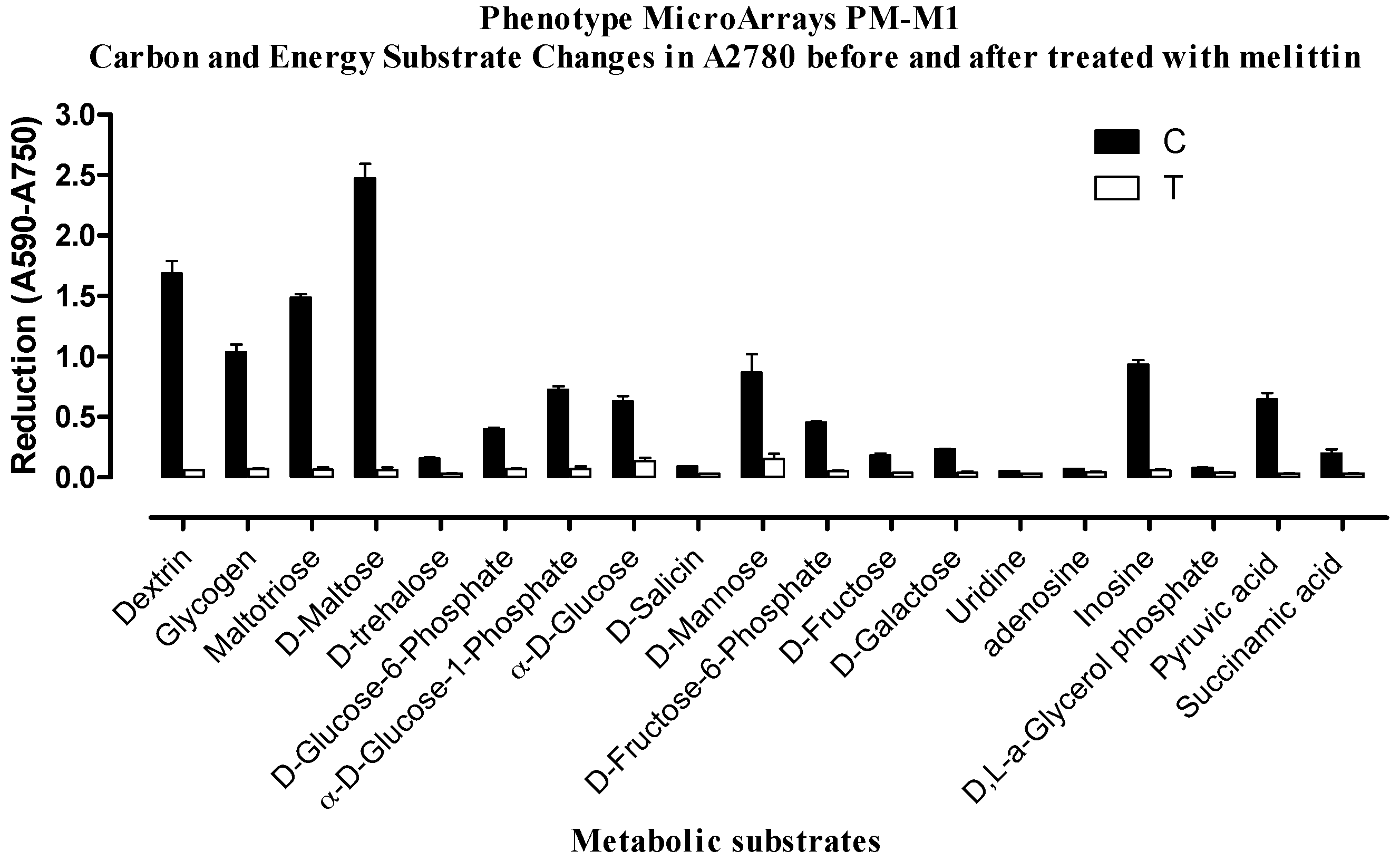
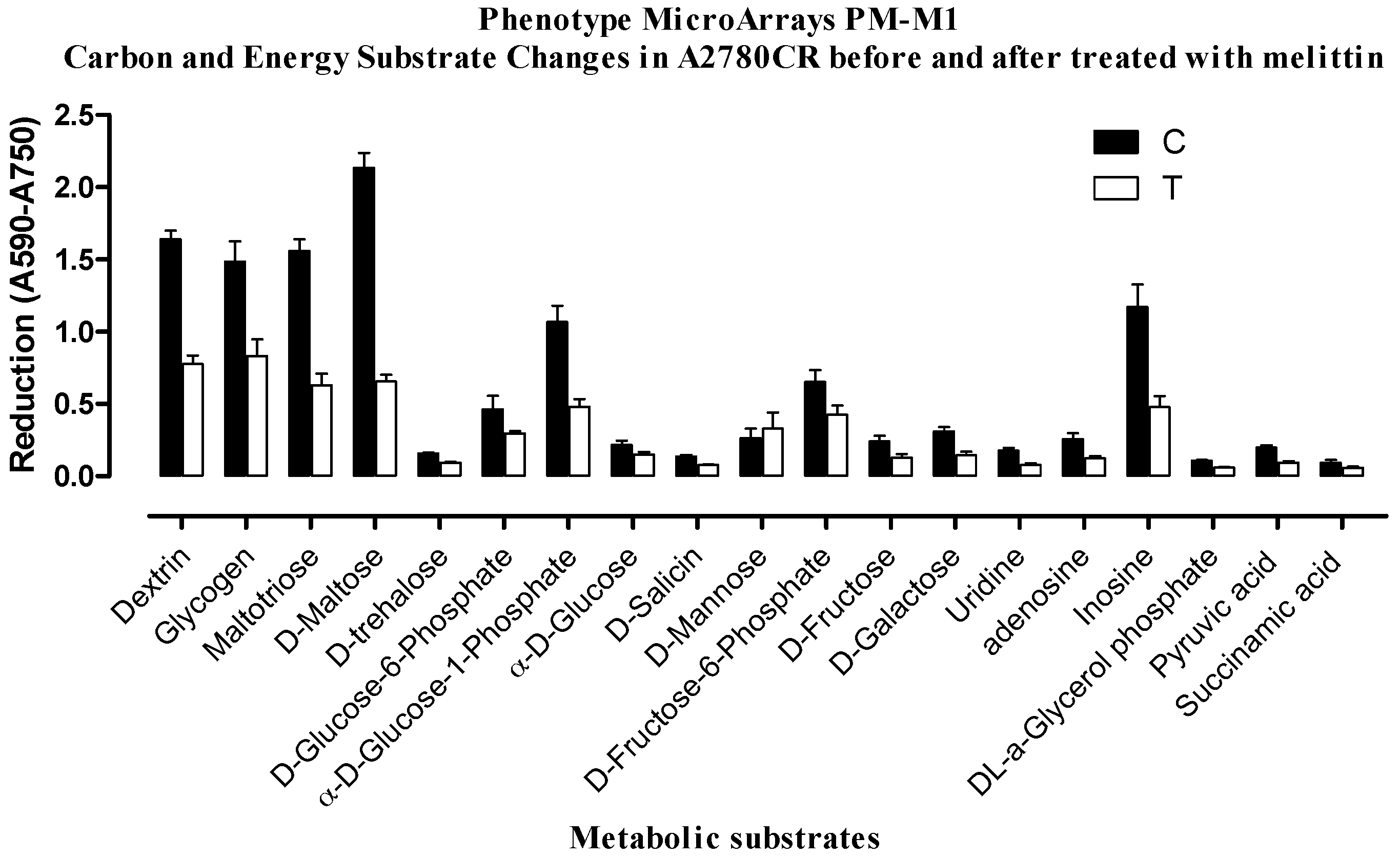
2.2. Phenotypic MicroArray (PM) Assay of Untreated and Melittin Treated A2780 and A2780CR Cells
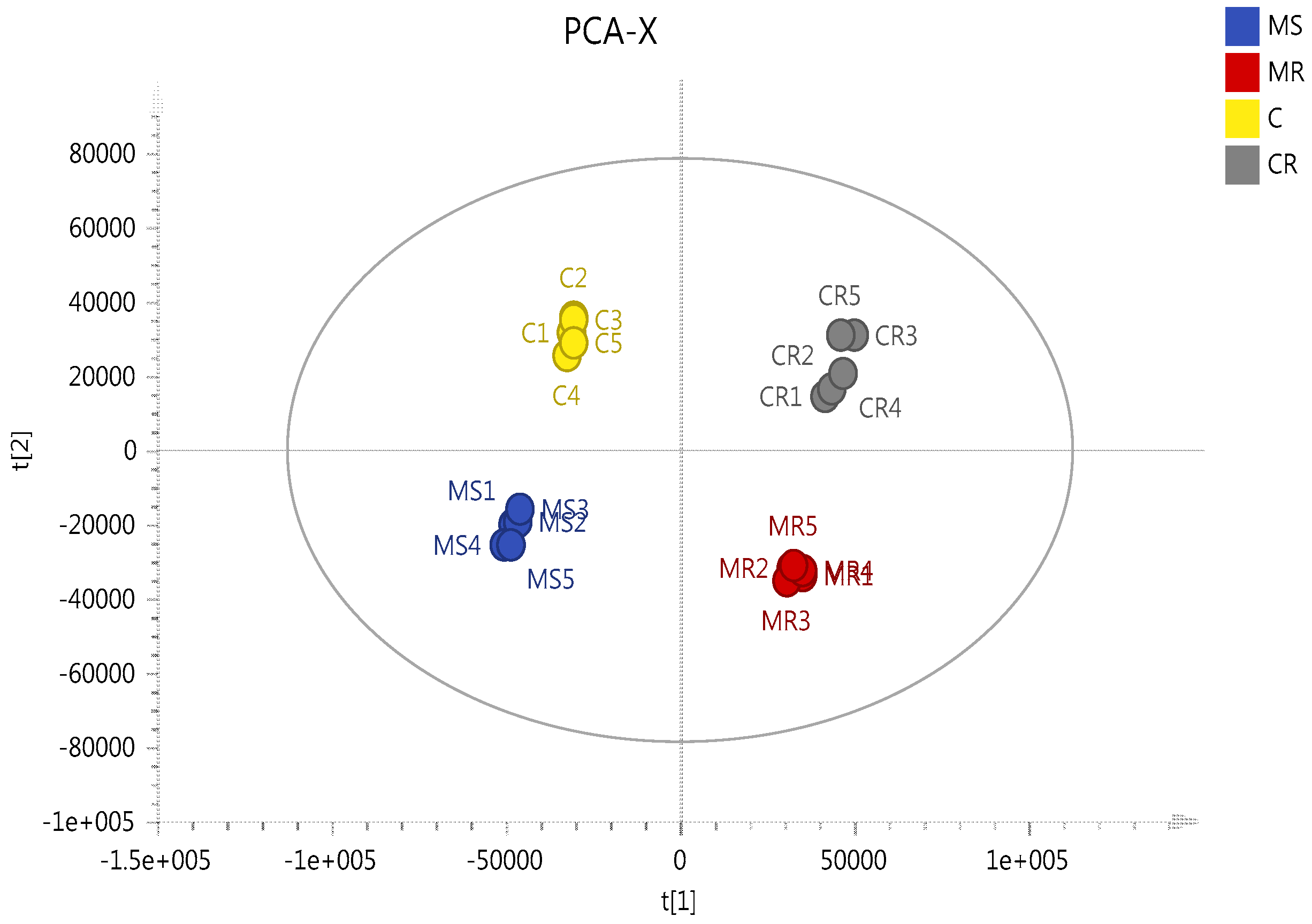
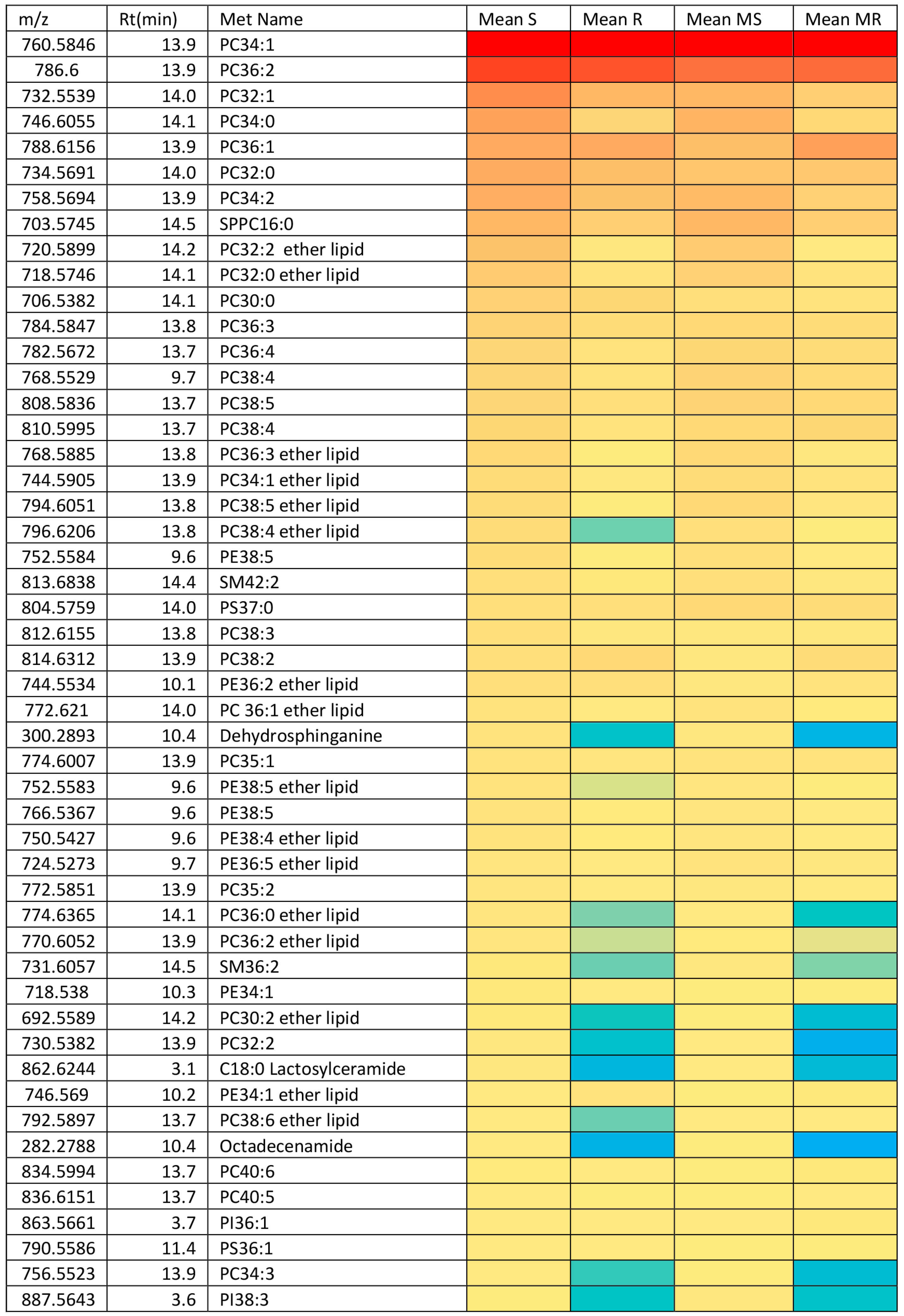
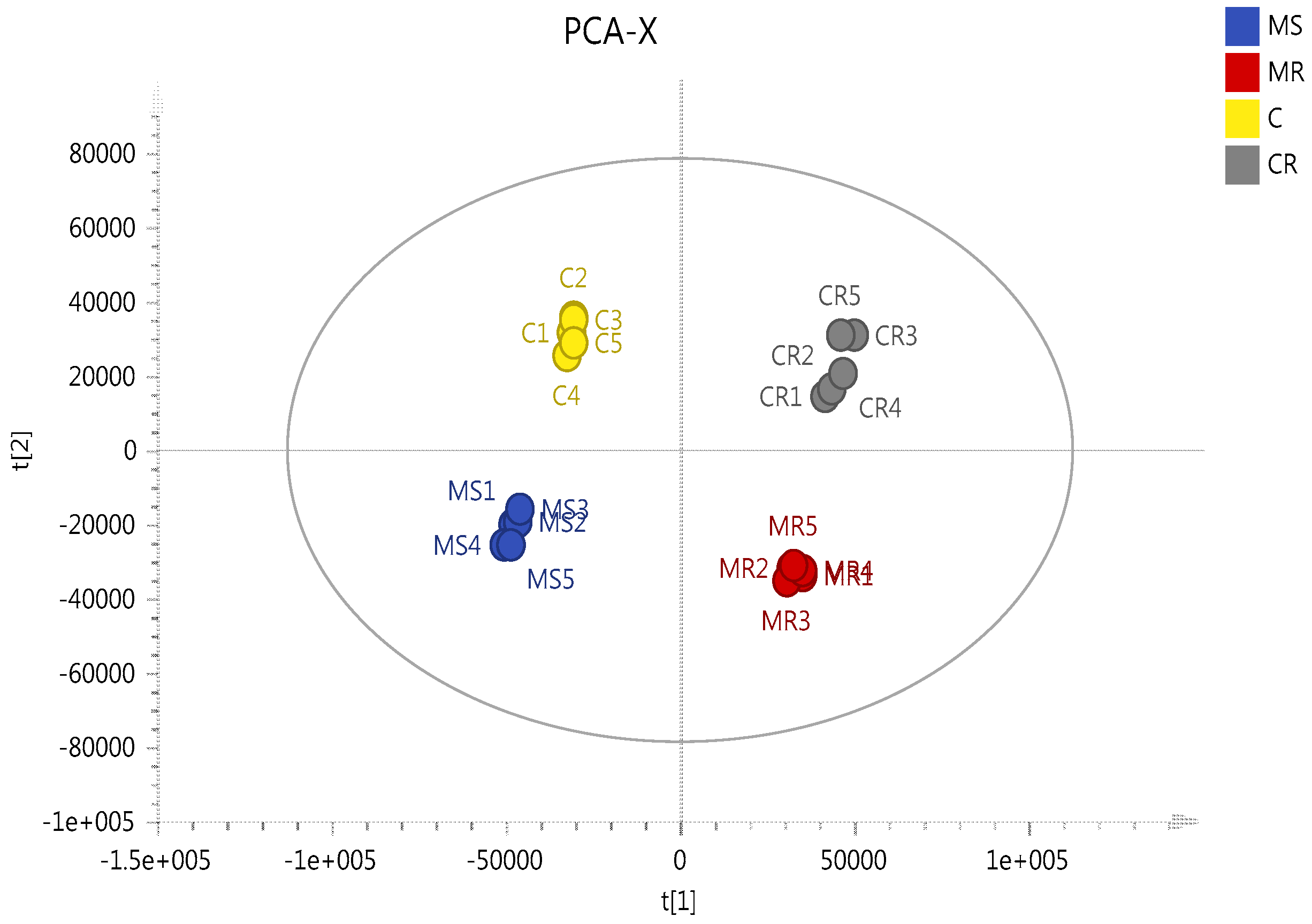
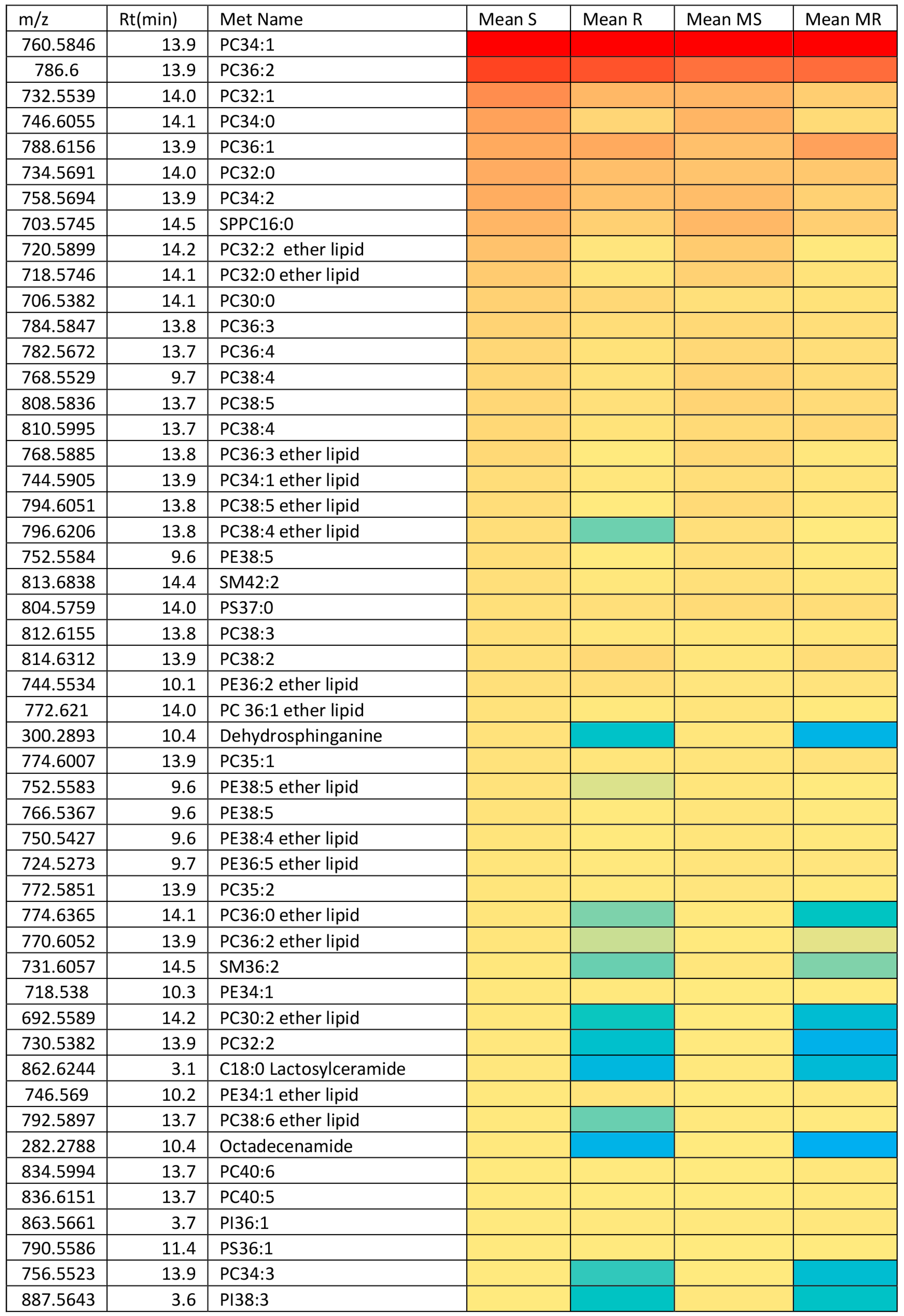
2.3. Effect of Melittin on the Metabolomes of Both Cell Lines
2.4. Assessment of Necrotic and Apoptotic Cell Death in A2780 and A2780CR Cells
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cultures
4.2. Cell Viability Assay against Melittin
4.3. Determination of IC50
4.4. Determination of Effect of Melittin on Cell Metabolomes
4.5. Optimisation of Phenotype Microarray Experiment Parameters
- (1)
- A2780 and A2780CR cells were cultured in a 75 cm2 culture flask containing 10 ml RPMI-1640 medium lacking phenol red but containing 5% (v/v) FBS, L-glutamine and Pen/Strep (Gibco™ by Life Technologies, Paisley, UK).
- (2)
- The medium was removed from the culture flask and saved in a 15 mL sterile conical tube. The remaining medium was aspirated and discarded from the culture flask. The adherent cells were washed twice with 10 mL of Dulbecco’s Phosphate-Buffered Saline (D-PBS) (Gibco, Paisley, UK) and any remaining D-PBS was aspirated and discarded.
- (3)
- The cells were then detached by adding 2 mL of 0.25 % (v/v) Trypsin-EDTA (Gibco, Paisley, UK) and incubated at 37 °C for 3 min.
- (4)
- Then, 3 mL of culture medium was taken from the 15 mL conical tube was added to quench the detachment reaction and the cell suspension mixed by gently pipetting up and down several times to disperse the cells.
- (5)
- The cells were harvested by transferring the cell suspension to the 15 mL conical tube containing the culture medium and centrifuged at 350× g for 5 min. After centrifugation, the medium was aspirated and 10 mL of D-PBS was added. After that, the cell pellet was suspended in the D-PBS by pipetting up and down several times, then centrifuged again at 350× g for 5 min.
- (6)
- After the second centrifugation, the medium was aspirated and 10 mL of pre-warmed MC-0 was added. The cell pellet in the MC-0 Assay Medium was suspended by pipetting up and down several times. The MC-0 medium was composed of IF-M1 (Technopath Distribution, Tipperary, Ireland) medium supplemented with 5.3% (v/v) dialysed foetal bovine serum (dFBS) (Gibco™ by Life Technologies, Paisley, UK), 1.1% of 100× Pen/Strep solution (Gibco™ by Life Technologies, Paisley, UK), and 0.16% (v/v) of 200 mM glutamine (final concentration 0.3 mM).
- (7)
- The cell number was determined and cell viability was assessed by trypan blue dye exclusion (Sigma-Aldrich, Dorset, UK).
- (8)
- The cells were suspended in enough MC-0 Assay Medium to fill the selected number of PM panels and to achieve a density of 4 × 105 cells/mL.
- (9)
- After that, 50 μL/well of the cell suspension was added on two sets of PM-M1 plates (Technopath Distribution, Tipperary, Ireland) so that each well had 20,000 cells. The first one was used as the control set, where untreated A2780 and A2780CR were cultured. A2780 and A2780CR seeded in the second set of plates were exposed to melittin. Both sets of PMs containing A2780 and A2780CR were first incubated for 24 h to allow cells to catabolise all nutrients in medium MC-0. The treated cells set was subsequently inoculated with 25 μL of Melittin/well of three PM-M1 plates at IC50 concentration, while 25 μL of MC-0 medium was added to each well in the control set of three PM-M1 plates.
- (10)
- Then, the PM plates were incubated at 37 °C in a humidified atmosphere with 95% Air-5% CO2 for 18 h, after which the Biolog Redox Dye Mix MA was added to all wells (15 µL/well to the plate). The plate was sealed with tape to prevent off-gassing of CO2.
- (11)
- The plates were incubated for an additional 6 h with Biolog Redox Dye Mix MA (Technopath Distribution, Tipperary, Ireland).
- (12)
- Tetrazolium reduction was determined with a microplate reader (SpectraMax M3, Molecular Devices, Sunnyvale, CA, USA). The endpoint read was performed at 590 nm with subtraction of a 750 nm reference reading (A590-750) which corrects for any background light scattering.
4.6. LC-MS Conditions
4.7. Data Extraction and Analysis
4.8. LDH Assay
4.9. Caspase Activity Assay
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Vermeersch, K.A.; Styczynski, M.P. Applications of metabolomics in cancer research. J. Carcinog. 2013, 12, 9. [Google Scholar] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Poisson, L.M.; Munkarah, A.; Madi, H.; Datta, I.; Hensley-Alford, S.; Tebbe, C.; Buekers, T.; Giri, S.; Rattan, R. A metabolomic approach to identifying platinum resistance in ovarian cancer. J. Ovarian Res. 2015, 8. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Lippard, S.J. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 2005, 4, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, K.; Eno, M.L.; Im, D.D.; Rosenshein, N.B.; Sood, A.K. Clinical relevance of extent of extreme drug resistance in epithelial ovarian carcinoma. Gynecol. Oncol. 2010, 116, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Zwelling, L.A.; Kohn, K.W. Mechanism of action of cis-dichlorodiammineplatinum (II). Cancer Treat. Rep. 1978, 63, 1439–1444. [Google Scholar]
- Siddik, Z.H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2012, 31, 1869–1883. [Google Scholar] [CrossRef] [PubMed]
- Mandic, A.; Hansson, J.; Linder, S.; Shoshan, M.C. Cisplatin induces endoplasmic reticulum stress and nucleus-independent apoptotic signaling. J. Biol. Chem. 2003, 278, 9100–9106. [Google Scholar] [CrossRef] [PubMed]
- Rabik, C.A.; Dolan, M.E. Molecular mechanisms of resistance and toxicity associated with platinating agents. Cancer Treat. Rev. 2007, 33, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Byun, S.S.; Kim, S.W.; Choi, H.; Lee, C.; Lee, E. Augmentation of cisplatin sensitivity in cisplatin-resistant human bladder cancer cells by modulating glutathione concentrations and glutathione-related enzyme activities. BJU Int. 2005, 95, 1086–1090. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.-J.; Choi, Y.; Shin, J.-M.; Cho, H.-J.; Kang, J.-H.; Park, K.-K.; Choe, J.-Y.; Bae, Y.-S.; Han, S.-M.; Kim, C.-H. Melittin suppresses EGF-induced cell motility and invasion by inhibiting PI3K/Akt/mTOR signaling pathway in breast cancer cells. Food Chem. Toxicol. 2014, 68, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Kohno, M.; Horibe, T.; Ohara, K.; Ito, S.; Kawakami, K. The Membrane-Lytic Peptides K8L9 and Melittin Enter Cancer Cells via Receptor Endocytosis following Subcytotoxic Exposure. Chem. Biol. 2014, 21, 1522–1532. [Google Scholar] [CrossRef] [PubMed]
- Mahmoodzadeh, A.; Morady, A.; Zarrinnahad, H.; Ghasemi-Dehkordi, P.; Mahdavi, M.; Shahbazzadeh, D.; Shahmorady, H. Isolation of melittin from bee venom and evaluation of its effect on proliferation of gastric cancer cells. Tehran Univ. Med. Sci. 2013, 70, 760–767. [Google Scholar]
- Mahmoodzadeh, A.; Zarrinnahad, H.; Bagheri, K.P.; Moradia, A.; Shahbazzadeh, D. First report on the isolation of melittin from Iranian honey bee venom and evaluation of its toxicity on gastric cancer AGS cells. J. Chin. Med. Assoc. 2015, 78, 574–583. [Google Scholar] [CrossRef] [PubMed]
- Jo, M.; Park, M.H.; Kollipara, P.S.; An, B.J.; Song, H.S.; Han, S.B.; Kim, J.H.; Song, M.J.; Hong, J.T. Anti-cancer effect of bee venom toxin and melittin in ovarian cancer cells through induction of death receptors and inhibition of JAK2/STAT3 pathway. Toxicol. Appl. Pharmacol. 2012, 258, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Zong, J.; Liu, Z.; Li, L.; Zheng, X.; Wang, B.; Sun, G. A novel melittin-MhIL-2 fusion protein inhibits the growth of human ovarian cancer SKOV3 cells in vitro and in vivo tumor growth. Cancer Immunol. Immunother. 2013, 62, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Qian, C.-Y.; Wang, K.-L.; Fang, F.-F.; Gu, W.; Huang, F.; Wang, F.-Z.; Li, B.; Wang, L.-N. Triple-controlled oncolytic adenovirus expressing melittin to exert inhibitory efficacy on hepatocellular carcinoma. Int. J. Clin. Exp. Pathol. 2015, 8, 10403. [Google Scholar] [PubMed]
- Wu, X.; Zhao, B.; Cheng, Y.; Yang, Y.; Huang, C.; Meng, X.; Wu, B.; Zhang, L.; Lv, X.; Li, J. Melittin induces PTCH1 expression by down-regulating MeCP2 in human hepatocellular carcinoma SMMC-7721 cells. Toxicol. Appl. Pharmacol. 2015, 288, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.-B.; Hwang, C.J.; Song, S.-Y.; Jung, Y.Y.; Yun, H.-M.; Sok, C.H.; Sung, H.C.; Yi, J.-M.; Park, D.H.; Ham, Y.W. Anti-cancer effect of tectochrysin in NSCLC cells through overexpression of death receptor and inactivation of STAT3. Cancer Lett. 2014, 353, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Gajski, G.; Garaj-Vrhovac, V. Melittin: A lytic peptide with anticancer properties. Environ. Toxicol. Pharmacol. 2013, 36, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.-M.; Jeong, Y.-J.; Cho, H.-J.; Park, K.-K.; Chung, I.-K.; Lee, I.-K.; Kwak, J.-Y.; Chang, H.-W.; Kim, C.-H.; Moon, S.-K. Melittin suppresses HIF-1α/VEGF expression through inhibition of ERK and mTOR/p70S6K pathway in human cervical carcinoma cells. PLoS ONE 2013, 8, e69380. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Watson, D.G.; Wang, L.; Abbas, M.; Murdoch, L.; Bashford, L.; Ahmad, I.; Lam, N.Y.; Ng, A.C.; Leung, H.Y. Application of Holistic Liquid Chromatography-High Resolution Mass Spectrometry Based Urinary Metabolomics for Prostate Cancer Detection and Biomarker Discovery. PLoS ONE 2013, 8, e65880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Watson, D.G. A short review of applications of liquid chromatography mass spectrometry based metabolomics techniques to the analysis of human urine. Analyst 2015, 140, 2907–2915. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Zhang, T.; Ali, A.M.; Al Washih, M.; Pickard, B.; Watson, D.G. Metabolomic Profiling of Post-Mortem Brain Reveals Changes in Amino Acid and Glucose Metabolism in Mental Illness Compared with Controls. Comput. Struct. Biotechnol. J. 2016, 14, 106–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frezza, C.; Zheng, L.; Tennant, D.A.; Papkovsky, D.B.; Hedley, B.A.; Kalna, G.; Watson, D.G.; Gottlieb, E. Metabolic profiling of hypoxic cells revealed a catabolic signature required for cell survival. PLoS ONE 2011, 6, e24411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beger, R.D. A review of applications of metabolomics in cancer. Metabolites 2013, 3, 552–574. [Google Scholar] [CrossRef] [PubMed]
- Griffin, J.L.; Shockcor, J.P. Metabolic profiles of cancer cells. Nat. Rev. Cancer 2004, 4, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Čuperlović-Culf, M.; Barnett, D.A.; Culf, A.S.; Chute, I. Cell culture metabolomics: Applications and future directions. Drug Discov. Today 2010, 15, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Bochner, B.R.; Siri, M.; Huang, R.H.; Noble, S.; Lei, X.H.; Clemons, P.A.; Wagner, B.K. Assay of the multiple energy-producing pathways of mammalian cells. PLoS ONE 2011, 6, e18147. [Google Scholar] [CrossRef] [PubMed]
- Vermeersch, K.A.; Wang, L.; McDonald, J.F.; Styczynski, M.P. Distinct metabolic responses of an ovarian cancer stem cell line. BMC Syst. Biol. 2014, 8. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Jin, L.; Li, X.; Deng, H.; Chen, Y.; Lian, Q.; Ge, R.; Deng, H. Gossypol induces apoptosis in ovarian cancer cells through oxidative stress. Mol. BioSyst. 2013, 9, 1489–1497. [Google Scholar] [CrossRef] [PubMed]
- Sasada, S.; Miyata, Y.; Tsutani, Y.; Tsuyama, N.; Masujima, T.; Hihara, J.; Okada, M. Metabolomic analysis of dynamic response and drug resistance of gastric cancer cells to 5-fluorouracil. Oncol. Rep. 2013, 29, 925–931. [Google Scholar] [PubMed]
- Schuber, F. Influence of polyamines on membrane functions. Biochem. J. 1989, 260, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Moinard, C.; Cynober, L.; de Bandt, J.-P. Polyamines: Metabolism and implications in human diseases. Clin.Nutr. 2005, 24, 184–197. [Google Scholar] [CrossRef] [PubMed]
- Morré, D.J.; Kartenbeck, J.; Franke, W.W. Membrane flow and interconversions among endomembranes. Biochim. Biophys. Acta Rev. Biomembr. 1979, 559, 71–152. [Google Scholar] [CrossRef]
- Seiler, N.; Raul, F. Polyamines and apoptosis. J. Cell. Mol. Med. 2005, 9, 623–642. [Google Scholar] [CrossRef] [PubMed]
- Uetaki, M.; Tabata, S.; Nakasuka, F.; Soga, T.; Tomita, M. Metabolomic alterations in human cancer cells by vitamin C-induced oxidative stress. Sci. Rep. 2015, 5, 13896. [Google Scholar] [CrossRef] [PubMed]
- Moore, Z.; Boothman, D.A. Tumor-specific targeting of the NAD metabolome with β-lapachone and NamPT inhibition. Cancer Res. 2014, 74 (Suppl. S19). [Google Scholar] [CrossRef]
- Komatsu, N.; Nakagawa, M.; Oda, T.; Muramatsu, T. Depletion of Intracellular NAD+ and ATP Levels during Ricin-Induced Apoptosis through the Specific Ribosomal Inactivation Results in the Cytolysis of U937 Cells. J. Biochem. 2000, 128, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Tolstikov, V.; Nikolayev, A.; Dong, S.; Zhao, G.; Kuo, M.-S. Metabolomics Analysis of Metabolic Effects of Nicotinamide Phosphoribosyltransferase (NAMPT) Inhibition on Human Cancer Cells. PLoS ONE 2014, 9, e114019. [Google Scholar] [CrossRef] [PubMed]
- Ben Sellem, D.; Elbayed, K.; Neuville, A.; Moussallieh, F.M.; Lang-Averous, G.; Piotto, M.; Bellocq, J.P.; Namer, I.J. Metabolomic Characterization of Ovarian Epithelial Carcinomas by HRMAS-NMR Spectroscopy. J. Oncol. 2011, 2011, 174019. [Google Scholar] [CrossRef] [PubMed]
- Denkert, C.; Budczies, J.; Kind, T.; Weichert, W.; Tablack, P.; Sehouli, J.; Niesporek, S.; Könsgen, D.; Dietel, M.; Fiehn, O. Mass spectrometry–based metabolic profiling reveals different metabolite patterns in invasive ovarian carcinomas and ovarian borderline tumors. Cancer Res. 2006, 66, 10795–10804. [Google Scholar] [CrossRef] [PubMed]
- Halama, A.; Guerrouahen, B.S.; Pasquier, J.; Diboun, I.; Karoly, E.D.; Suhre, K.; Rafii, A. Metabolic signatures differentiate ovarian from colon cancer cell lines. J. Transl. Med. 2015, 13, 223. [Google Scholar] [CrossRef] [PubMed]
- Zammit, V.A.; Ramsay, R.R.; Bonomini, M.; Arduini, A. Carnitine, mitochondrial function and therapy. Adv. Drug Deliv. Rev. 2009, 61, 1353–1362. [Google Scholar] [CrossRef] [PubMed]
- Veldman, R.J.; Klappe, K.; Hinrichs, J.; Hummel, I.; van der Schaaf, G.; Sietsma, H.; Kok, J.W. Altered sphingolipid metabolism in multidrug-resistant ovarian cancer cells is due to uncoupling of glycolipid biosynthesis in the Golgi apparatus. FASEB J. 2002, 16, 1111–1113. [Google Scholar] [CrossRef] [PubMed]
- Lavie, Y.; Cao, H.-T.; Bursten, S.L.; Giuliano, A.E.; Cabot, M.C. Accumulation of Glucosylceramides in Multidrug-resistant Cancer Cells. J. Biol. Chem. 1996, 271, 19530–19536. [Google Scholar] [CrossRef] [PubMed]
- May, G.L.; Wright, L.C.; Dyne, M.; Mackinnon, W.B.; Fox, R.M.; Mountford, C.E. Plasma membrane lipid composition of vinblastine sensitive and resistant human leukaemic lymphoblasts. Int. J. Cancer 1988, 42, 728–733. [Google Scholar] [CrossRef] [PubMed]
- Yonezawa, A.; Inui, K.-I. Organic cation transporter OCT/SLC22A and H+/organic cation antiporter MATE/SLC47A are key molecules for nephrotoxicity of platinum agents. Biochem. Pharmacol. 2011, 81, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Tusiimire, J.; Wallace, J.; Dufton, M.; Parkinson, J.; Clements, C.J.; Young, L.; Park, J.K.; Jeon, J.W.; Watson, D.G. An LCMS method for the assay of melittin in cosmetic formulations containing bee venom. Anal. Bioanal. Chem. 2015, 407, 3627–3635. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Watson, D.G.; Wang, L.; Westrop, G.D.; Coombs, G.H.; Zhang, T. Evaluation of mobile phase characteristics on three zwitterionic columns in hydrophilic interaction liquid chromatography mode for liquid chromatography-high resolution mass spectrometry based untargeted metabolite profiling of Leishmania parasites. J. Chromatogr. A 2014, 1362, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; T’Kind, R.; Decuypere, S.; von Freyend, S.J.; Coombs, G.H.; Watson, D.G. Profiling of lipids in Leishmania donovani using hydrophilic interaction chromatography in combination with Fourier transform mass spectrometry. Rapid Commun. Mass Spectrom. 2010, 24, 2074–2082. [Google Scholar] [CrossRef] [PubMed]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Oresic, M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinform. 2010, 11, 395. [Google Scholar] [CrossRef] [PubMed]
- Katajamaa, M.; Orešič, M. Processing methods for differential analysis of LC/MS profile data. BMC Bioinform. 2005, 6, 179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrasco, R.A.; Stamm, N.B.; Patel, B.K. One-step cellular caspase-3/7 assay. Biotechniques 2003, 34, 1064–1067. [Google Scholar] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| m/z | RT | Metabolite Name | S/R | MS/MR | MR/R | MS/S | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| p-Value | Ratio | p-Value | Ratio | p-Value | Ratio | p-Value | Ratio | |||
| Proline/glutamate/arginine/polyamine metabolism | ||||||||||
| 116.071 | 12.8 | * Proline | <0.01 | 0.837 | <0.001 | 0.409 | ns | 1.056 | <0.001 | 0.516 |
| 128.035 | 10.3 | 1-Pyrroline-3-hydroxy-5-carboxylate | <0.05 | 0.839 | <0.001 | 0.364 | ns | 1.114 | <0.001 | 0.453 |
| 130.051 | 14.4 | Glutamate-5-semialdehyde | ns | 0.944 | <0.001 | 0.607 | ns | 1.065 | <0.001 | 0.658 |
| 131.083 | 11.4 | * Ornithine | <0.001 | 4.774 | <0.001 | 3.573 | ns | 1.001 | <0.001 | 0.749 |
| 132.030 | 15.2 | * Aspartate | ns | 1.091 | <0.001 | 1.229 | ns | 1.055 | <0.01 | 1.159 |
| 146.046 | 10.8 | Glutamate | <0.001 | 0.608 | <0.001 | 0.113 | ns | 0.999 | <0.001 | 0.187 |
| 147.076 | 14.9 | * Glutamine | <0.001 | 0.299 | <0.001 | 0.503 | <0.001 | 1.204 | <0.001 | 1.906 |
| 173.104 | 24.6 | * Arginine | <0.001 | 0.155 | <0.001 | 0.004 | ns | 1.023 | <0.05 | 0.026 |
| 188.057 | 14.4 | * N-Acetyl-l-glutamate | <0.001 | 0.637 | <0.001 | 0.146 | <0.001 | 1.348 | <0.001 | 0.313 |
| 89.107 | 15.4 | ** Putrescine | <0.001 | 2.339 | <0.001 | 1.490 | ns | 1.051 | <0.001 | 0.670 |
| 131.118 | 8.24 | N-Acetylputrescine | <0.001 | 5.175 | <0.001 | 1.603 | ns | 1.021 | <0.001 | 0.316 |
| 146.165 | 26.2 | ** Spermidine | <0.001 | 2.354 | ns | 1.150 | ns | 1.127 | <0.001 | 0.550 |
| 150.058 | 11.4 | * Methionine | <0.001 | 0.422 | <0.001 | 0.669 | <0.01 | 1.127 | <0.001 | 1.594 |
| 298.096 | 6.42 | * 5′-Methylthioadenosine | 0.003 | 1.92 | ns | 0.830 | ns | 0.839 | <0.001 | 0.361 |
| TCA cycle/glycolysis | ||||||||||
| 133.014 | 16.4 | * Malate | <0.001 | 0.647 | <0.001 | 0.238 | <0.001 | 1.129 | <0.001 | 0.418 |
| 145.014 | 15.9 | * 2-Oxoglutarate | ns | 0.972 | <0.001 | 0.173 | <0.001 | 1.217 | <0.001 | 0.221 |
| 191.020 | 18.4 | * Citrate | <0.001 | 2.207 | <0.001 | 0.534 | ns | 1.098 | <0.001 | 0.265 |
| 508.003 | 16.6 | * ATP | <0.001 | 0.267 | <0.001 | 0.118 | ns | 0.963 | <0.001 | 0.415 |
| Carnitine metabolism/fatty acid metabolism | ||||||||||
| 162.112 | 13.3 | * Carnitine | <0.001 | 0.253 | <0.001 | 0.164 | ns | 1.065 | <0.001 | 0.676 |
| 204.123 | 11.0 | * Acetylcarnitine | <0.001 | 0.273 | <0.001 | 0.050 | ns | 1.016 | <0.001 | 0.190 |
| 232.154 | 8.7 | Butanoylcarnitine | <0.001 | 14.083 | ns | 1.992 | ns | 0.903 | <0.001 | 0.131 |
| 664.117 | 14.3 | * NAD+ | <0.001 | 0.487 | <0.001 | 0.135 | <0.001 | 1.228 | <0.001 | 0.336 |
| Miscellaneous | ||||||||||
| 104.106 | 19.6 | * Choline | <0.001 | 0.019 | <0.001 | 0.231 | <0.001 | 1.423 | <0.001 | 7.270 |
| 166.086 | 10.0 | * Phenylalanine | <0.001 | 0.381 | <0.05 | 0.873 | <0.01 | 1.157 | <0.001 | 2.358 |
| 118.086 | 12.4 | * Valine | <0.001 | 1.148 | <0.01 | 2.257 | <0.01 | 1.148 | <0.001 | 2.257 |
| 120.065 | 14.3 | * Threonine | <0.001 | 0.610 | ns | 0.948 | <0.05 | 1.140 | <0.001 | 1.704 |
| 88.040 | 14.7 | * Alanine | ns | 0.965 | <0.001 | 0.385 | ns | 1.098 | <0.001 | 0.442 |
| 179.056 | 17.1 | * Hexose | <0.001 | 0.564 | <0.001 | 0.364 | <0.01 | 0.931 | <0.001 | 0.605 |
| 195.051 | 13.7 | * Gluconic acid | <0.001 | 0.559 | <0.001 | 0.103 | <0.001 | 0.837 | <0.001 | 0.154 |
| 258.110 | 14.4 | * Glycerophosphocholine | <0.001 | 0.020 | <0.001 | 0.031 | <0.001 | 1.529 | <0.001 | 2.342 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alonezi, S.; Tusiimire, J.; Wallace, J.; Dufton, M.J.; Parkinson, J.A.; Young, L.C.; Clements, C.J.; Park, J.K.; Jeon, J.W.; Ferro, V.A.; et al. Metabolomic Profiling of the Effects of Melittin on Cisplatin Resistant and Cisplatin Sensitive Ovarian Cancer Cells Using Mass Spectrometry and Biolog Microarray Technology. Metabolites 2016, 6, 35. https://doi.org/10.3390/metabo6040035
Alonezi S, Tusiimire J, Wallace J, Dufton MJ, Parkinson JA, Young LC, Clements CJ, Park JK, Jeon JW, Ferro VA, et al. Metabolomic Profiling of the Effects of Melittin on Cisplatin Resistant and Cisplatin Sensitive Ovarian Cancer Cells Using Mass Spectrometry and Biolog Microarray Technology. Metabolites. 2016; 6(4):35. https://doi.org/10.3390/metabo6040035
Chicago/Turabian StyleAlonezi, Sanad, Jonans Tusiimire, Jennifer Wallace, Mark J. Dufton, John A. Parkinson, Louise C. Young, Carol J. Clements, Jin Kyu Park, Jong Woon Jeon, Valerie A. Ferro, and et al. 2016. "Metabolomic Profiling of the Effects of Melittin on Cisplatin Resistant and Cisplatin Sensitive Ovarian Cancer Cells Using Mass Spectrometry and Biolog Microarray Technology" Metabolites 6, no. 4: 35. https://doi.org/10.3390/metabo6040035