
Microscale Quantitative Analysis of Polyhydroxybutyrate in Prokaryotes Using IDMS

Abstract
:1. Introduction
2. Materials and Methods
2.1. Strain and Preculture
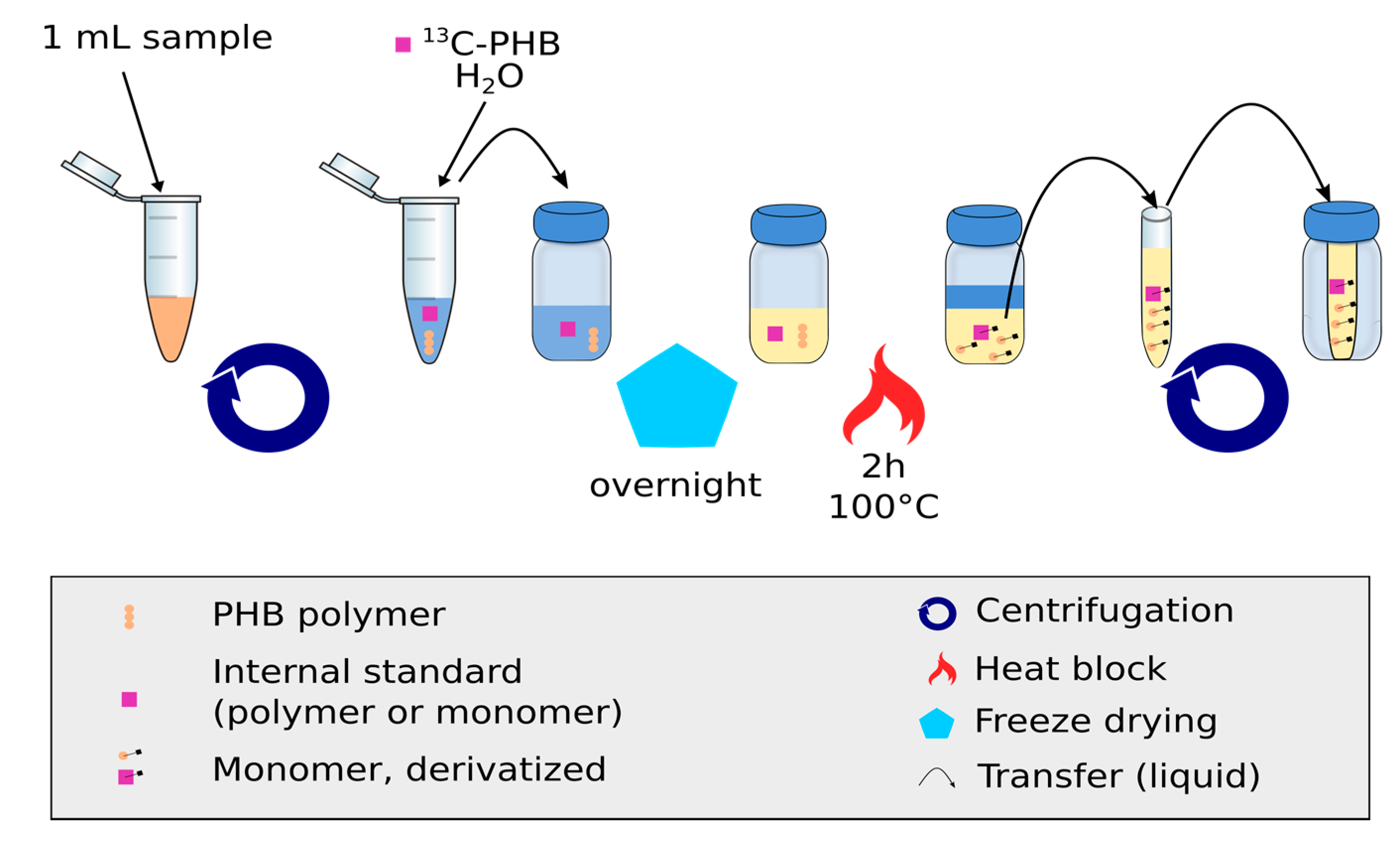
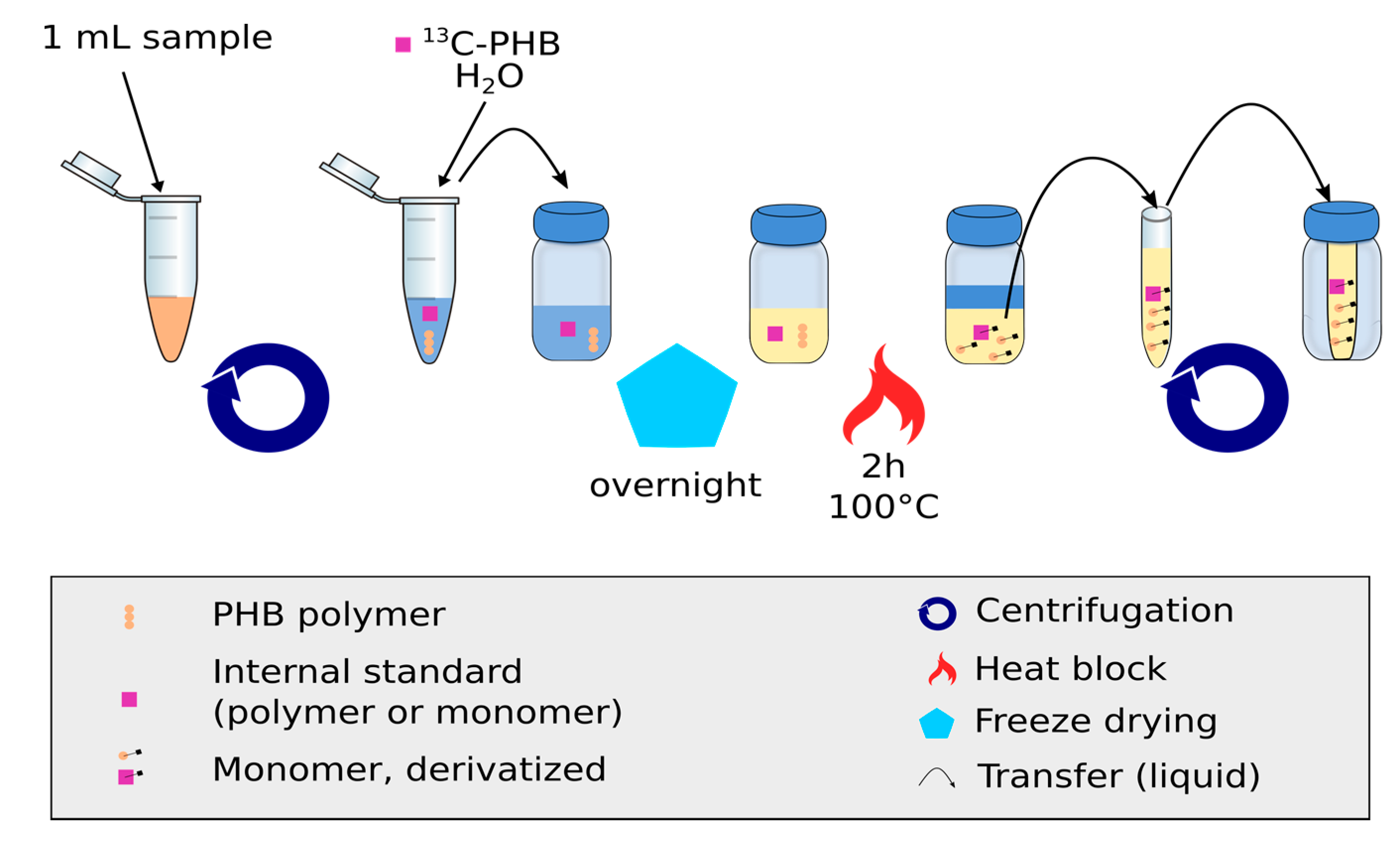
2.2. Production of the Internal Standard, 13C Labeled Cell Suspension Containing PHB
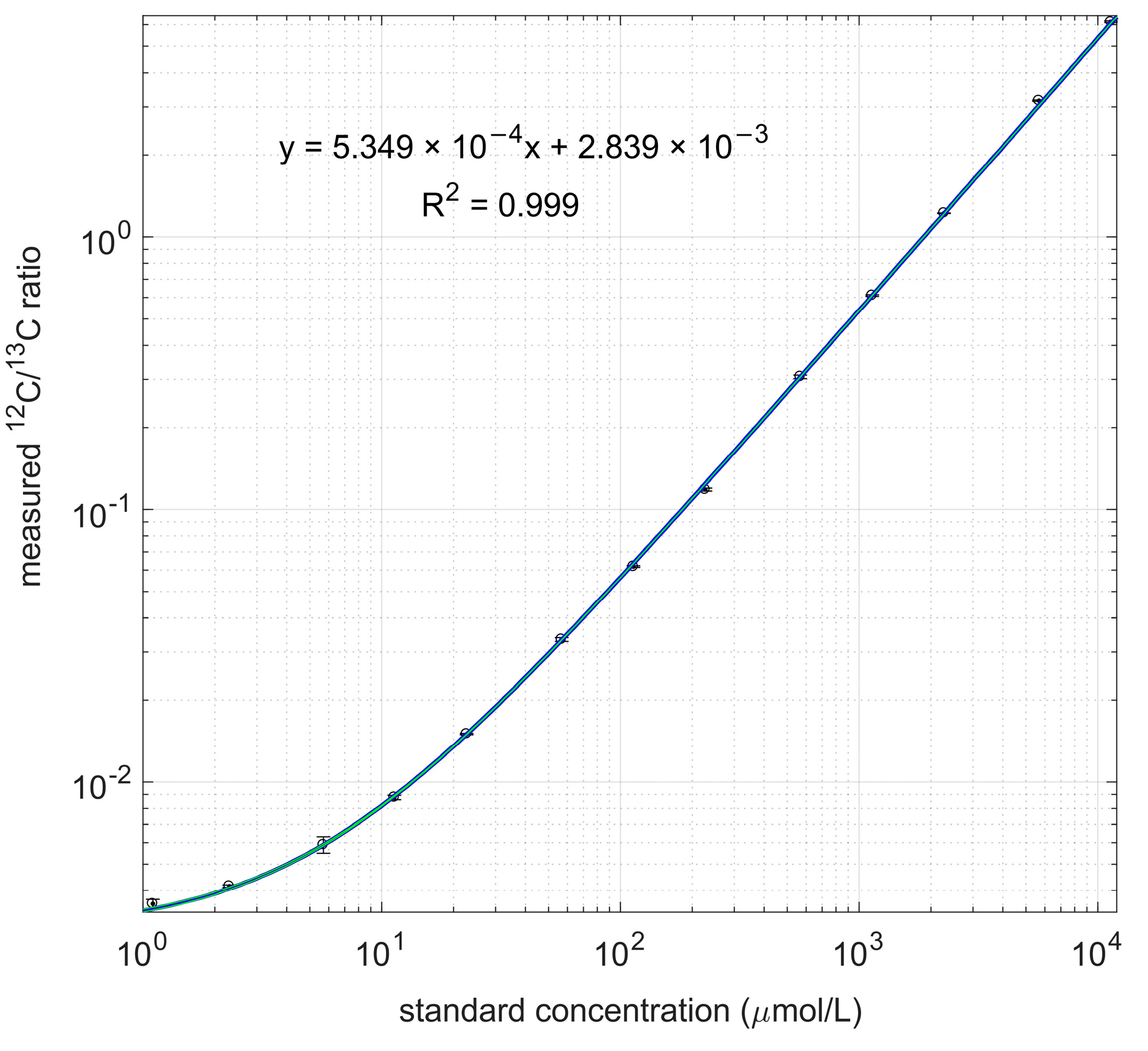
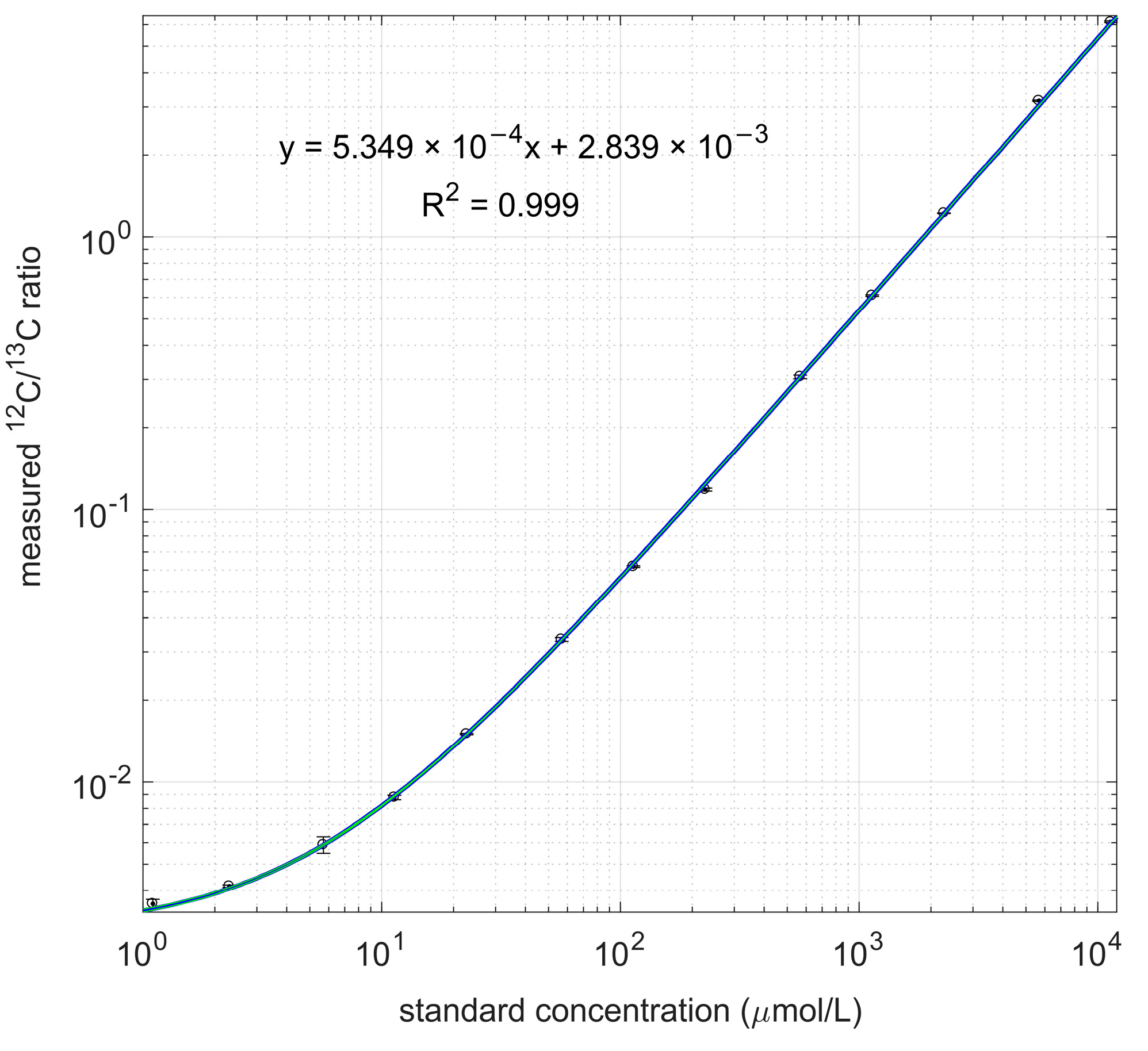
2.3. Calibration Lines of 3-HB
2.4. Cell Dry Weight Measurements
2.5. Quantification of PHB
2.6. GC-IDMS Condition Analysis
3. Results and Discussion
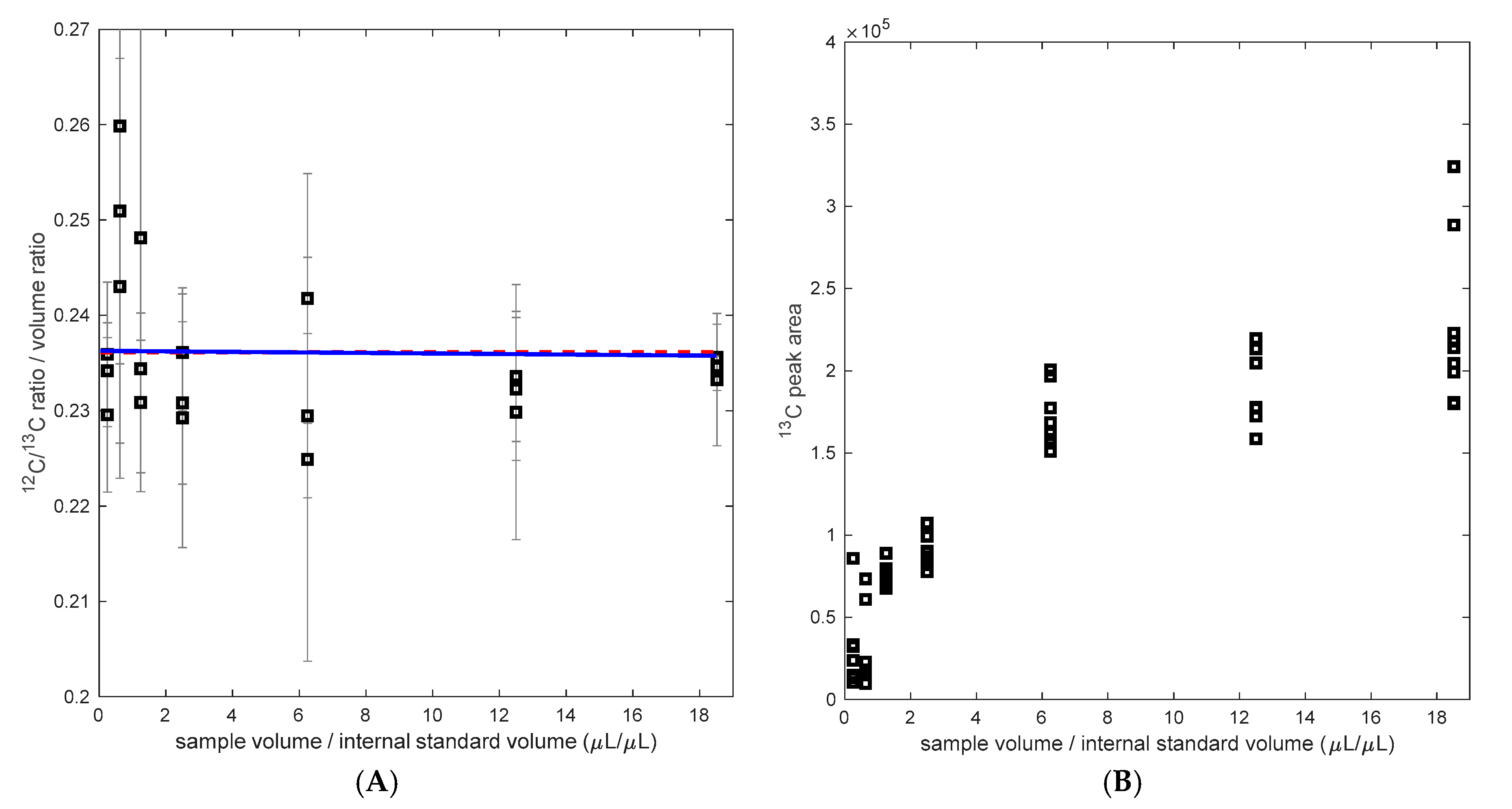
3.1. PHB Determination from Small Sample Volume
3.2. Calibration with Labeled PHB as Internal Standard
4. Validation Experiments
- Technical reproducibility
- Sample background impact (matrix effect)
- Putative influence of the cell matrix
4.1. Technical Reproducibility Including Sample Processing
4.2. Putative Influence of Sample Background (Matrix Effect)
4.3. Putative Influence of the Cell Matrix
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- De Rijk, T.C.; van de Meer, P.; Eggink, G.; Weusthuis, R.A. Methods for Analysis of Poly(3-hydroxyalkanoate) (PHA) Composition. In Biopolymers Online; Wiley: New York, NY, USA, 2005. [Google Scholar] [CrossRef]
- Koller, M.; Rodríguez-Contreras, A. Techniques for tracing PHA-producing organisms and for qualitative and quantitative analysis of intra- and extracellular PHA. Eng. Life Sci. 2015, 15, 558–581. [Google Scholar] [CrossRef]
- Lemoigne, M. Produits de dehydration et de polymerisation de l’acide ß-oxobutyrique. Bull. Soc. Chim. Biol. (Paris) 1926, 8, 770–778. (In French) [Google Scholar]
- Braunegg, G.; Sonnleitner, B.; Lafferty, R.M.; Springer-veriag, I. A Rapid Gas Chromatographic Method for the Determination of Poly-β-hydroxybutyric Acid in Microbial Biomass. Eur. J. Appl. Microbiol. Biotechnol. 1978, 6, 29–37. [Google Scholar] [CrossRef]
- Werker, A.; Lind, P.; Bengtsson, S.; Nordström, F. Chlorinated-solvent-free gas chromatographic analysis of biomass containing polyhydroxyalkanoates. Water Res. 2008, 42, 2517–2526. [Google Scholar] [CrossRef]
- Riis, V.; Mai, W. Gas chromatographic determination microbial biomass after hydrochloric of poly-beta-hydroxybutyric acid propanolysis. J. Chromatogr. 1988, 445, 285–289. [Google Scholar] [CrossRef]
- Carlson, R.; Wlaschin, A.; Srienc, F. Kinetic Studies and Biochemical Pathway Analysis of Anaerobic Poly-(R)-3-Hydroxybutyric Acid Synthesis in Escherichia coli. Appl. Environ. Microbiol. 2005, 71, 713–720. [Google Scholar] [CrossRef]
- Kacmar, J.; Carlson, R.; Balogh, S.J.; Srienc, F. Staining and quantification of poly-3-hydroxybutyrate in Saccharomyces cerevisiae and Cupriavidus necator cell populations using automated flow cytometry. Cytom. Part A 2006, 69, 27–35. [Google Scholar] [CrossRef]
- Mahishi, L.H.; Tripathi, G.; Rawal, S.K. Poly(3-hydroxybutyrate) (PHB) synthesis by recombinant Escherichia coli harbouring Streptomyces aureofaciens PHB biosynthesis genes: Effect of various carbon and nitrogen sources. Microbiol. Res. 2003, 158, 19–27. [Google Scholar] [CrossRef]
- Baetens, D.; Aurola, A.M.; Foglia, A.; Dionisi, D.; Van Loosdrecht, M.C.M. Gas chromatographic analysis of polyhydroxybutyrate in activated sludge: A round-robin test. Water Sci. Technol. 2002, 46, 357–361. [Google Scholar] [CrossRef]
- Du, G.C.; Chen, J.; Yu, J.; Lun, S. Continuous production of poly-3-hydrobutyrate by Ralstonia eutropha in a two-stage culture system. J. Biotechnol. 2001, 88, 59–65. [Google Scholar] [CrossRef]
- Elhottová, D.; Tríska, J.; Petersen, S.O.; Santrůcková, H. Analysis of poly-beta-hydroxybutyrate in environmental samples by GC-MS/MS. Fresenius J. Anal. Chem. 2000, 367, 157–164. [Google Scholar] [CrossRef]
- Karr, D.B.; Waters, J.K.; Emerich, D.W. Analysis of Poly-β-Hydroxybutyrate in Rhizobium japonicum Bacteroids by Ion-Exclusion High-Pressure Liquid Chromatography and UV Detectiont. Appl. Environ. Microbiol. 1983, 46, 1339–1344. [Google Scholar]
- Hesselmann, R.P.X.; Fleischmann, T.; Hany, R.; Zehnder, A.J.B. Determination of polyhydroxyalkanoates in activated sludge by ion chromatographic and enzymatic methods. J. Microbiol. Methods 1999, 35, 111–119. [Google Scholar] [CrossRef]
- Samek, O.; Obruča, S.; Šiler, M.; Sedláček, P.; Benešová, P.; Kučera, D.; Márova, I.; Ježek, J.; Bernatová, S.; Zemánek, P. Quantitative raman spectroscopy analysis of polyhydroxyalkanoates produced by Cupriavidus necator H16. Sensors 2016, 16, 1808. [Google Scholar] [CrossRef]
- Wu, L.; Mashego, M.R.; van Dam, J.C.; Proell, A.M.; Vinke, J.L.; Ras, C.; van Winden, W.A.; van Gulik, W.M.; Heijnen, J.J. Quantitative analysis of the microbial metabolome by isotope dilution mass spectrometry using uniformly 13C-labeled cell extracts as internal standards. Anal. Biochem. 2005, 336, 164–171. [Google Scholar] [CrossRef]
- Noack, S.; Wiechert, W. Quantitative metabolomics: A phantom? Trends Biotechnol. 2014, 32, 238–244. [Google Scholar] [CrossRef]
- Mashego, M.R.; van Gulik, W.M.; Vinke, J.L.; Visser, D.; Heijnen, J.J. In vivo kinetics with rapid perturbation experiments in Saccharomyces cerevisiae using a second-generation BioScope. Metab. Eng. 2006, 8, 370–383. [Google Scholar] [CrossRef]
- Niedenführ, S.; ten Pierick, A.; van Dam, P.T.N.; Suarez-Mendez, C.A.; Nöh, K.; Wahl, S.A. Natural isotope correction of MS/MS measurements for metabolomics and 13C fluxomics. Biotechnol. Bioeng. 2016, 113, 1137–1147. [Google Scholar] [CrossRef]
- De Sousa Gomes, R. Obtenção de Mutantes Deficientes No Acúmulo de PHA E Construção de Linhagens Recombinantes Para O Controle da Composição Monomérica. Ph.D. Thesis, Universidade de São Paulo, São Paulo, Brazil, 19 May 2009. (In Portuguese). [Google Scholar]
- Taymaz-Nikerel, H.; de Mey, M.; Ras, C.; ten Pierick, A.; Seifar, R.M.; van Dam, J.C.; Heijnen, J.J.; van Gulik, W.M. Development and application of a differential method for reliable metabolome analysis in Escherichia coli. Anal. Biochem. 2009, 386, 9–19. [Google Scholar] [CrossRef]
- Niessen, W.M.A.; Manini, P.; Andreoli, R. Matrix effects in quantitative pesticide analysis using liquid chromatography-mass spectrometry. Mass Spectrom. Rev. 2009, 25, 881–899. [Google Scholar] [CrossRef]
- Wahl, S.A.; Seifar, R.M.; ten Pierick, A.; Ras, C.; van Dam, J.C.; Heijnen, J.J.; van Gulik, W.M. Quantitative Metabolomics Using ID-MS. Methods Mol. Biol. 2014, 1191, 91–105. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Method | ||||||
|---|---|---|---|---|---|---|
| GC-FID [6,12] | GC-MS/MS [12] | HPLC [13] | IC [14] | GC-IDMS [This Study] | Raman Spectroscopy [15] | |
| Sampled biomass amount (mgDW) | 50 | 50 | 1000 | 10 | 1 | 1 |
| Lowest (PHB)1 concentration (nmol/L) | 1.16 | 0.0012 | 140 (a) | 106 (a) | 0.011 | 1395 (a) |
| Injection volume | 0.1 µL | 0.1 µL | 10 µL | − | 1 µL | NA |
| Used split | 1:100 | SL (b) | − | − | 1:500 | NA |
| Internal standard | Benzoic acid | β-hydroxyoctanoic acid | Adipic acid | NA | U 13C-PHB | DNA/Amide I |
| External standard | PHB | 3-HBNa (c) | Crotonic acid | 3.3:1 PHB:PHV (d) | 3-HBNa | PHB |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Velasco Alvarez, M.I.; Ten Pierick, A.; Van Dam, P.T.N.; Maleki Seifar, R.; Van Loosdrecht, M.C.M.; Wahl, S.A. Microscale Quantitative Analysis of Polyhydroxybutyrate in Prokaryotes Using IDMS. Metabolites 2017, 7, 19. https://doi.org/10.3390/metabo7020019
Velasco Alvarez MI, Ten Pierick A, Van Dam PTN, Maleki Seifar R, Van Loosdrecht MCM, Wahl SA. Microscale Quantitative Analysis of Polyhydroxybutyrate in Prokaryotes Using IDMS. Metabolites. 2017; 7(2):19. https://doi.org/10.3390/metabo7020019
Chicago/Turabian StyleVelasco Alvarez, Mariana Itzel, Angela Ten Pierick, Patricia T. N. Van Dam, Reza Maleki Seifar, Mark C. M. Van Loosdrecht, and S. Aljoscha Wahl. 2017. "Microscale Quantitative Analysis of Polyhydroxybutyrate in Prokaryotes Using IDMS" Metabolites 7, no. 2: 19. https://doi.org/10.3390/metabo7020019