Endoplasmic Reticulum Calcium Pumps and Cancer Cell Differentiation

Abstract
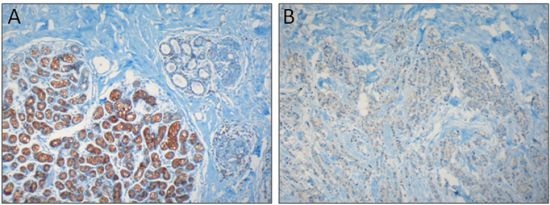
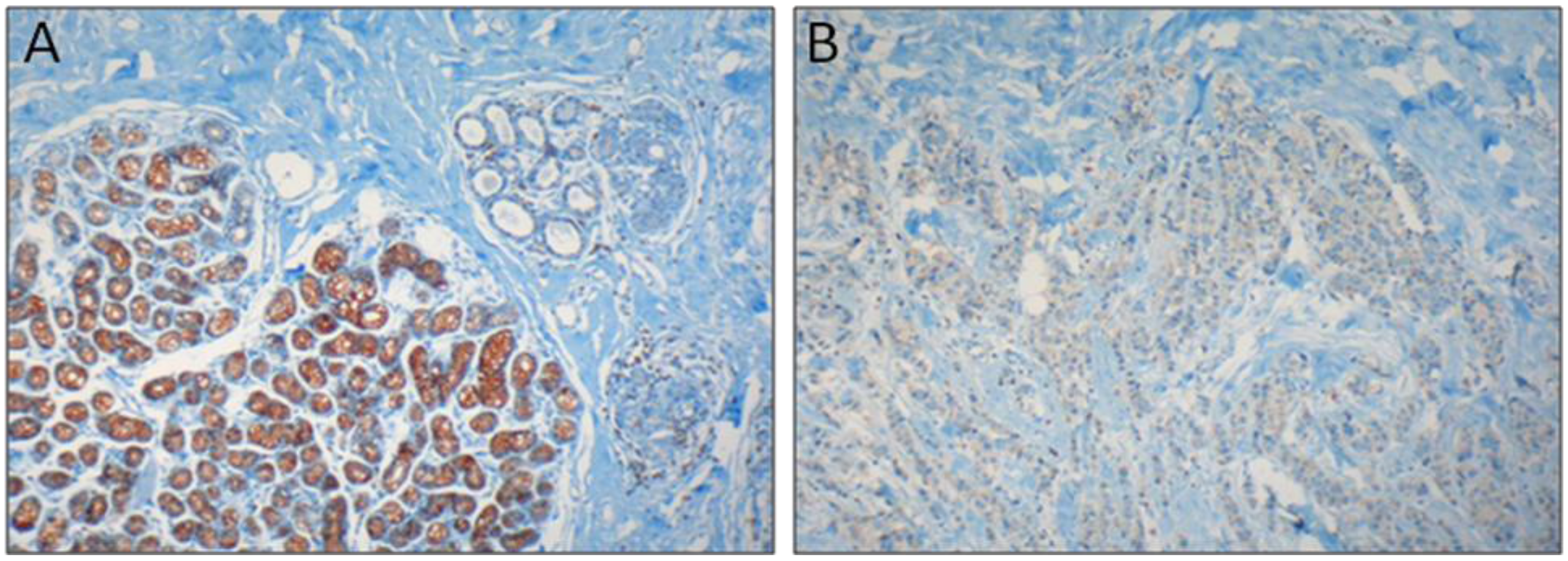
:
{kind=link}
{kind=link}
{kind=link}
1. ER Calcium Sequestration: An Essential Component and Key Modulator of Cell Activation and Survival
2. The SERCA Multigene Family, Co-Expression of SERCA2 and SERCA3 Proteins
2.1. Myeloid Leukemia
2.2. Colon Carcinoma
2.3. Breast Cancer

2.4. T Lymphocyte Activation
2.5. B Lymphocyte Immortalization
3. Discussion
3.1. SERCA3: A New Marker of Cell Differentiation
3.2. Remodeling of ER Calcium Homeostasis during Differentiation
3.3. Cross-Talk between SERCA Function and the Control of Differentiation
3.4. Cellular Calcium Homeostasis: A Heavily Interconnected System
4. Conclusions
Acknowledgements
References
- Bygrave, F.L.; Benedetti, A. What is the concentration of calcium ions in the endoplasmic reticulum? Cell Calcium 1996, 19, 547–551. [Google Scholar] [CrossRef]
- Solovyova, N.; Verkhratsky, A. Monitoring of free calcium in the neuronal endoplasmic reticulum: an overview of modern approaches. J. Neurosci. Methods 2002, 122, 1–12. [Google Scholar] [CrossRef]
- Berridge, M.J. Inositol trisphosphate and calcium signalling mechanisms. Biochim. Biophys. Acta 2009, 1793, 933–940. [Google Scholar]
- Laurent-Puig, P.; Lièvre, A.; Blons, H. Mutations and response to epidermal growth factor receptor inhibitors. Clin. Cancer Res. 2009, 15, 1133–1139. [Google Scholar] [CrossRef]
- de Mello, R.A.; Marques, D.S.; Medeiros, R.; Araujo, A.M. Epidermal growth factor receptor and K-Ras in non-small cell lung cancer-molecular pathways involved and targeted therapies. World J. Clin. Oncol. 2011, 2, 367–376. [Google Scholar] [CrossRef]
- Feske, S.; Gwack, Y.; Prakriya, M.; Srikanth, S.; Puppel, S.H.; Tanasa, B.; Hogan, P.G.; Lewis, R.S.; Daly, M.; Rao, A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 2006, 441, 179–185. [Google Scholar]
- Cahalan, M.D. STIMulating store-operated Ca2+ entry. Nat. Cell Biol. 2009, 11, 669–677. [Google Scholar] [CrossRef]
- Barr, V.A.; Bernot, K.M.; Shaffer, M.H.; Burkhardt, J.K.; Samelson, L.E. Formation of STIM and Orai complexes: puncta and distal caps. Immunol. Rev. 2009, 231, 148–159. [Google Scholar] [CrossRef]
- Petersen, O.H.; Michalak, M.; Verkhratsky, A. Calcium signalling: Past, present and future. Cell Calcium 2005, 38, 161–169. [Google Scholar] [CrossRef]
- Liu, J.O. Calmodulin-dependent phosphatase, kinases, and transcriptional corepressors involved in T-cell activation. Immunol. Rev. 2009, 228, 184–198. [Google Scholar] [CrossRef]
- Zhang, S.; Fritz, N.; Ibarra, C.; Uhlen, P. Inositol 1,4,5-trisphosphate receptor subtype-specific regulation of calcium oscillations. Neurochem. Res. 2011, 36, 1175–1185. [Google Scholar] [CrossRef]
- Collins, S.R.; Meyer, T. Evolutionary origins of STIM1 and STIM2 within ancient Ca2+ signaling systems. Trends Cell Biol. 2011, 21, 202–211. [Google Scholar] [CrossRef]
- Johnstone, L.S.; Graham, S.J.; Dziadek, M.A. STIM proteins: Integrators of signalling pathways in development, differentiation and disease. J. Cell. Mol. Med. 2010, 14, 1890–1903. [Google Scholar] [CrossRef]
- Bertram, R.; Arceo, R.C., 2nd. A mathematical study of the differential effects of two SERCA isoforms on Ca2+ oscillations in pancreatic islets. Bull. Math. Biol. 2008, 70, 1251–1271. [Google Scholar] [CrossRef]
- Higgins, E.R.; Cannell, M.B.; Sneyd, J. A buffering SERCA pump in models of calcium dynamics. Biophys. J. 2006, 91, 151–163. [Google Scholar] [CrossRef]
- Dellen, B.K.; Barber, M.J.; Ristig, M.L.; Hescheler, J.; Sauer, H.; Wartenberg, M. Ca2+ oscillations in a model of energy-dependent Ca2+ uptake by the endoplasmic reticulum. J. Theor. Biol. 2005, 237, 279–290. [Google Scholar] [CrossRef]
- Juska, A. Calcium fluxes into and out of cytosol in human platelets: Analysis of experimental data. Biochem. Biophys. Res. Commun. 2011, 412, 537–542. [Google Scholar] [CrossRef]
- Juska, A. Dynamics of calcium fluxes in nonexcitable cells: Mathematical modeling. J. Membr. Biol. 2006, 211, 89–99. [Google Scholar] [CrossRef]
- Juska, A.; Redondo, P.C.; Rosado, J.A.; Salido, G.M. Dynamics of calcium fluxes in human platelets assessed in calcium-free medium. Biochem. Biophys. Res. Commun. 2005, 334, 779–786. [Google Scholar] [CrossRef]
- Bakowski, D.; Parekh, A.B. Sarcoplasmic/endoplasmic-reticulum-Ca2+-ATPase-mediated Ca2+ reuptake, and not Ins(1,4,5)P3 receptor inactivation, prevents the activation of macroscopic Ca2+ release-activated Ca2+ current in the presence of physiological Ca2+ buffer in rat basophilic leukemia-1 cells. Biochem. J. 2001, 353, 561–567. [Google Scholar] [CrossRef]
- Dolmetsch, R.E.; Lewis, R.S.; Goodnow, C.C.; Healy, J.I. Differential activation of transcription factors induced by Ca2+ response amplitude and duration. Nature 1997, 386, 855–858. [Google Scholar]
- Dolmetsch, R.E.; Xu, K.; Lewis, R.S. Calcium oscillations increase the efficiency and specificity of gene expression. Nature 1998, 392, 933–936. [Google Scholar]
- Coe, H.; Michalak, M. Calcium binding chaperones of the endoplasmic reticulum. Gen. Physiol. Biophys. 2009, Focus Issue. 28, F96–F103. [Google Scholar]
- Brostrom, M.A.; Brostrom, C.O. Calcium dynamics and endoplasmic reticular function in the regulation of protein synthesis: Implications for cell growth and adaptability. Cell Calcium 2003, 34, 345–363. [Google Scholar] [CrossRef]
- Burdakov, D.; Petersen, O.H.; Verkhratsky, A. Intraluminal calcium as a primary regulator of endoplasmic reticulum function. Cell Calcium 2005, 38, 303–310. [Google Scholar] [CrossRef]
- Bedard, K.; Szabó, É.; Michalak, M.; Opas, M. Cellular functions of endoplasmic reticulum chaperones calreticulin, calnexin, and ERp57. Int. Rev. Cytol. 2005, 245, 91–121. [Google Scholar] [CrossRef]
- Michalak, M.; Groenendyk, J.; Szabó, É.; Gold, L.I.; Opas, M. Calreticulin, a multi-process calcium-buffering chaperone of the endoplasmic reticulum. Biochem. J. 2009, 417, 651–666. [Google Scholar] [CrossRef]
- Paschen, W. Dependence of vital cell function on endoplasmic reticulum calcium levels: Implications for the mechanisms underlying neuronal cell injury in different pathological states. Cell Calcium 2001, 29, 1–11. [Google Scholar] [CrossRef]
- Malhotra, J.D.; Kaufman, R.J. The endoplasmic reticulum and the unfolded protein response. Semin. Cell Dev. Biol. 2007, 18, 716–731. [Google Scholar] [CrossRef]
- Lai, E.; Teodoro, T.; Volchuk, A. Endoplasmic reticulum stress: Signaling the unfolded protein response. Physiology (Bethesda) 2007, 22, 193–201. [Google Scholar] [CrossRef]
- Mekahli, D.; Bultynck, G.; Parys, J.B.; De Smedt, H.; Missiaen, L. Endoplasmic-reticulum calcium depletion and disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a004317. [Google Scholar] [CrossRef]
- Høyer-Hansen, M.; Jäättelä, M. Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ. 2007, 14, 1576–1582. [Google Scholar] [CrossRef]
- Christensen, S.B.; Skytte, D.M.; Denmeade, S.R.; Dionne, C.; Møller, J.V.; Nissen, P.; Isaacs, J.T. A Trojan horse in drug development: Targeting of thapsigargins towards prostate cancer cells. Anticancer Agents Med. Chem. 2009, 9, 276–294. [Google Scholar]
- Identifier: NCT 01056029 Dose-Escalation Phase A Study of G-202 in Patients With Advanced Solid Tumors. Available online: http://clinicaltrials.gov (accessed on 04 March 2012).
- Vandecaetsbeek, I.; Vangheluwe, P.; Raeymaekers, L.; Wuytack, F.; Vanoevelen, J. The Ca2+ pumps of the endoplasmic reticulum and Golgi apparatus. Cold Spring Harb. Perspect. Biol. 2011, 3, a004184. [Google Scholar] [CrossRef]
- Vangheluwe, P.; Sepulveda, M.R.; Missiaen, L.; Raeymaekers, L.; Wuytack, F.; Vanoevelen, J. Intracellular Ca2+- and Mn2+-transport ATPases. Chem. Rev. 2009, 109, 4733–4759. [Google Scholar] [CrossRef]
- Wuytack, F.; Raeymaekers, L.; Missiaen, L. Molecular physiology of the SERCA and SPCA pumps. Cell Calcium 2002, 32, 279–305. [Google Scholar] [CrossRef]
- Baba-Aissa, F.; Raeymaekers, L.; Wuytack, F.; Dode, L.; Casteels, R. Distribution and isoform diversity of the organellar Ca2+ pumps in the brain. Mol. Chem. Neuropathol. 1998, 33, 199–208. [Google Scholar] [CrossRef]
- Wuytack, F.; Dode, L.; Baba-Aissa, F.; Raeymaekers, L. The SERCA3-type of organellar Ca2+ pumps. Biosci. Rep. 1995, 15, 299–306. [Google Scholar] [CrossRef]
- Gélébart, P.; Martin, V.; Enouf, J.; Papp, B. Identification of a new SERCA2 splice variant regulated during monocytic differentiation. Biochem. Biophys. Res. Commun. 2003, 303, 676–684. [Google Scholar] [CrossRef]
- Burk, S.E.; Lytton, J.; MacLennan, D.H.; Shull, G.E. cDNA cloning, functional expression, and mRNA tissue distribution of a third organellar Ca2+ pump. J. Biol. Chem. 1989, 264, 18561–18568. [Google Scholar]
- Bobe, R.; Bredoux, R.; Corvazier, E.; Lacabaratz-Porret, C.; Martin, V.; Kovács, T.; Enouf, J. How many Ca2+ATPase isoforms are expressed in a cell type? A growing family of membrane proteins illustrated by studies in platelets. Platelets 2005, 16, 133–150. [Google Scholar] [CrossRef]
- Dally, S.; Corvazier, E.; Bredoux, R.; Bobe, R.; Enouf, J. Multiple and diverse coexpression, location, and regulation of additional SERCA2 and SERCA3 isoforms in nonfailing and failing human heart. J. Mol. Cell. Cardiol. 2010, 48, 633–644. [Google Scholar] [CrossRef]
- Inesi, G.; Lewis, D.; Ma, H.; Prasad, A.; Toyoshima, C. Concerted conformational effects of Ca2+ and ATP are required for activation of sequential reactions in the Ca2+ ATPase (SERCA) catalytic cycle. Biochemistry 2006, 45, 13769–13778. [Google Scholar]
- Sugita, Y.; Ikeguchi, M.; Toyoshima, C. Relationship between Ca2+-affinity and shielding of bulk water in the Ca2+-pump from molecular dynamics simulations. Proc. Natl. Acad. Sci. USA 2010, 107, 21465–21469. [Google Scholar]
- Espinoza-Fonseca, L.M.; Thomas, D.D. Atomic-level characterization of the activation mechanism of SERCA by calcium. PLoS One 2011, 6, e26936. [Google Scholar] [CrossRef]
- Chandrasekera, P.C.; Kargacin, M.E.; Deans, J.P.; Lytton, J. Determination of apparent calcium affinity for endogenously expressed human sarco(endo)plasmic reticulum calcium-ATPase isoform SERCA3. Am. J. Physiol. Cell Physiol. 2009, 296, C1105–C1114. [Google Scholar] [CrossRef]
- Poch, E.; Leach, S.; Snape, S.; Cacic, T.; MacLennan, D.H.; Lytton, J. Functional characterization of alternatively spliced human SERCA3 transcripts. Am. J. Physiol. 1998, 275, C1449–C1458. [Google Scholar]
- Dode, L.; Vilsen, B.; Van Baelen, K.; Wuytack, F.; Clausen, J.D.; Andersen, J.P. Dissection of the functional differences between sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA) 1 and 3 isoforms by steady-state and transient kinetic analyses. J. Biol. Chem. 2002, 277, 45579–45591. [Google Scholar]
- Periasamy, M.; Kalyanasundaram, A. SERCA pump isoforms: their role in calcium transport and disease. Muscle Nerve 2007, 35, 430–442. [Google Scholar] [CrossRef]
- Lytton, J.; Westlin, M.; Burk, S.E.; Shull, G.E.; MacLennan, D.H. Functional comparisons between isoforms of the sarcoplasmic or endoplasmic reticulum family of calcium pumps. J. Biol. Chem. 1992, 267, 14483–14489. [Google Scholar]
- Papp, B.; Enyedi, A.; Pászty, K.; Kovács, T.; Sarkadi, B.; Gárdos, G.; Magnier, C.; Wuytack, F.; Enouf, J. Simultaneous presence of two distinct endoplasmic-reticulum-type calcium-pump isoforms in human cells. Characterization by radio-immunoblotting and inhibition by 2,5-di-(t-butyl)-1,4-benzohydroquinone. Biochem. J. 1992, 288, 297–302. [Google Scholar]
- Papp, B.; Enyedi, Á.; Kovács, T.; Sarkadi, B.; Wuytack, F.; Thastrup, O.; Gárdos, G.; Bredoux, R.; Lévy-Tolédano, S.; Enouf, J. Demonstration of two forms of calcium pumps by thapsigargin inhibition and radioimmunoblotting in platelet membrane vesicles. J. Biol. Chem. 1991, 266, 14593–14596. [Google Scholar]
- Mountian, I.; Manolopoulos, V.G.; De Smedt, H.; Parys, J.B.; Missiaen, L.; Wuytack, F. Expression patterns of sarco/endoplasmic reticulum Ca2+-ATPase and inositol 1,4,5-trisphosphate receptor isoforms in vascular endothelial cells. Cell Calcium 1999, 25, 371–380. [Google Scholar] [CrossRef]
- Møller, J.V.; Olesen, C.; Winther, A.M.; Nissen, P. The sarcoplasmic Ca2+-ATPase: design of a perfect chemi-osmotic pump. Q. Rev. Biophys. 2010, 43, 501–566. [Google Scholar] [CrossRef]
- Vangheluwe, P.; Raeymaekers, L.; Dode, L.; Wuytack, F. Modulating sarco(endo)plasmic reticulum Ca2+-ATPase 2 (SERCA2) activity: Cell biological implications. Cell Calcium 2005, 38, 291–302. [Google Scholar] [CrossRef]
- Strehler, E.E.; Treiman, M. Calcium pumps of plasma membrane and cell interior. Curr. Mol. Med. 2004, 4, 323–335. [Google Scholar] [CrossRef]
- Toyoshima, C. How Ca2+-ATPase pumps ions across the sarcoplasmic reticulum membrane. Biochim. Biophys. Acta 2009, 1793, 941–946. [Google Scholar]
- Toyoshima, C. Ion pumping by calcium ATPase of sarcoplasmic reticulum. Adv. Exp. Med. Biol. 2007, 592, 295–303. [Google Scholar] [CrossRef]
- Brini, M.; Carafoli, E. Calcium pumps in health and disease. Physiol. Rev. 2009, 89, 1341–1378. [Google Scholar] [CrossRef]
- Inesi, G.; Hua, S.; Xu, C.; Ma, H.; Seth, M.; Prasad, A.M.; Sumbilla, C. Studies of Ca2+ ATPase (SERCA) inhibition. J. Bioenerg. Biomembr. 2005, 37, 365–368. [Google Scholar] [CrossRef]
- Prasad, V.; Okunade, G.W.; Miller, M.L.; Shull, G.E. Phenotypes of SERCA and PMCA knockout mice. Biochem. Biophys. Res. Commun. 2004, 322, 1192–1203. [Google Scholar] [CrossRef]
- Shull, G.E. Gene knockout studies of Ca2+-transporting ATPases. Eur. J. Biochem. 2000, 267, 5284–5290. [Google Scholar] [CrossRef]
- Hovnanian, A. SERCA pumps and human diseases. Subcell. Biochem. 2007, 45, 337–363. [Google Scholar] [CrossRef]
- Cooper, S.M.; Burge, S.M. Darier’s disease: Epidemiology, pathophysiology, and management. Am. J. Clin. Dermatol. 2003, 4, 97–105. [Google Scholar] [CrossRef]
- Gommans, I.M.; Vlak, M.H.; de Haan, A.; van Engelen, B.G. Calcium regulation and muscle disease. J. Muscle Res. Cell Motil. 2002, 23, 59–63. [Google Scholar] [CrossRef]
- MacLennan, D.H. Ca2+ signalling and muscle disease. Eur. J. Biochem. 2000, 267, 5291–5297. [Google Scholar] [CrossRef]
- Monteith, G.R.; McAndrew, D.; Faddy, H.M.; Roberts-Thomson, S.J. Calcium and cancer: targeting Ca2+ transport. Nat. Rev. Cancer 2007, 7, 519–530. [Google Scholar] [CrossRef]
- Roderick, H.L.; Cook, S.J. Ca2+ signalling checkpoints in cancer: remodelling Ca2+ for cancer cell proliferation and survival. Nat. Rev. Cancer 2008, 8, 361–375. [Google Scholar] [CrossRef]
- Nasr, R.; de Thé, H. Eradication of acute promyelocytic leukemia-initiating cells by PML/RARA-targeting. Int. J. Hematol. 2010, 91, 742–747. [Google Scholar] [CrossRef]
- Nasr, R.; Guillemin, M.-C.; Ferhi, O.; Soilihi, H.; Peres, L.; Berthier, C.; Rousselot, P.; Robledo-Sarmiento, M.; Lallemand-Breitenbach, V.; Gourmel, B.; Vitoux, D.; Pandolfi, P.P.; Rochette-Egly, C.; Zhu, J.; de Thé, H. Eradication of acute promyelocytic leukemia-initiating cells through PML-RARA degradation. Nat. Med. 2008, 14, 1333–1342. [Google Scholar]
- Nasr, R.; Lallemand-Breitenbach, V.; Zhu, J.; Guillemin, M.-C.; de Thé, H. Therapy-induced PML/RARA proteolysis and acute promyelocytic leukemia cure. Clin. Cancer Res. 2009, 15, 6321–6326. [Google Scholar] [CrossRef]
- Chomienne, C.; Fenaux, P.; Degos, L. Retinoid differentiation therapy in promyelocytic leukemia. FASEB J. 1996, 10, 1025–1030. [Google Scholar]
- Launay, S.; Giannì, M.; Kovács, T.; Bredoux, R.; Bruel, A.; Gélébart, P.; Zassadowski, F.; Chomienne, C.; Enouf, J.; Papp, B. Lineage-specific modulation of calcium pump expression during myeloid differentiation. Blood 1999, 93, 4395–4405. [Google Scholar]
- Lacabaratz-Porret, C.; Launay, S.; Corvazier, E.; Bredoux, R.; Papp, B.; Enouf, J. Biogenesis of endoplasmic reticulum proteins involved in Ca2+ signalling during megakaryocytic differentiation: An in vitro study. Biochem. J. 2000, 350, 723–734. [Google Scholar] [CrossRef]
- Lacabaratz-Porret, C.; Corvazier, E.; Kovács, T.; Bobe, R.; Bredoux, R.; Launay, S.; Papp, B.; Enouf, J. Platelet sarco/endoplasmic reticulum Ca2+ATPase isoform 3b and Rap 1b: interrelation and regulation in physiopathology. Biochem. J. 1998, 332, 173–181. [Google Scholar]
- Papp, B.; Pászty, K.; Kovács, T.; Sarkadi, B.; Gárdos, G.; Enouf, J.; Enyedi, Á. Characterization of the inositol trisphosphate-sensitive and insensitive calcium stores by selective inhibition of the endoplasmic reticulum-type calcium pump isoforms in isolated platelet membrane vesicles. Cell Calcium 1993, 14, 531–538. [Google Scholar] [CrossRef]
- Humphries, A.; Wright, N.A. Colonic crypt organization and tumorigenesis. Nat. Rev. Cancer 2008, 8, 415–424. [Google Scholar] [CrossRef]
- Arends, J.W. Molecular interactions in the Vogelstein model of colorectal carcinoma. J. Pathol. 2000, 190, 412–416. [Google Scholar] [CrossRef]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Bright-Thomas, R.M.; Hargest, R. APC, beta-Catenin and hTCF-4; an unholy trinity in the genesis of colorectal cancer. Eur. J. Surg. Oncol. 2003, 29, 107–117. [Google Scholar] [CrossRef]
- Aaltonen, L.A.; Hamilton, S.R. Pathology and Genetics of Tumors of the Digestive System; IARC Press: Lyon, France; Oxford University Press: Oxford, UK, 2000; pp. 1–314. [Google Scholar]
- Frazin, G.; Zamboni, G.; Scarpa, A.; Dina, R.; Iannucci, A.; Novelli, P. Hyperplastic (metaplastic) polyps of the colon. A histologic and histochemical study. Am. J. Surg. Pathol. 1984, 8, 687–698. [Google Scholar] [CrossRef]
- Scholzel, S.; Zimmermann, W.; Schwarzkopf, G.; Grunert, F.; Rogaczewski, B.; Thompson, J. Carcinoembryonic antigen family members CEACAM6 and CEACAM7 are differentially expressed in normal tissues and oppositely deregulated in hyperplastic colorectal polyps and early adenomas. Am. J. Pathol. 2000, 156, 595–605. [Google Scholar] [CrossRef]
- Brouland, J.-P.; Gélébart, P.; Kovács, T.; Enouf, J.; Grossmann, J.; Papp, B. The loss of sarco/endoplasmic reticulum calcium transport ATPase 3 expression is an early event during the multistep process of colon carcinogenesis. Am. J. Pathol. 2005, 167, 233–242. [Google Scholar] [CrossRef]
- Gélébart, P.; Kovács, T.; Brouland, J.-P.; van Gorp, R.; Grossmann, J.; Rivard, N.; Panis, Y.; Martin, V.; Bredoux, R.; Enouf, J.; Papp, B. Expression of endomembrane calcium pumps in colon and gastric cancer cells. Induction of SERCA3 expression during differentiation. J. Biol. Chem. 2002, 277, 26310–26320. [Google Scholar]
- Lipkin, M.; Reddy, B.; Newmark, H.; Lamprecht, S.A. Dietary factors in human colorectal cancer. Annu. Rev. Nutr. 1999, 19, 545–586. [Google Scholar] [CrossRef]
- Aune, D.; Chan, D.S.; Lau, R.; Vieira, R.; Greenwood, D.C.; Kampman, E.; Norat, T. Dietary fibre, whole grains, and risk of colorectal cancer: Systematic review and dose-response meta-analysis of prospective studies. BMJ 2011, 343, d6617. [Google Scholar]
- Artursson, P. Epithelial transport of drugs in cell culture. I: A model for studying the passive diffusion of drugs over intestinal absorptive (Caco-2) cells. J. Pharm. Sci. 1990, 79, 476–482. [Google Scholar] [CrossRef]
- Reis-Filho, J.S.; Simpson, P.T.; Gale, T.; Lakhani, S.R. The molecular genetics of breast cancer: the contribution of comparative genomic hybridization. Pathol. Res. Pract. 2005, 201, 713–725. [Google Scholar]
- Simpson, P.T.; Gale, T.; Reis-Filho, J.S.; Jones, C.; Parry, S.; Sloane, J.P.; Handby, A.; Lee, A.H.; Humphreys, S.; Ellis, I.O.; Lakhani, S.R. Columnar cell lesions of the breast: the missing link in breast cancer progression? A morphological and molecular analysis. Am. J. Surg. Pathol. 2005, 29, 734–746. [Google Scholar] [CrossRef]
- Simpson, P.T.; Reis-Filho, J.S.; Gale, T.; Lakhani, S.R. Molecular evolution of breast cancer. J. Pathol. 2005, 205, 248–254. [Google Scholar] [CrossRef]
- Sørlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Thorsen, T.; Quist, H.; Matese, J.C.; Brown, P.O.; Botstein, D.; Eystein-Lønning, P.; Børresen-Dale, A.L. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar]
- Tavassoli, F.A.; Devilee, P. Pathology and Genetics of Tumors of the Breast and Female Genital Organs; IARC Press: Lyon, France, 2003; pp. 1–432. [Google Scholar]
- Perou, C.M.; Borresen-Dale, A.L. Systems biology and genomics of breast cancer. Cold Spring Harb. Perspect. Biol. 2011, 3, a003293. [Google Scholar] [CrossRef]
- Papp, B.; Brouland, J.-P. Altered endoplasmic reticulum calcium pump expression during breast tumorigenesis. Breast Cancer (Auckl) 2011, 5, 163–174. [Google Scholar]
- Smith-Garvin, J.E.; Koretzky, G.A.; Jordan, M.S. T cell activation. Annu. Rev. Immunol. 2009, 27, 591–619. [Google Scholar] [CrossRef]
- Launay, S.; Bobe, R.; Lacabaratz-Porret, C.; Bredoux, R.; Kovács, T.; Enouf, J.; Papp, B. Modulation of endoplasmic reticulum calcium pump expression during T lymphocyte activation. J. Biol. Chem. 1997, 272, 10746–10750. [Google Scholar]
- Penninga, L.; Møller, C.H.; Gustafsson, F.; Steinbrüchel, D.A.; Gluud, C. Tacrolimus versus cyclosporine as primary immunosuppression after heart transplantation: Systematic review with meta-analyses and trial sequential analyses of randomised trials. Eur. J. Clin. Pharmacol. 2010, 66, 1177–1187. [Google Scholar] [CrossRef]
- Bornkamm, G.W.; Hammerschmidt, W. Molecular virology of Epstein-Barr virus. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2001, 356, 437–459. [Google Scholar] [CrossRef]
- Rowe, D.T. Epstein-Barr virus immortalization and latency. Front. Biosci. 1999, 4, D346–D371. [Google Scholar] [CrossRef]
- Pattle, S.B.; Farrell, P.J. The role of Epstein-Barr virus in cancer. Expert Opin. Biol. Ther. 2006, 6, 1193–1205. [Google Scholar] [CrossRef]
- Thompson, M.P.; Kurzrock, R. pstein-Barr virus and cancer. Clin. Cancer Res. 2004, 10, 803–821. [Google Scholar] [CrossRef]
- Eliopoulos, A.G.; Young, L.S. LMP-1 structure and signal transduction. Semin. Cancer Biol. 2001, 11, 435–444. [Google Scholar] [CrossRef]
- Farrell, P.J. Signal transduction from the Epstein-Barr virus LMP-1 transforming protein. Trends Microbiol. 1998, 6, 175–177. [Google Scholar] [CrossRef]
- Farrell, P.J.; Cludts, I.; Stuhler, A. Epstein-Barr virus genes and cancer cells. Biomed. Pharmacother. 1997, 51, 258–267. [Google Scholar] [CrossRef]
- Klein, E.; Teramoto, N.; Gogolák, P.; Nagy, N.; Björkholm, M. LMP-1, the Epstein-Barr virus-encoded oncogene with a B cell activating mechanism similar to CD40. Immunol. Lett. 1999, 68, 147–154. [Google Scholar] [CrossRef]
- Wang, D.; Liebowitz, D.; Wang, F.; Gregory, C.; Rickinson, A.; Larson, R.; Springer, T.; Kieff, E. Epstein-Barr virus latent infection membrane protein alters the human B-lymphocyte phenotype: deletion of the amino terminus abolishes activity. J. Virol. 1988, 62, 4173–4184. [Google Scholar]
- Lenoir, G.M.; Vuillaume, M.; Bonnardel, C. The use of lymphomatous and lymphoblastoid cell lines in the study of Burkitt’s lymphoma. IARC Sci. Publ. 1985, 60, 309–318. [Google Scholar]
- Calender, A.; Billaud, M.; Aubry, J.-P.; Banchereau, J.; Vuillaume, M.; Lenoir, G.M. Epstein-Barr virus (EBV) induces expression of B-cell activation markers on in vitro infection of EBV-negative B-lymphoma cells. Proc. Natl. Acad. Sci. USA 1987, 84, 8060–8064. [Google Scholar] [CrossRef]
- Wang, F.; Tsang, S.F.; Kurilla, M.G.; Cohen, J.I.; Kieff, E. Epstein-Barr virus nuclear antigen 2 transactivates latent membrane protein LMP1. J. Virol. 1990, 64, 3407–3416. [Google Scholar]
- Dellis, O.; Arbabian, A.; Brouland, J.-P.; Kovács, T.; Rowe, M.; Chomienne, C.; Joab, I.; Papp, B. Modulation of B-cell endoplasmic reticulum calcium homeostasis by Epstein-Barr virus latent membrane protein-1. Mol. Cancer 2009, 8, 59. [Google Scholar] [CrossRef]
- Laude, A.J.; Simpson, A.W. Compartmentalized signalling: Ca2+ compartments, microdomains and the many facets of Ca2+ signalling. FEBS J. 2009, 276, 1800–1816. [Google Scholar] [CrossRef]
- Parekh, A.B. Ca2+ microdomains near plasma membrane Ca2+ channels: impact on cell function. J. Physiol. 2008, 586, 3043–3054. [Google Scholar] [CrossRef]
- Rizzuto, R.; Pozzan, T. Microdomains of intracellular Ca2+: Molecular determinants and functional consequences. Physiol. Rev. 2006, 86, 369–408. [Google Scholar] [CrossRef]
- Chandrasekera, C.P.; Lytton, J. Inhibition of human SERCA3 by PL/IM430. Molecular analysis of the interaction. J. Biol. Chem. 2003, 278, 12482–12488. [Google Scholar] [CrossRef]
- Hack, N.; Authi, K.S.; Crawford, N. Introduction of antibody (PL/IM 430) to a 100 kDa protein into permeabilised platelets inhibits intracellular sequestration of Ca2+. Biosci. Rep. 1988, 8, 379–388. [Google Scholar] [CrossRef]
- Quintana, A.; Pasche, M.; Junker, C.; Al-Ansary, D.; Rieger, H.; Kummerow, C.; Nuñez, L.; Villalobos, C.; Meraner, P.; Becherer, U.; Rettig, J.; Niemeyer, B.A.; Hoth, M. Calcium microdomains at the immunological synapse: How ORAI channels, mitochondria and calcium pumps generate local calcium signals for efficient T-cell activation. EMBO J. 2011, 30, 3895–3912. [Google Scholar] [CrossRef]
- Launay, S.; Giannì, M.; Diomede, L.; Machesky, L.M.; Enouf, J.; Papp, B. Enhancement of ATRA-induced cell differentiation by inhibition of calcium accumulation into the endoplasmic reticulum: Cross-talk between RAR alpha and calcium-dependent signaling. Blood 2003, 101, 3220–3228. [Google Scholar] [CrossRef]
- Apáti, Á.; Jánossy, J.; Brózik, A.; Bauer, P.I.; Magócsi, M. Calcium induces cell survival and proliferation through the activation of the MAPK pathway in a human hormone-dependent leukemia cell line, TF-1. J. Biol. Chem. 2003, 278, 9235–9243. [Google Scholar]
- Papp, B.; Byrn, R.A. Stimulation of HIV expression by intracellular calcium pump inhibition. J. Biol. Chem. 1995, 270, 10278–10283. [Google Scholar] [CrossRef]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef]
- Abell, E.; Ahrends, R.; Bandara, S.; Park, B.O.; Teruel, M.N. Parallel adaptive feedback enhances reliability of the Ca2+ signaling system. Proc. Natl. Acad. Sci. USA 2011, 108, 14485–14490. [Google Scholar]
- Tsai, T.Y.; Choi, Y.S.; Ma, W.; Pomerening, J.R.; Tang, C.; Ferrell, J.E, Jr. Robust, tunable biological oscillations from interlinked positive and negative feedback loops. Science 2008, 321, 126–129. [Google Scholar] [CrossRef]
- Strehler, E.E.; Caride, A.J.; Filoteo, A.G.; Xiong, Y.; Penniston, J.T.; Enyedi, Á. Plasma membrane Ca2+ ATPases as dynamic regulators of cellular calcium handling. Ann. NY Acad. Sci. 2007, 1099, 226–236. [Google Scholar] [CrossRef]
- Guerrero-Hernandez, A.; Dagnino-Acosta, A.; Verkhratsky, A. An intelligent sarco-endoplasmic reticulum Ca2+ store: Release and leak channels have differential access to a concealed Ca2+ pool. Cell Calcium 2010, 48, 143–149. [Google Scholar] [CrossRef]
- Godic, A.; Strazisar, M.; Zupan, A.; Korosec, B.; Kansky, A.; Glavac, D. Darier disease in Slovenia: Spectrum of ATP2A2 mutations and relation to patients' phenotypes. Eur. J. Dermatol. 2010, 20, 271–275. [Google Scholar]
- Sakuntabhai, A.; Ruiz-Perez, V.; Carter, S.; Jacobsen, N.; Burge, S.; Monk, S.; Smith, M.; Munro, C.S.; O'Donovan, M.; Craddock, N.; Kucherlapati, R.; Rees, J.L.; Owen, M.; Lathrop, G.M.; Monaco, A.P.; Strachan, T.; Hovnanian, A. Mutations in ATP2A2, encoding a Ca2+ pump, cause Darier disease. Nat. Genet. 1999, 21, 271–277. [Google Scholar] [CrossRef]
- Hakii, H.; Fujiki, H.; Suganuma, M.; Nakayasu, M.; Tahira, T.; Sugimura, T.; Scheuer, P.J.; Christensen, S.B. Thapsigargin, a histamine secretagogue, is a non-12-O-tetradecanoylphorbol-13-acetate (TPA) type tumor promoter in two-stage mouse skin carcinogenesis. J. Cancer Res. Clin. Oncol. 1986, 111, 177–181. [Google Scholar] [CrossRef]
- Sakai, A.; Teshima, R. 2,5-di-tert-butyl-1,4-hydroquinone enhances cell transformation accompanied by an increase in intracellular free calcium ion concentration. Cancer Lett. 2001, 168, 183–190. [Google Scholar] [CrossRef]
- Korošec, B.; Glavac, D.; Volavsek, M.; Ravnik-Glavac, M. ATP2A3 gene is involved in cancer susceptibility. Cancer Genet. Cytogenet. 2009, 188, 88–94. [Google Scholar] [CrossRef]
- Korošec, B.; Glavac, D.; Volavsek, M.; Ravnik-Glavac, M. Alterations in genes encoding sarcoplasmic-endoplasmic reticulum Ca2+ pumps in association with head and neck squamous cell carcinoma. Cancer Genet. Cytogenet. 2008, 181, 112–118. [Google Scholar] [CrossRef]
- Korošec, B.; Glavac, D.; Rott, T.; Ravnik-Glavac, M. Alterations in the ATP2A2 gene in correlation with colon and lung cancer. Cancer Genet. Cytogenet. 2006, 171, 105–111. [Google Scholar] [CrossRef]
- Juska, A.; Jardin, I.; Rosado, J.A. Physical properties of two types of calcium stores and SERCAs in human platelets. Mol. Cell. Biochem. 2008, 311, 9–18. [Google Scholar] [CrossRef]
- Means, S.; Smith, A.J.; Shepherd, J.; Shadid, J.; Fowler, J.; Wojcikiewicz, R.J.; Mazel, T.; Smith, G.D.; Wilson, B.S. Reaction diffusion modeling of calcium dynamics with realistic ER geometry. Biophys. J. 2006, 91, 537–557. [Google Scholar]
- Aung, C.S.; Ye, W.; Plowman, G.; Peters, A.A.; Monteith, G.R.; Roberts-Thomson, S.J. Plasma membrane calcium ATPase 4 and the remodeling of calcium homeostasis in human colon cancer cells. Carcinogenesis 2009, 30, 1962–1969. [Google Scholar] [CrossRef]
- Ribiczey, P.; Tordai, A.; Andrikovics, H.; Filoteo, A.G.; Penniston, J.T.; Enouf, J.; Enyedi, Á.; Papp, B.; Kovács, T. Isoform-specific up-regulation of plasma membrane Ca2+ATPase expression during colon and gastric cancer cell differentiation. Cell Calcium 2007, 42, 590–605. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Papp, B.; Brouland, J.-P.; Arbabian, A.; Gélébart, P.; Kovács, T.; Bobe, R.; Enouf, J.; Varin-Blank, N.; Apáti, Á. Endoplasmic Reticulum Calcium Pumps and Cancer Cell Differentiation. Biomolecules 2012, 2, 165-186. https://doi.org/10.3390/biom2010165
Papp B, Brouland J-P, Arbabian A, Gélébart P, Kovács T, Bobe R, Enouf J, Varin-Blank N, Apáti Á. Endoplasmic Reticulum Calcium Pumps and Cancer Cell Differentiation. Biomolecules. 2012; 2(1):165-186. https://doi.org/10.3390/biom2010165
Chicago/Turabian StylePapp, Béla, Jean-Philippe Brouland, Atousa Arbabian, Pascal Gélébart, Tünde Kovács, Régis Bobe, Jocelyne Enouf, Nadine Varin-Blank, and Ágota Apáti. 2012. "Endoplasmic Reticulum Calcium Pumps and Cancer Cell Differentiation" Biomolecules 2, no. 1: 165-186. https://doi.org/10.3390/biom2010165