New Perspectives on Oxidized Genome Damage and Repair Inhibition by Pro-Oxidant Metals in Neurological Diseases

 ,
,  ,
, 
Abstract
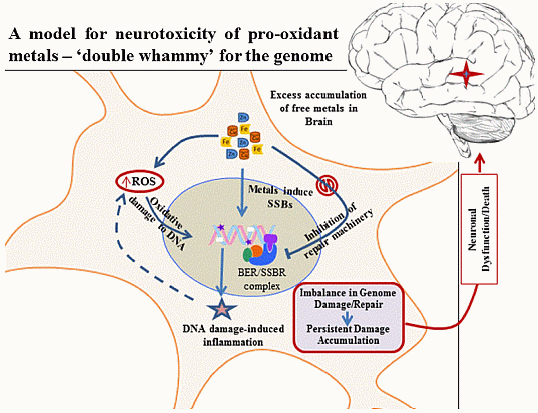
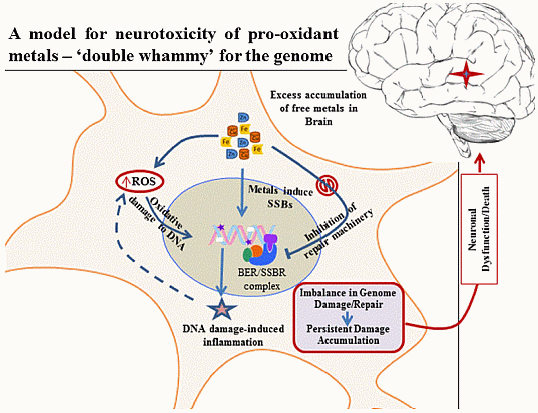
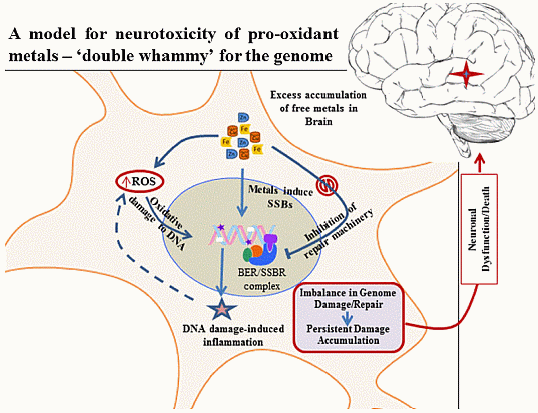
:
1. Introduction
2. Oxidative Insults and Repair in Human Genome
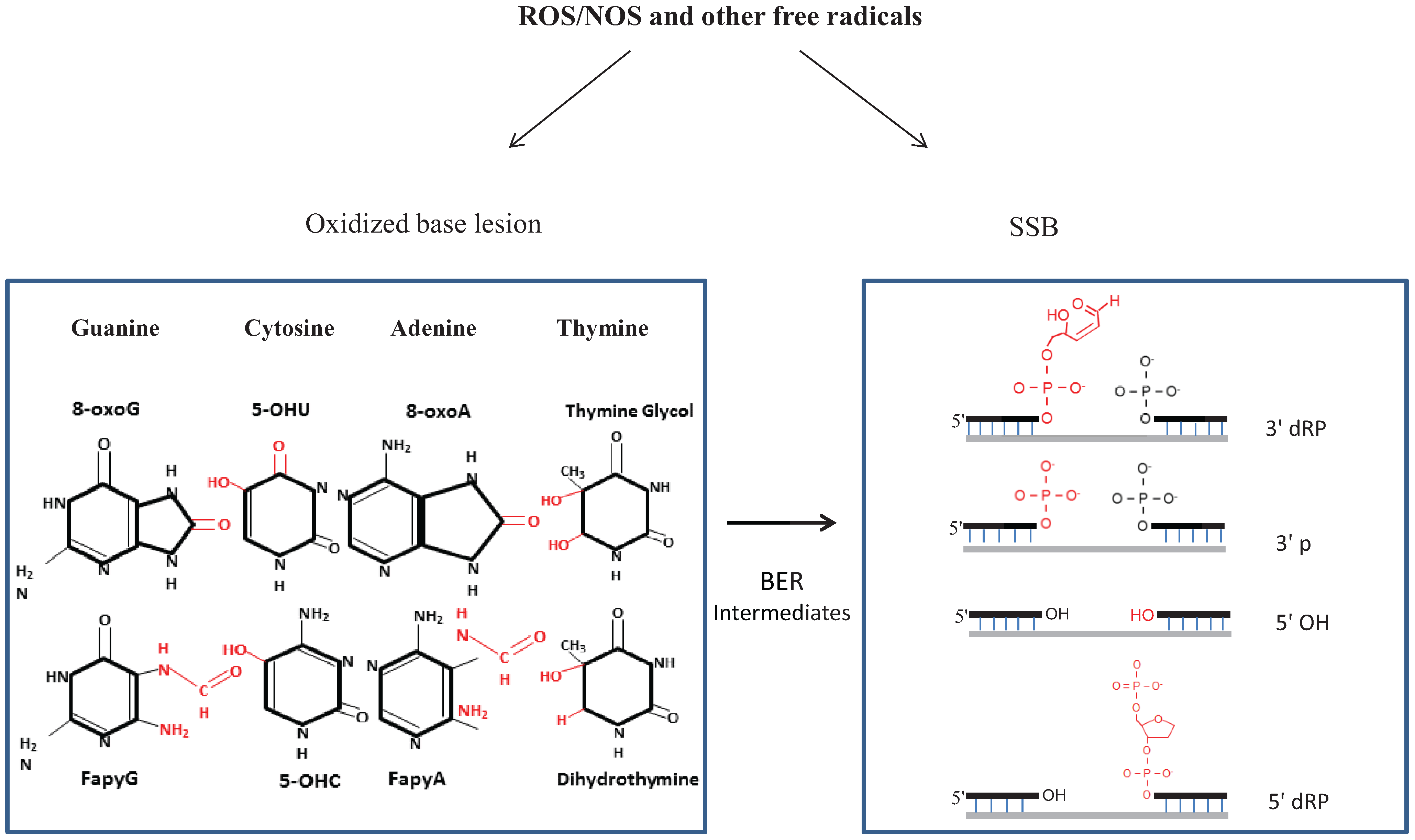
2.1. Types of Oxidative Damage in DNA

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Linked Metal Toxicity | References |
|---|---|---|
| Alzheimer’s Disease | Fe, Cu, Zn, Al | [30,31,32,33,34] |
| Parkinson’s Disease | Fe, Zn, Al, Cu, Mn | [1,10,34,35,36,37,38,39,40] |
| Huntington’s Disease | Cu, Fe | [41,42,43,44,45,46,47] |
| Wilson’s Disease | Cu, Fe | [48,49,50,51] |
| Amyotrophic Lateral Sclerosis | Fe, Cu | [52,53] |
| Friedreich’s Ataxia | Fe, Cu, Zn | [54,55,56] |
| Xeroderma Pigmentosum | Co, Cd, Ni | [57,58,59] |
| Cancer | Fe, Pb, Cd, Ni, Hg, Co | [20,60,61,62,63,64,65] |
2.2. Repair of Oxidative Genome Damage: Basic Mechanism(s) and Complexity
2.3. SSBR, a Variant of BER with Diverse End-Processing Reactions
2.4. Complexity of BER/SSBR Pathway
2.5. Mitochondrial (Mt) BER/SSBR
3. Current Understanding of BER/SSBR in the Central Nervous System (CNS)
4. Revisiting Metal Toxicity Diseases
4.1. Metal Accumulation in CNS Etiologically Linked to Neurological Diseases
4.1.1. Essential Transition Metals
4.1.2. Non-Essential Heavy Metals’ Impact on Human Health
4.2. Complex Nature of Metal Toxicity in Neurodegenerative Diseases
4.2.1. Environmental/Occupational Exposure versus Internal Redistribution
4.2.2. Transition Metals’ Charge-Dependent Changes with Disease Progression
4.3. Metal Toxicity as Homeostatic Imbalance: A New Perspective
5. Metals Induce Genome Damage, Both Directly and Circuitously
6. Inhibition of DNA Repair by Redox Metals: A Double Whammy
6.1. Pro-Oxidant Metals Inhibit BER Pathways
6.2. Mechanism of Repair Inhibition: Oxidation of Cysteines in NEIL DNA Glycosylases
6.3. Summary of Metal Toxicity Impact on Other DNA Repair Enzymes/Pathways
| Repair Protein Affected by Metal(s) | Repair Pathway | Inhibiting Metal | References |
|---|---|---|---|
| NEIL1 | BER | Fe, Cu | [9,175] |
| NEIL2 | BER | Fe, Cu | [9,175] |
| APE1 | BER/SSBR | Fe, Cd, Pb | [189] |
| PNKP | BER/SSBR/DSBR | Cd, Cu | [188] |
| FEN-1 | BER/SSBR/DSBR | Fe | [11] |
| LigIII | BER/SSBR/DSBR | Fe | [11] |
| MPG | BER | Cd, Ni, Zn | [186,187] |
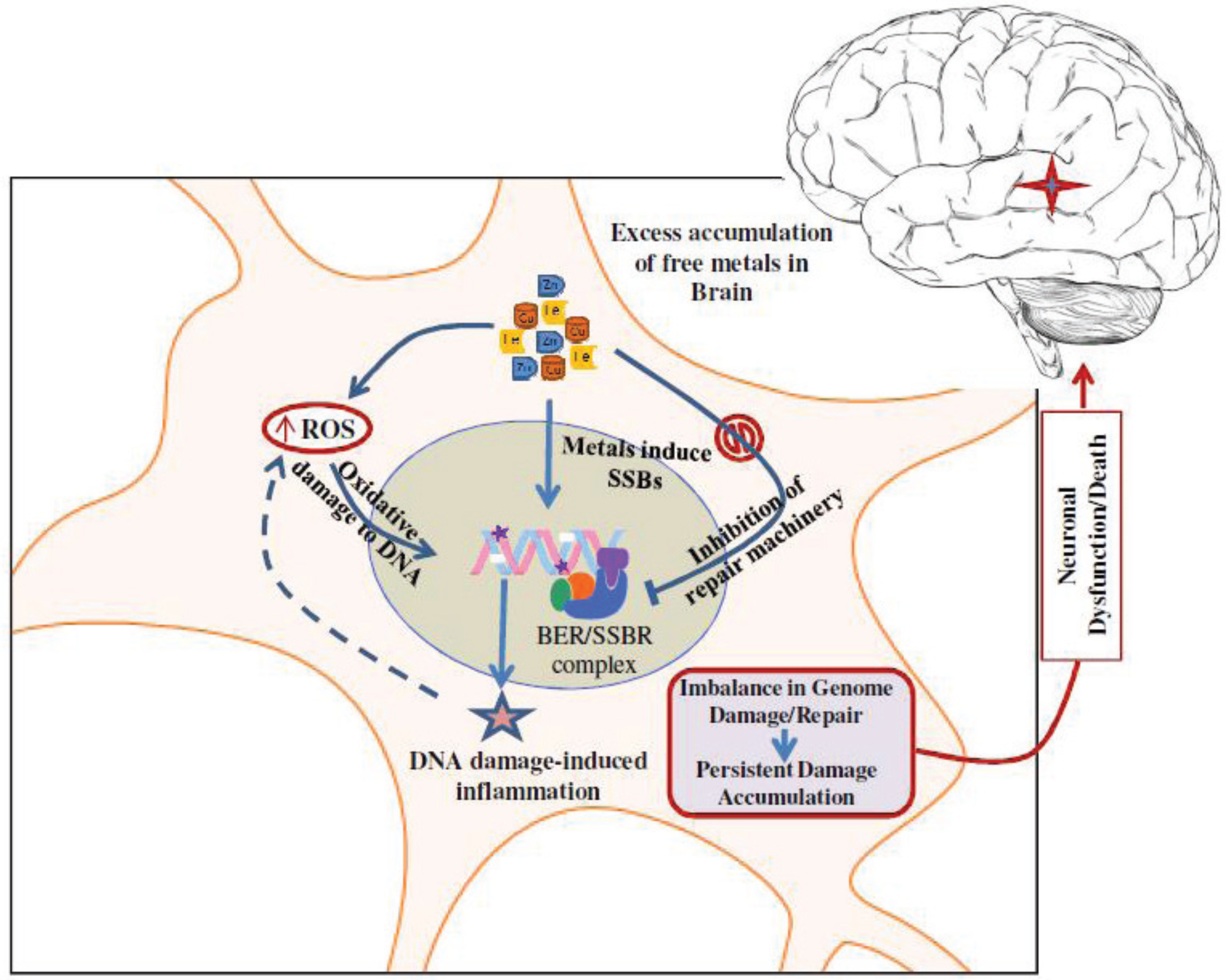
7. Conclusions and Future Perspectives
Molecular Understanding of the Complex, Multi-Targeted Nature of Metal Toxicity Is Important for Intervention Strategies
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hegde, M.L.; Shanmugavelu, P.; Vengamma, B.; Rao, T.S.; Menon, R.B.; Rao, R.V.; Rao, K.S. Serum trace element levels and the complexity of inter-element relations in patients with parkinson’s disease. J. Trace Elem. Med. Biol. 2004, 18, 163–171. [Google Scholar] [CrossRef]
- Faller, P.; Hureau, C.; Berthoumieu, O. Role of metal ions in the self-assembly of the Alzheimer’s amyloid-beta peptide. Inorg. Chem. 2013, 52, 12193–12206. [Google Scholar] [CrossRef]
- Stork, C.J.; Li, Y.V. Rising zinc: A significant cause of ischemic neuronal death in the CA1 region of rat hippocampus. J. Cereb. Blood Flow Metab. 2009, 29, 1399–1408. [Google Scholar] [CrossRef]
- Hanawalt, P.C. Emerging links between premature ageing and defective DNA repair. Mech. Ageing Dev. 2008, 129, 503–505. [Google Scholar] [CrossRef]
- Tchounwou, P.B.; Yedjou, C.G.; Sutton, D.J. Heavy metal toxicity and the environment. Mol. Clin. Environ. Toxicol. 2012, 101, 133–164. [Google Scholar] [CrossRef]
- Hartmann, A.; Speit, G. Effect of arsenic and cadmium on the persistence of mutagen-induced DNA lesions in human cells. Environ. Mol. Mutagen. 1996, 27, 98–104. [Google Scholar] [CrossRef]
- Hegde, M.L.; Bharathi, P.; Suram, A.; Venugopal, C.; Jagannathan, R.; Poddar, P.; Srinivas, P.; Sambamurti, K.; Rao, K.J.; Scancar, J.; et al. Challenges associated with metal chelation therapy in Alzheimer’s disease. J. Alzheimers Dis. 2009, 17, 457–468. [Google Scholar]
- Kim, S.; Cheon, H.S.; Kim, S.Y.; Juhnn, Y.S.; Kim, Y.Y. Cadmium induces neuronal cell death through reactive oxygen species activated by GADD153. BMC Cell Biol. 2013. [Google Scholar] [CrossRef]
- Hegde, M.L.; Hegde, P.M.; Holthauzen, L.M.; Hazra, T.K.; Rao, K.S.; Mitra, S. Specific inhibition of NEIL-initiated repair of oxidized base damage in human genome by copper and iron: Potential etiological linkage to neurodegenerative diseases. J. Biol. Chem. 2010, 285, 28812–28825. [Google Scholar] [CrossRef]
- Zatta, P.; Lucchini, R.; van Rensburg, S.J.; Taylor, A. The role of metals in neurodegenerative processes: Aluminum, manganese, and zinc. Brain Res. Bull. 2003, 62, 15–28. [Google Scholar] [CrossRef]
- Li, H.; Swiercz, R.; Englander, E.W. Elevated metals compromise repair of oxidative DNA damage via the base excision repair pathway: Implications of pathologic iron overload in the brain on integrity of neuronal DNA. J. Neurochem. 2009, 110, 1774–1783. [Google Scholar] [CrossRef]
- Breen, A.P.; Murphy, J.A. Reactions of oxyl radicals with DNA. Free Radic. Biol. Med. 1995, 18, 1033–1077. [Google Scholar] [CrossRef]
- Janssen, Y.M.; van Houten, B.; Borm, P.J.; Mossman, B.T. Cell and tissue responses to oxidative damage. Lab. Investig. 1993, 69, 261–274. [Google Scholar]
- Church, D.F.; Pryor, W.A. Free-radical chemistry of cigarette smoke and its toxicological implications. Environ. Health Perspect. 1985, 64, 111–126. [Google Scholar] [CrossRef]
- Comhair, S.A.; Thomassen, M.J.; Erzurum, S.C. Differential induction of extracellular glutathione peroxidase and nitric oxide synthase 2 in airways of healthy individuals exposed to 100% O2 or cigarette smoke. Am. J. Respir. Cell Mol. Biol. 2000, 23, 350–354. [Google Scholar] [CrossRef]
- Narayanan, P.K.; Goodwin, E.H.; Lehnert, B.E. Alpha particles initiate biological production of superoxide anions and hydrogen peroxide in human cells. Cancer Res. 1997, 57, 3963–3971. [Google Scholar]
- Friedberg, E.C.; Walker, G.C.; Siede, W.; Wood, R.D.; Schultz, R.A.; Ellenberger, T. DNA Repair and Mutagenesis, 2nd ed.; ASM Press: Washington, DC, USA, 2006. [Google Scholar]
- Pardo, B.; Gomez-Gonzalez, B.; Aguilera, A. DNA repair in mammalian cells: DNA double-strand break repair: How to fix a broken relationship. Cell. Mol. Life Sci. 2009, 66, 1039–1056. [Google Scholar] [CrossRef]
- Lloyd, R.V.; Hanna, P.M.; Mason, R.P. The origin of the hydroxyl radical oxygen in the fenton reaction. Free Radic. Biol. Med. 1997, 22, 885–888. [Google Scholar] [CrossRef]
- Toyokuni, S. Iron and carcinogenesis: From fenton reaction to target genes. Redox Rep. 2002, 7, 189–197. [Google Scholar] [CrossRef]
- Valko, M.; Izakovic, M.; Mazur, M.; Rhodes, C.J.; Telser, J. Role of oxygen radicals in DNA damage and cancer incidence. Mol. Cell. Biochem. 2004, 266, 37–56. [Google Scholar] [CrossRef]
- Mitra, S.; Izumi, T.; Boldogh, I.; Bhakat, K.K.; Hill, J.W.; Hazra, T.K. Choreography of oxidative damage repair in mammalian genomes. Free Radic. Biol. Med. 2002, 33, 15–28. [Google Scholar] [CrossRef]
- Hegde, M.L.; Hazra, T.K.; Mitra, S. Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res. 2008, 18, 27–47. [Google Scholar] [CrossRef]
- Lyras, L.; Cairns, N.J.; Jenner, A.; Jenner, P.; Halliwell, B. An assessment of oxidative damage to proteins, lipids, and DNA in brain from patients with Alzheimer’s disease. J. Neurochem. 1997, 68, 2061–2069. [Google Scholar]
- Halliwell, B. Antioxidant defence mechanisms: From the beginning to the end (of the beginning). Free Radic. Res. 1999, 31, 261–272. [Google Scholar] [CrossRef]
- Tsuzuki, K.; Sugiyama, M.; Haramaki, N. DNA single-strand breaks and cytotoxicity induced by chromate(VI), cadmium(II), and mercury(II) in hydrogen peroxide-resistant cell lines. Environ. Health Perspect. 1994, 102, 341–342. [Google Scholar] [CrossRef]
- Lai, H.; Singh, N.P. Magnetic-field-induced DNA strand breaks in brain cells of the rat. Environ. Health Perspect. 2004, 112, 687–694. [Google Scholar]
- Chen, Q.; Marsh, J.; Ames, B.; Mossman, B. Detection of 8-oxo-2'-deoxyguanosine, a marker of oxidative DNA damage, in culture medium from human mesothelial cells exposed to crocidolite asbestos. Carcinogenesis 1996, 17, 2525–2527. [Google Scholar] [CrossRef]
- Radak, Z.; Boldogh, I. 8-oxo-7,8-dihydroguanine: Links to gene expression, aging, and defense against oxidative stress. Free Radic. Biol. Med. 2010, 49, 587–596. [Google Scholar] [CrossRef]
- Bourassa, M.W.; Leskovjan, A.C.; Tappero, R.V.; Farquhar, E.R.; Colton, C.A.; van Nostrand, W.E.; Miller, L.M. Elevated copper in the amyloid plaques and iron in the cortex are observed in mouse models of Alzheimer’s disease that exhibit neurodegeneration. Biomed. Spectrosc. Imaging 2013, 2, 129–139. [Google Scholar]
- Tiiman, A.; Palumaa, P.; Tougu, V. The missing link in the amyloid cascade of Alzheimer’s disease—Metal ions. Neurochem. Int. 2013, 62, 367–378. [Google Scholar] [CrossRef]
- Lovell, M.A.; Robertson, J.D.; Teesdale, W.J.; Campbell, J.L.; Markesbery, W.R. Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurol. Sci. 1998, 158, 47–52. [Google Scholar] [CrossRef]
- Strong, M.J. Aluminum neurotoxicity: An experimental approach to the induction of neurofilamentous inclusions. J. Neurol. Sci. 1994, 124, 20–26. [Google Scholar] [CrossRef]
- Zecca, L.; Youdim, M.B.; Riederer, P.; Connor, J.R.; Crichton, R.R. Iron, brain ageing and neurodegenerative disorders. Nat. Rev. Neurosci. 2004, 5, 863–873. [Google Scholar] [CrossRef]
- Sofic, E.; Riederer, P.; Heinsen, H.; Beckmann, H.; Reynolds, G.P.; Hebenstreit, G.; Youdim, M.B. Increased iron (III) and total iron content in post mortem substantia nigra of parkinsonian brain. J. Neural. Transm. 1988, 74, 199–205. [Google Scholar] [CrossRef]
- Schipper, H.M. Astrocytes, brain aging, and neurodegeneration. Neurobiol. Aging 1996, 17, 467–480. [Google Scholar] [CrossRef]
- Forte, G.; Bocca, B.; Senofonte, O.; Petrucci, F.; Brusa, L.; Stanzione, P.; Zannino, S.; Violante, N.; Alimonti, A.; Sancesario, G. Trace and major elements in whole blood, serum, cerebrospinal fluid and urine of patients with Parkinson’s disease. J. Neural. Transm. 2004, 111, 1031–1040. [Google Scholar]
- Rasia, R.M.; Bertoncini, C.W.; Marsh, D.; Hoyer, W.; Cherny, D.; Zweckstetter, M.; Griesinger, C.; Jovin, T.M.; Fernandez, C.O. Structural characterization of copper(II) binding to alpha-synuclein: Insights into the bioinorganic chemistry of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2005, 102, 4294–4299. [Google Scholar] [CrossRef]
- Lucchini, R.G.; Martin, C.J.; Doney, B.C. From manganism to manganese-induced parkinsonism: A conceptual model based on the evolution of exposure. Neuromol. Med. 2009, 11, 311–321. [Google Scholar] [CrossRef]
- Milatovic, D.; Zaja-Milatovic, S.; Gupta, R.C.; Yu, Y.; Aschner, M. Oxidative damage and neurodegeneration in manganese-induced neurotoxicity. Toxicol. Appl. Pharmacol. 2009, 240, 219–225. [Google Scholar] [CrossRef]
- Fox, J.H.; Kama, J.A.; Lieberman, G.; Chopra, R.; Dorsey, K.; Chopra, V.; Volitakis, I.; Cherny, R.A.; Bush, A.I.; Hersch, S. Mechanisms of copper ion mediated Huntington’s disease progression. PLoS One 2007, 2, e334. [Google Scholar] [CrossRef]
- Xiao, B.G.; Link, H. Immune regulation within the central nervous system. J. Neurol. Sci. 1998, 157, 1–12. [Google Scholar] [CrossRef]
- Dexter, D.T.; Jenner, P.; Schapira, A.H.; Marsden, C.D. Alterations in levels of iron, ferritin, and other trace metals in neurodegenerative diseases affecting the basal ganglia. The royal kings and queens Parkinson’s disease research group. Ann. Neurol. 1992, 32, S94–S100. [Google Scholar] [CrossRef]
- Boll, M.C.; Alcaraz-Zubeldia, M.; Montes, S.; Rios, C. Free copper, ferroxidase and SOD1 activities, lipid peroxidation and NOx content in the CS. A different marker profile in four neurodegenerative diseases. Neurochem. Res. 2008, 33, 1717–1723. [Google Scholar] [CrossRef]
- Loeffler, D.A.; LeWitt, P.A.; Juneau, P.L.; Sima, A.A.; Nguyen, H.U.; DeMaggio, A.J.; Brickman, C.M.; Brewer, G.J.; Dick, R.D.; Troyer, M.D.; et al. Increased regional brain concentrations of ceruloplasmin in neurodegenerative disorders. Brain Res. 1996, 738, 265–274. [Google Scholar] [CrossRef]
- Simmons, D.A.; Casale, M.; Alcon, B.; Pham, N.; Narayan, N.; Lynch, G. Ferritin accumulation in dystrophic microglia is an early event in the development of Huntington’s disease. Glia 2007, 55, 1074–1084. [Google Scholar] [CrossRef]
- Bartzokis, G.; Cummings, J.; Perlman, S.; Hance, D.B.; Mintz, J. Increased basal ganglia iron levels in Huntington disease. Arch. Neurol. 1999, 56, 569–574. [Google Scholar] [CrossRef]
- Fatemi, N.; Sarkar, B. Molecular mechanism of copper transport in wilson disease. Environ. Health Perspect. 2002, 110, 695–698. [Google Scholar] [CrossRef]
- Litwin, T.; Gromadzka, G.; Szpak, G.M.; Jablonka-Salach, K.; Bulska, E.; Czlonkowska, A. Brain metal accumulation in Wilson’s disease. J. Neurol. Sci. 2013, 329, 55–58. [Google Scholar] [CrossRef]
- DiDonato, M.; Hsu, H.F.; Narindrasorasak, S.; Que, L., Jr.; Sarkar, B. Copper-induced conformational changes in the N-terminal domain of the Wilson disease copper-transporting ATPase. Biochemistry 2000, 39, 1890–1896. [Google Scholar] [CrossRef]
- Kanwar, P.; Kowdley, K.V. Metal storage disorders: Wilson disease and hemochromatosis. Med. Clin. North Am. 2014, 98, 87–102. [Google Scholar] [CrossRef]
- Jeong, S.Y.; Rathore, K.I.; Schulz, K.; Ponka, P.; Arosio, P.; David, S. Dysregulation of iron homeostasis in the CNS contributes to disease progression in a mouse model of amyotrophic lateral sclerosis. J. Neurosci. 2009, 29, 610–619. [Google Scholar] [CrossRef]
- Kasarskis, E.J.; Tandon, L.; Lovell, M.A.; Ehmann, W.D. Aluminum, calcium, and iron in the spinal cord of patients with sporadic amyotrophic lateral sclerosis using laser microprobe mass spectroscopy: A preliminary study. J. Neurol. Sci. 1995, 130, 203–208. [Google Scholar] [CrossRef]
- Koeppen, A.H.; Ramirez, R.L.; Yu, D.; Collins, S.E.; Qian, J.; Parsons, P.J.; Yang, K.X.; Chen, Z.; Mazurkiewicz, J.E.; Feustel, P.J. Friedreich’s ataxia causes redistribution of iron, copper, and zinc in the dentate nucleus. Cerebellum 2012, 11, 845–860. [Google Scholar] [CrossRef]
- Hu, K.H.; Friede, R.L. Topographic determination of zinc in human brain by atomic absorption spectrophotometry. J. Neurochem. 1968, 15, 677–685. [Google Scholar] [CrossRef]
- Cossee, M.; Puccio, H.; Gansmuller, A.; Koutnikova, H.; Dierich, A.; LeMeur, M.; Fischbeck, K.; Dolle, P.; Koenig, M. Inactivation of the friedreich ataxia mouse gene leads to early embryonic lethality without iron accumulation. Hum. Mol. Genet. 2000, 9, 1219–1226. [Google Scholar] [CrossRef]
- Kopera, E.; Schwerdtle, T.; Hartwig, A.; Bal, W. Co(II) and Cd(II) substitute for Zn(II) in the zinc finger derived from the DNA repair protein XPA, demonstrating a variety of potential mechanisms of toxicity. Chem. Res. Toxicol. 2004, 17, 1452–1458. [Google Scholar] [CrossRef]
- Hartwig, A.; Schwerdtle, T.; Bal, W. Biophysical analysis of the interaction of toxic metal ions and oxidants with the zinc finger domain of XPA. Methods Mol. Biol. 2010, 649, 399–410. [Google Scholar] [CrossRef]
- Bal, W.; Schwerdtle, T.; Hartwig, A. Mechanism of NICKEl assault on the zinc finger of DNA repair protein XPA. Chem. Res. Toxicol. 2003, 16, 242–248. [Google Scholar] [CrossRef]
- Zhou, Z.; Wang, C.; Liu, H.; Huang, Q.; Wang, M.; Lei, Y. Cadmium induced cell apoptosis, DNA damage, decreased DNA repair capacity, and genomic instability during malignant transformation of human bronchial epithelial cells. Int. J. Med. Sci. 2013, 10, 1485–1496. [Google Scholar] [CrossRef]
- Smith, E.B. Primary cancer of the lung in women. J. Natl. Med. Assoc. 1989, 81, 945–948. [Google Scholar]
- Lam, T.V.; Agovino, P.; Niu, X.; Roche, L. Linkage study of cancer risk among lead-exposed workers in new jersey. Sci. Total Environ. 2007, 372, 455–462. [Google Scholar] [CrossRef]
- Beveridge, R.; Pintos, J.; Parent, M.E.; Asselin, J.; Siemiatycki, J. Lung cancer risk associated with occupational exposure to NICKEl, chromium VI, and cadmium in two population-based case-control studies in montreal. Am. J. Ind. Med. 2010, 53, 476–485. [Google Scholar]
- Koedrith, P.; Kim, H.; Weon, J.I.; Seo, Y.R. Toxicogenomic approaches for understanding molecular mechanisms of heavy metal mutagenicity and carcinogenicity. Int. J. Hyg. Environ. Health 2013, 216, 587–598. [Google Scholar] [CrossRef]
- Aquino, N.B.; Sevigny, M.B.; Sabangan, J.; Louie, M.C. The role of cadmium and NICKEl in estrogen receptor signaling and breast cancer: Metalloestrogens or not? J. Environ. Sci. Health C 2012, 30, 189–224. [Google Scholar] [CrossRef]
- McCullough, A.K.; Dodson, M.L.; Lloyd, R.S. Initiation of base excision repair: Glycosylase mechanisms and structures. Annu. Rev. Biochem. 1999, 68, 255–285. [Google Scholar] [CrossRef]
- Bandaru, V.; Sunkara, S.; Wallace, S.S.; Bond, J.P. A novel human DNA glycosylase that removes oxidative DNA damage and is homologous to Escherichia coli endonuclease VIII. DNA Repair 2002, 1, 517–529. [Google Scholar] [CrossRef]
- Hazra, T.K.; Izumi, T.; Boldogh, I.; Imhoff, B.; Kow, Y.W.; Jaruga, P.; Dizdaroglu, M.; Mitra, S. Identification and characterization of a human DNA glycosylase for repair of modified bases in oxidatively damaged DNA. Proc. Natl. Acad. Sci. USA 2002, 99, 3523–3528. [Google Scholar]
- Ellenberger, T.; Tomkinson, A.E. Eukaryotic DNA ligases: Structural and functional insights. Annu. Rev. Biochem. 2008, 77, 313–338. [Google Scholar] [CrossRef]
- Prakash, A.; Doublie, S.; Wallace, S.S. The Fpg/Nei family of DNA glycosylases: Substrates, structures, and search for damage. Prog. Mol. Biol. Transl. Sci. 2012, 110, 71–91. [Google Scholar] [CrossRef]
- Hazra, T.K.; Kow, Y.W.; Hatahet, Z.; Imhoff, B.; Boldogh, I.; Mokkapati, S.K.; Mitra, S.; Izumi, T. Identification and characterization of a novel human DNA glycosylase for repair of cytosine-derived lesions. J. Biol. Chem. 2002, 277, 30417–30420. [Google Scholar]
- Liu, M.; Bandaru, V.; Bond, J.P.; Jaruga, P.; Zhao, X.; Christov, P.P.; Burrows, C.J.; Rizzo, C.J.; Dizdaroglu, M.; Wallace, S.S. The mouse ortholog of NEIL3 is a functional DNA glycosylase in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 4925–4930. [Google Scholar] [CrossRef]
- Wallace, S.S.; Bandaru, V.; Kathe, S.D.; Bond, J.P. The enigma of endonuclease VIII. DNA Repair 2003, 2, 441–453. [Google Scholar] [CrossRef]
- Wallace, S.S. Base excision repair: A critical player in many games. DNA Repair 2014, 19, 14–26. [Google Scholar] [CrossRef]
- Wiederhold, L.; Leppard, J.B.; Kedar, P.; Karimi-Busheri, F.; Rasouli-Nia, A.; Weinfeld, M.; Tomkinson, A.E.; Izumi, T.; Prasad, R.; Wilson, S.H.; et al. AP endonuclease-independent DNA base excision repair in human cells. Mol. Cell 2004, 15, 209–220. [Google Scholar] [CrossRef]
- Sobol, R.W.; Prasad, R.; Evenski, A.; Baker, A.; Yang, X.P.; Horton, J.K.; Wilson, S.H. The lyase activity of the DNA repair protein beta-polymerase protects from DNA-damage-induced cytotoxicity. Nature 2000, 405, 807–810. [Google Scholar] [CrossRef]
- Hegde, M.L.; Hegde, P.M.; Bellot, L.J.; Mandal, S.M.; Hazra, T.K.; Li, G.M.; Boldogh, I.; Tomkinson, A.E.; Mitra, S. Prereplicative repair of oxidized bases in the human genome is mediated by neil1 DNA glycosylase together with replication proteins. Proc. Natl. Acad. Sci. USA 2013, 110, E3090–E3099. [Google Scholar] [CrossRef]
- Hegde, M.L.; Theriot, C.A.; Das, A.; Hegde, P.M.; Guo, Z.; Gary, R.K.; Hazra, T.K.; Shen, B.; Mitra, S. Physical and functional interaction between human oxidized base-specific DNA glycosylase NEIL1 and flap endonuclease 1. J. Biol. Chem. 2008, 283, 27028–27037. [Google Scholar] [CrossRef]
- Liu, Y.; Kao, H.I.; Bambara, R.A. Flap endonuclease 1: A central component of DNA metabolism. Annu. Rev. Biochem. 2004, 73, 589–615. [Google Scholar] [CrossRef]
- Tomkinson, A.E.; Chen, L.; Dong, Z.; Leppard, J.B.; Levin, D.S.; Mackey, Z.B.; Motycka, T.A. Completion of base excision repair by mammalian DNA ligases. Prog. Nucleic Acid Res. Mol. Biol. 2001, 68, 151–164. [Google Scholar] [CrossRef]
- Simsek, D.; Furda, A.; Gao, Y.; Artus, J.; Brunet, E.; Hadjantonakis, A.K.; van Houten, B.; Shuman, S.; McKinnon, P.J.; Jasin, M. Crucial role for DNA ligase III in mitochondria but not in XRCC1-dependent repair. Nature 2011, 471, 245–248. [Google Scholar] [CrossRef]
- Montecucco, A.; Biamonti, G.; Savini, E.; Focher, F.; Spadari, S.; Ciarrocchi, G. DNA ligase I gene expression during differentiation and cell proliferation. Nucleic Acids Res. 1992, 20, 6209–6214. [Google Scholar] [CrossRef]
- Dou, H.; Theriot, C.A.; Das, A.; Hegde, M.L.; Matsumoto, Y.; Boldogh, I.; Hazra, T.K.; Bhakat, K.K.; Mitra, S. Interaction of the human DNA glycosylase NEIL1 with proliferating cell nuclear antigen. The potential for replication-associated repair of oxidized bases in mammalian genomes. J. Biol. Chem. 2008, 283, 3130–3140. [Google Scholar] [CrossRef]
- Yang, S.W.; Burgin, A.B., Jr.; Huizenga, B.N.; Robertson, C.A.; Yao, K.C.; Nash, H.A. A eukaryotic enzyme that can disjoin dead-end covalent complexes between DNA and type I topoisomerases. Proc. Natl. Acad. Sci. USA 1996, 93, 11534–11539. [Google Scholar] [CrossRef]
- Pouliot, J.J.; Yao, K.C.; Robertson, C.A.; Nash, H.A. Yeast gene for a Tyr-DNA phosphodiesterase that repairs topoisomerase I complexes. Science 1999, 286, 552–555. [Google Scholar] [CrossRef]
- El-Khamisy, S.F.; Saifi, G.M.; Weinfeld, M.; Johansson, F.; Helleday, T.; Lupski, J.R.; Caldecott, K.W. Defective DNA single-strand break repair in spinocerebellar ataxia with axonal neuropathy-1. Nature 2005, 434, 108–113. [Google Scholar] [CrossRef]
- Ahel, I.; Rass, U.; El-Khamisy, S.F.; Katyal, S.; Clements, P.M.; McKinnon, P.J.; Caldecott, K.W.; West, S.C. The neurodegenerative disease protein aprataxin resolves abortive DNA ligation intermediates. Nature 2006, 443, 713–716. [Google Scholar] [CrossRef]
- Rass, U.; Ahel, I.; West, S.C. Actions of aprataxin in multiple DNA repair pathways. J. Biol. Chem. 2007, 282, 9469–9474. [Google Scholar] [CrossRef]
- Banerjee, D.; Mandal, S.M.; Das, A.; Hegde, M.L.; Das, S.; Bhakat, K.K.; Boldogh, I.; Sarkar, P.S.; Mitra, S.; Hazra, T.K. Preferential repair of oxidized base damage in the transcribed genes of mammalian cells. J. Biol. Chem. 2011, 286, 6006–6016. [Google Scholar] [CrossRef]
- Schreiber, V.; Dantzer, F.; Ame, J.C.; de Murcia, G. Poly(ADP-ribose): Novel functions for an old molecule. Nat. Rev. Mol. Cell Biol. 2006, 7, 517–528. [Google Scholar] [CrossRef]
- Caldecott, K.W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 2008, 9, 619–631. [Google Scholar]
- Dianova, I.I.; Sleeth, K.M.; Allinson, S.L.; Parsons, J.L.; Breslin, C.; Caldecott, K.W.; Dianov, G.L. XRCC1-DNA polymerase beta interaction is required for efficient base excision repair. Nucleic Acids Res. 2004, 32, 2550–2555. [Google Scholar] [CrossRef]
- El-Khamisy, S.F.; Hartsuiker, E.; Caldecott, K.W. TDP1 facilitates repair of ionizing radiation-induced DNA single-strand breaks. DNA Repair 2007, 6, 1485–1495. [Google Scholar] [CrossRef]
- Vidal, A.E.; Boiteux, S.; Hickson, I.D.; Radicella, J.P. XRCC1 coordinates the initial and late stages of DNA abasic site repair through protein-protein interactions. EMBO J. 2001, 20, 6530–6539. [Google Scholar] [CrossRef]
- Chattopadhyay, R.; Das, S.; Maiti, A.K.; Boldogh, I.; Xie, J.; Hazra, T.K.; Kohno, K.; Mitra, S.; Bhakat, K.K. Regulatory role of human AP-endonuclease (APE1/Ref-1) in YB-1-mediated activation of the multidrug resistance gene MDR1. Mol. Cell. Biol. 2008, 28, 7066–7080. [Google Scholar] [CrossRef]
- Marenstein, D.R.; Ocampo, M.T.; Chan, M.K.; Altamirano, A.; Basu, A.K.; Boorstein, R.J.; Cunningham, R.P.; Teebor, G.W. Stimulation of human endonuclease III by Y box-binding protein 1 (DNA-binding protein β). Interaction between a base excision repair enzyme and a transcription factor. J. Biol. Chem. 2001, 276, 21242–21249. [Google Scholar] [CrossRef]
- Hegde, M.L.; Banerjee, S.; Hegde, P.M.; Bellot, L.J.; Hazra, T.K.; Boldogh, I.; Mitra, S. Enhancement of NEIL1 protein-initiated oxidized DNA base excision repair by heterogeneous nuclear ribonucleoprotein U (hnRNP-U) via direct interaction. J. Biol. Chem. 2012, 287, 34202–34211. [Google Scholar]
- Bhakat, K.K.; Hazra, T.K.; Mitra, S. Acetylation of the human DNA glycosylase NEIL2 and inhibition of its activity. Nucleic Acids Res. 2004, 32, 3033–3039. [Google Scholar] [CrossRef]
- Bhakat, K.K.; Mokkapati, S.K.; Boldogh, I.; Hazra, T.K.; Mitra, S. Acetylation of human 8-oxoguanine-DNA glycosylase by p300 and its role in 8-oxoguanine repair in vivo. Mol. Cell. Biol. 2006, 26, 1654–1665. [Google Scholar] [CrossRef]
- Sengupta, S.; Mantha, A.K.; Mitra, S.; Bhakat, K.K. Human AP endonuclease (APE1/Ref-1) and its acetylation regulate YB-1-p300 recruitment and RNA polymerase II loading in the drug-induced activation of multidrug resistance gene MDR1. Oncogene 2010, 30, 482–493. [Google Scholar]
- Tini, M.; Benecke, A.; Um, S.J.; Torchia, J.; Evans, R.M.; Chambon, P. Association of CBP/p300 acetylase and thymine DNA glycosylase links DNA repair and transcription. Mol. Cell 2002, 9, 265–277. [Google Scholar] [CrossRef]
- Yacoub, A.; Kelley, M.R.; Deutsch, W.A. The DNA repair activity of human redox/repair protein APE/Ref-1 is inactivated by phosphorylation. Cancer Res. 1997, 57, 5457–5459. [Google Scholar]
- Fritz, G.; Kaina, B. Phosphorylation of the DNA repair protein APE/Ref-1 by CKII affects redox regulation of AP-1. Oncogene 1999, 18, 1033–1040. [Google Scholar] [CrossRef]
- Huang, E.; Qu, D.; Zhang, Y.; Venderova, K.; Haque, M.E.; Rousseaux, M.W.; Slack, R.S.; Woulfe, J.M.; Park, D.S. The role of CDK5-mediated apurinic/apyrimidinic endonuclease 1 phosphorylation in neuronal death. Nat. Cell Biol. 2010, 12, 563–571. [Google Scholar] [CrossRef]
- Parsons, J.L.; Tait, P.S.; Finch, D.; Dianova, I.I.; Edelmann, M.J.; Khoronenkova, S.V.; Kessler, B.M.; Sharma, R.A.; McKenna, W.G.; Dianov, G.L. Ubiquitin ligase ARF-BP1/mule modulates base excision repair. EMBO J. 2009, 28, 3207–3215. [Google Scholar] [CrossRef]
- Parsons, J.L.; Dianova, I.I.; Finch, D.; Tait, P.S.; Strom, C.E.; Helleday, T.; Dianov, G.L. XRCC1 phosphorylation by CK2 is required for its stability and efficient DNA repair. DNA Repair 2010, 9, 835–841. [Google Scholar] [CrossRef]
- Wang, T.; Simbulan-Rosenthal, C.M.; Smulson, M.E.; Chock, P.B.; Yang, D.C. Polyubiquitylation of PARP-1 through ubiquitin K48 is modulated by activated DNA, NAD+, and dipeptides. J. Cell. Biochem. 2008, 104, 318–328. [Google Scholar] [CrossRef]
- Takahashi, H.; Hatakeyama, S.; Saitoh, H.; Nakayama, K.I. Noncovalent SUMO-1 binding activity of thymine DNA glycosylase (TDG) is required for its SUMO-1 modification and colocalization with the promyelocytic leukemia protein. J. Biol. Chem. 2005, 280, 5611–5621. [Google Scholar]
- Hardeland, U.; Steinacher, R.; Jiricny, J.; Schar, P. Modification of the human thymine-DNA glycosylase by ubiquitin-like proteins facilitates enzymatic turnover. EMBO J. 2002, 21, 1456–1464. [Google Scholar] [CrossRef]
- Steinacher, R.; Schar, P. Functionality of human thymine DNA glycosylase requires SUMO-regulated changes in protein conformation. Curr. Biol. 2005, 15, 616–623. [Google Scholar] [CrossRef]
- El-Andaloussi, N.; Valovka, T.; Toueille, M.; Hassa, P.O.; Gehrig, P.; Covic, M.; Hubscher, U.; Hottiger, M.O. Methylation of DNA polymerase beta by protein arginine methyltransferase 1 regulates its binding to proliferating cell nuclear antigen. FASEB J. 2007, 21, 26–34. [Google Scholar]
- Kedar, P.S.; Kim, S.J.; Robertson, A.; Hou, E.; Prasad, R.; Horton, J.K.; Wilson, S.H. Direct interaction between mammalian DNA polymerase beta and proliferating cell nuclear antigen. J. Biol. Chem. 2002, 277, 31115–31123. [Google Scholar]
- Hakme, A.; Wong, H.K.; Dantzer, F.; Schreiber, V. The expanding field of poly(ADP-ribosyl)ation reactions. “Protein modifications: Beyond the usual suspects” review series. EMBO Rep. 2008, 9, 1094–1100. [Google Scholar] [CrossRef]
- El-Khamisy, S.F.; Masutani, M.; Suzuki, H.; Caldecott, K.W. A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res. 2003, 31, 5526–5533. [Google Scholar] [CrossRef]
- Huang, J.Y.; Chen, W.H.; Chang, Y.L.; Wang, H.T.; Chuang, W.T.; Lee, S.C. Modulation of nucleosome-binding activity of fact by poly(ADP-ribosyl)ation. Nucleic Acids Res. 2006, 34, 2398–2407. [Google Scholar] [CrossRef]
- Chang, D.Y.; Shi, G.; Durand-Dubief, M.; Ekwall, K.; Lu, A.L. The role of MutY homolog (Myh1) in controlling the histone deacetylase Hst4 in the fission yeast Schizosaccharomyces pombe. J. Mol. Biol. 2011, 405, 653–665. [Google Scholar] [CrossRef]
- Hegde, M.L.; Izumi, T.; Mitra, S. Oxidized base damage and single-strand break repair in mammalian genomes: Role of disordered regions and posttranslational modifications in early enzymes. Prog. Mol. Biol. Transl. Sci. 2012, 110, 123–153. [Google Scholar] [CrossRef]
- Hegde, M.L.; Hazra, T.K.; Mitra, S. Functions of disordered regions in mammalian early base excision repair proteins. Cell. Mol. Life Sci. 2010, 67, 3573–3587. [Google Scholar] [CrossRef]
- Haynes, C.; Oldfield, C.J.; Ji, F.; Klitgord, N.; Cusick, M.E.; Radivojac, P.; Uversky, V.N.; Vidal, M.; Iakoucheva, L.M. Intrinsic disorder is a common feature of hub proteins from four eukaryotic interactomes. PLoS Comput. Biol. 2006, 2, e100. [Google Scholar] [CrossRef]
- Pesole, G.; Gissi, C.; de Chirico, A.; Saccone, C. Nucleotide substitution rate of mammalian mitochondrial genomes. J. Mol. Evol. 1999, 48, 427–434. [Google Scholar] [CrossRef]
- Yakes, F.M.; van Houten, B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. USA 1997, 94, 514–519. [Google Scholar] [CrossRef]
- Caldecott, K.W.; McKeown, C.K.; Tucker, J.D.; Ljungquist, S.; Thompson, L.H. An interaction between the mammalian DNA repair protein XRCC1 and DNA ligase III. Mol. Cell. Biol. 1994, 14, 68–76. [Google Scholar]
- Szczesny, B.; Tann, A.W.; Longley, M.J.; Copeland, W.C.; Mitra, S. Long patch base excision repair in mammalian mitochondrial genomes. J. Biol. Chem. 2008, 283, 26349–26356. [Google Scholar] [CrossRef]
- Wunderlich, V.; Tetzlaff, I.; Graffi, A. Studies on nitrosodimethylamine: Preferential methylation of mitochondrial DNA in rats and hamsters. Chem. Biol. Interact. 1972, 4, 81–89. [Google Scholar] [CrossRef]
- Gavin, C.E.; Gunter, K.K.; Gunter, T.E. Mn2+ sequestration by mitochondria and inhibition of oxidative phosphorylation. Toxicol. Appl. Pharmacol. 1992, 115, 1–5. [Google Scholar] [CrossRef]
- Johnson, F.O.; Atchison, W.D. The role of environmental mercury, lead and pesticide exposure in development of amyotrophic lateral sclerosis. Neurotoxicology 2009, 30, 761–765. [Google Scholar] [CrossRef]
- Sokolova, I.M.; Ringwood, A.H.; Johnson, C. Tissue-specific accumulation of cadmium in subcellular compartments of eastern oysters Crassostrea virginica Gmelin (Bivalvia: Ostreidae). Aquat. Toxicol. 2005, 74, 218–228. [Google Scholar] [CrossRef]
- Karahalil, B.; Hogue, B.A.; de Souza-Pinto, N.C.; Bohr, V.A. Base excision repair capacity in mitochondria and nuclei: Tissue-specific variations. FASEB J. 2002, 16, 1895–1902. [Google Scholar] [CrossRef]
- Szczesny, B.; Tann, A.W.; Mitra, S. Age- and tissue-specific changes in mitochondrial and nuclear DNA base excision repair activity in mice: Susceptibility of skeletal muscles to oxidative injury. Mech. Ageing Dev. 2010, 131, 330–337. [Google Scholar] [CrossRef]
- Englander, E.W. Brain capacity for repair of oxidatively damaged DNA and preservation of neuronal function. Mech. Ageing Dev. 2008, 129, 475–482. [Google Scholar] [CrossRef]
- Raffoul, J.J.; Cabelof, D.C.; Nakamura, J.; Meira, L.B.; Friedberg, E.C.; Heydari, A.R. Apurinic/apyrimidinic endonuclease (APE/Ref-1) haploinsufficient mice display tissue-specific differences in DNA polymerase beta-dependent base excision repair. J. Biol. Chem. 2004, 279, 18425–18433. [Google Scholar]
- Canugovi, C.; Yoon, J.S.; Feldman, N.H.; Croteau, D.L.; Mattson, M.P.; Bohr, V.A. Endonuclease VIII-like 1 (NEIL1) promotes short-term spatial memory retention and protects from ischemic stroke-induced brain dysfunction and death in mice. Proc. Natl. Acad. Sci. USA 2012, 109, 14948–14953. [Google Scholar]
- Torisu, K.; Tsuchimoto, D.; Ohnishi, Y.; Nakabeppu, Y. Hematopoietic tissue-specific expression of mouse NEIL3 for endonuclease VIII-like protein. J. Biochem. 2005, 138, 763–772. [Google Scholar] [CrossRef]
- Hildrestrand, G.A.; Neurauter, C.G.; Diep, D.B.; Castellanos, C.G.; Krauss, S.; Bjoras, M.; Luna, L. Expression patterns of NEIL3 during embryonic brain development and neoplasia. BMC Neurosci. 2009. [Google Scholar] [CrossRef]
- Regnell, C.E.; Hildrestrand, G.A.; Sejersted, Y.; Medin, T.; Moldestad, O.; Rolseth, V.; Krokeide, S.Z.; Suganthan, R.; Luna, L.; Bjoras, M.; et al. Hippocampal adult neurogenesis is maintained by NEIL3-dependent repair of oxidative DNA lesions in neural progenitor cells. Cell Rep. 2012, 2, 503–510. [Google Scholar] [CrossRef]
- Rolseth, V.; Krokeide, S.Z.; Kunke, D.; Neurauter, C.G.; Suganthan, R.; Sejersted, Y.; Hildrestrand, G.A.; Bjoras, M.; Luna, L. Loss of NEIL3, the major DNA glycosylase activity for removal of hydantoins in single stranded DNA, reduces cellular proliferation and sensitizes cells to genotoxic stress. Biochim. Biophys. Acta 2013, 1833, 1157–1164. [Google Scholar] [CrossRef]
- Uhlen, M.; Oksvold, P.; Fagerberg, L.; Lundberg, E.; Jonasson, K.; Forsberg, M.; Zwahlen, M.; Kampf, C.; Wester, K.; Hober, S.; et al. Towards a knowledge-based human protein atlas. Nat. Biotechnol. 2010, 28, 1248–1250. [Google Scholar] [CrossRef]
- Swain, U.; Subba Rao, K. Study of DNA damage via the comet assay and base excision repair activities in rat brain neurons and astrocytes during aging. Mech. Ageing Dev. 2011, 132, 374–381. [Google Scholar] [CrossRef]
- Fukae, J.; Mizuno, Y.; Hattori, N. Mitochondrial dysfunction in Parkinson’s disease. Mitochondrion 2007, 7, 58–62. [Google Scholar] [CrossRef]
- Sheng, Z.; Oka, S.; Tsuchimoto, D.; Abolhassani, N.; Nomaru, H.; Sakumi, K.; Yamada, H.; Nakabeppu, Y. 8-oxoguanine causes neurodegeneration during mutyh-mediated DNA base excision repair. J. Clin. Investig. 2012, 122, 4344–4361. [Google Scholar] [CrossRef]
- Jiang, Y.; Guo, C.; Fishel, M.L.; Wang, Z.Y.; Vasko, M.R.; Kelley, M.R. Role of APE1 in differentiated neuroblastoma SH-SY5Y cells in response to oxidative stress: Use of APE1 small molecule inhibitors to delineate APE1 functions. DNA Repair 2009, 8, 1273–1282. [Google Scholar] [CrossRef]
- Vasko, M.R.; Guo, C.; Kelley, M.R. The multifunctional DNA repair/redox enzyme APE1/Ref-1 promotes survival of neurons after oxidative stress. DNA Repair 2005, 4, 367–379. [Google Scholar] [CrossRef]
- Jiang, Y.; Guo, C.; Vasko, M.R.; Kelley, M.R. Implications of apurinic/apyrimidinic endonuclease in reactive oxygen signaling response after cisplatin treatment of dorsal root ganglion neurons. Cancer Res. 2008, 68, 6425–6434. [Google Scholar] [CrossRef]
- Breslin, C.; Caldecott, K.W. DNA 3'-phosphatase activity is critical for rapid global rates of single-strand break repair following oxidative stress. Mol. Cell. Biol. 2009, 29, 4653–4662. [Google Scholar] [CrossRef]
- Wilson, S.H. Mammalian base excision repair and DNA polymerase beta. Mutat. Res. 1998, 407, 203–215. [Google Scholar] [CrossRef]
- Wilson, D.M., 3rd; McNeill, D.R. Base excision repair and the central nervous system. Neuroscience 2007, 145, 1187–1200. [Google Scholar] [CrossRef]
- Akbari, M.; Pena-Diaz, J.; Andersen, S.; Liabakk, N.B.; Otterlei, M.; Krokan, H.E. Extracts of proliferating and non-proliferating human cells display different base excision pathways and repair fidelity. DNA Repair 2009, 8, 834–843. [Google Scholar] [CrossRef]
- Wei, W.; Englander, E.W. DNA polymerase beta-catalyzed-PCNA independent long patch base excision repair synthesis: A mechanism for repair of oxidatively damaged DNA ends in post-mitotic brain. J. Neurochem. 2008, 107, 734–744. [Google Scholar] [CrossRef]
- Kenche, V.B.; Barnham, K.J. Alzheimer’s disease & metals: Therapeutic opportunities. Br. J. Pharmacol. 2011, 163, 211–219. [Google Scholar] [CrossRef]
- Raven, E.P.; Lu, P.H.; Tishler, T.A.; Heydari, P.; Bartzokis, G. Increased iron levels and decreased tissue integrity in hippocampus of Alzheimer’s disease detected in vivo with magnetic resonance imaging. J. Alzheimers Dis. 2013, 37, 127–136. [Google Scholar]
- Lee, H.P.; Zhu, X.; Liu, G.; Chen, S.G.; Perry, G.; Smith, M.A.; Lee, H.G. Divalent metal transporter, iron, and Parkinson’s disease: A pathological relationship. Cell Res. 2010, 20, 397–399. [Google Scholar] [CrossRef]
- Pamphlett, R.; Kum Jew, S. Heavy metals in locus ceruleus and motor neurons in motor neuron disease. Acta Neuropathol. Commun. 2013. [Google Scholar] [CrossRef]
- Barbeito, A.G.; Levade, T.; Delisle, M.B.; Ghetti, B.; Vidal, R. Abnormal iron metabolism in fibroblasts from a patient with the neurodegenerative disease hereditary ferritinopathy. Mol. Neurodegener. 2010. [Google Scholar] [CrossRef]
- Texel, S.J.; Xu, X.; Harris, Z.L. Ceruloplasmin in neurodegenerative diseases. Biochem. Soc. Trans. 2008, 36, 1277–1281. [Google Scholar] [CrossRef]
- Koeppen, A.H. A brief history of brain iron research. J. Neurol. Sci. 2003, 207, 95–97. [Google Scholar] [CrossRef]
- Rao, G.N.; Ney, E.; Herbert, R.A. Changes associated with delay of mammary cancer by retinoid analogues in transgenic mice bearing c-neu oncogene. Breast Cancer Res. Treat. 1999, 58, 241–254. [Google Scholar]
- Molina-Holgado, F.; Hider, R.C.; Gaeta, A.; Williams, R.; Francis, P. Metals ions and neurodegeneration. Biometals 2007, 20, 639–654. [Google Scholar] [CrossRef]
- Huang, X.; Atwood, C.S.; Moir, R.D.; Hartshorn, M.A.; Vonsattel, J.P.; Tanzi, R.E.; Bush, A.I. Zinc-induced Alzheimer’s Abeta1-40 aggregation is mediated by conformational factors. J. Biol. Chem. 1997, 272, 26464–26470. [Google Scholar]
- Youdim, M.B.; Ben-Shachar, D.; Riederer, P. Iron in brain function and dysfunction with emphasis on Parkinson’s disease. Eur. Neurol. 1991, 31, 34–40. [Google Scholar] [CrossRef]
- Bowman, A.B.; Kwakye, G.F.; Herrero Hernandez, E.; Aschner, M. Role of manganese in neurodegenerative diseases. J. Trace Elem. Med. Biol. 2011, 25, 191–203. [Google Scholar] [CrossRef]
- Shaw, C.A.; Tomljenovic, L. Aluminum in the central nervous system (CNS): Toxicity in humans and animals, vaccine adjuvants, and autoimmunity. Immunol. Res. 2013, 56, 304–316. [Google Scholar] [CrossRef]
- Walton, N.M.; Shin, R.; Tajinda, K.; Heusner, C.L.; Kogan, J.H.; Miyake, S.; Chen, Q.; Tamura, K.; Matsumoto, M. Adult neurogenesis transiently generates oxidative stress. PLoS One 2012, 7, e35264. [Google Scholar] [CrossRef]
- Zatta, P. Biological models for the study of aluminum neurotoxicity. Acta Med. Romana 1997, 35, 592–600. [Google Scholar]
- Rao, K.S.; Hegde, M.L.; Anitha, S.; Musicco, M.; Zucca, F.A.; Turro, N.J.; Zecca, L. Amyloid beta and neuromelanin—Toxic or protective molecules? The cellular context makes the difference. Prog. Neurobiol. 2006, 78, 364–373. [Google Scholar] [CrossRef]
- Wright, R.O.; Tsaih, S.W.; Schwartz, J.; Spiro, A., 3rd; McDonald, K.; Weiss, S.T.; Hu, H. Lead exposure biomarkers and mini-mental status exam scores in older men. Epidemiology 2003, 14, 713–718. [Google Scholar] [CrossRef]
- Tavakoli-Nezhad, M.; Barron, A.J.; Pitts, D.K. Postnatal inorganic lead exposure decreases the number of spontaneously active midbrain dopamine neurons in the rat. Neurotoxicology 2001, 22, 259–269. [Google Scholar] [CrossRef]
- Basha, M.R.; Murali, M.; Siddiqi, H.K.; Ghosal, K.; Siddiqi, O.K.; Lashuel, H.A.; Ge, Y.W.; Lahiri, D.K.; Zawia, N.H. Lead (PB) exposure and its effect on APP proteolysis and Abeta aggregation. FASEB J. 2005, 19, 2083–2084. [Google Scholar]
- Wu, J.; Basha, M.R.; Brock, B.; Cox, D.P.; Cardozo-Pelaez, F.; McPherson, C.A.; Harry, J.; Rice, D.C.; Maloney, B.; Chen, D.; et al. Alzheimer’s disease (AD)-like pathology in aged monkeys after infantile exposure to environmental metal lead (PB): Evidence for a developmental origin and environmental link for AD. J. Neurosci. 2008, 28, 3–9. [Google Scholar] [CrossRef]
- Bourdineaud, J.P.; Fujimura, M.; Laclau, M.; Sawada, M.; Yasutake, A. Deleterious effects in mice of fish-associated methylmercury contained in a diet mimicking the western populations’ average fish consumption. Environ. Int. 2011, 37, 303–313. [Google Scholar] [CrossRef]
- Yee, S.; Choi, B.H. Oxidative stress in neurotoxic effects of methylmercury poisoning. Neurotoxicology 1996, 17, 17–26. [Google Scholar]
- Hegde, M.L.; Anitha, S.; Latha, K.S.; Mustak, M.S.; Stein, R.; Ravid, R.; Rao, K.S. First evidence for helical transitions in supercoiled DNA by amyloid beta peptide (1–42) and aluminum: A new insight in understanding Alzheimer’s disease. J. Mol. Neurosci. 2004, 22, 19–31. [Google Scholar] [CrossRef]
- Rao, K.S.J.; Rao, R.V.; Shanmugavelu, P.; Menon, R.B. Trace elements in Alzheimer’s disease brain: A new hypothesis. Alzheimers Rep. 1999, 2, 241–246. [Google Scholar]
- Pande, M.B.; Nagabhushan, P.; Hegde, M.L.; Rao, T.S.; Rao, K.S. An algorithmic approach to understand trace elemental homeostasis in serum samples of parkinson disease. Comput. Biol. Med. 2005, 35, 475–493. [Google Scholar] [CrossRef]
- Pfaender, S.; Grabrucker, A.M. Characterization of biometal profiles in neurological disorders. Metallomics 2014, 6, 960–977. [Google Scholar] [CrossRef]
- Hegde, M.L.; Hegde, P.M.; Rao, K.S.; Mitra, S. Oxidative genome damage and its repair in neurodegenerative diseases: Function of transition metals as a double-edged sword. J. Alzheimers Dis. 2011, 24, 183–198. [Google Scholar]
- Robison, S.H.; Cantoni, O.; Costa, M. Analysis of metal-induced DNA lesions and DNA-repair replication in mammalian cells. Mutat. Res. 1984, 131, 173–181. [Google Scholar]
- Giles, N.M.; Watts, A.B.; Giles, G.I.; Fry, F.H.; Littlechild, J.A.; Jacob, C. Metal and redox modulation of cysteine protein function. Chem. Biol. 2003, 10, 677–693. [Google Scholar] [CrossRef]
- Himmelfarb, J.; McMonagle, E.; McMenamin, E. Plasma protein thiol oxidation and carbonyl formation in chronic renal failure. Kidney Int. 2000, 58, 2571–2578. [Google Scholar] [CrossRef]
- Hegde, M.L.; Gupta, V.B.; Anitha, M.; Harikrishna, T.; Shankar, S.K.; Muthane, U.; Subba Rao, K.; Jagannatha Rao, K.S. Studies on genomic DNA topology and stability in brain regions of Parkinson’s disease. Arch. Biochem. Biophys. 2006, 449, 143–156. [Google Scholar] [CrossRef]
- Jeppesen, D.K.; Bohr, V.A.; Stevnsner, T. DNA repair deficiency in neurodegeneration. Prog. Neurobiol. 2011, 94, 166–200. [Google Scholar] [CrossRef]
- Rass, U.; Ahel, I.; West, S.C. Defective DNA repair and neurodegenerative disease. Cell 2007, 130, 991–1004. [Google Scholar] [CrossRef]
- Maynard, S.; Schurman, S.H.; Harboe, C.; de Souza-Pinto, N.C.; Bohr, V.A. Base excision repair of oxidative DNA damage and association with cancer and aging. Carcinogenesis 2009, 30, 2–10. [Google Scholar]
- Gredilla, R.; Garm, C.; Stevnsner, T. Nuclear and mitochondrial DNA repair in selected eukaryotic aging model systems. Oxid. Med. Cell. Longev. 2012. [Google Scholar] [CrossRef]
- Hartwig, A. Role of DNA repair inhibition in lead- and cadmium-induced genotoxicity: A review. Environ. Health Perspect. 1994, 102, 45–50. [Google Scholar] [CrossRef]
- Hengstler, J.G.; Bolm-Audorff, U.; Faldum, A.; Janssen, K.; Reifenrath, M.; Gotte, W.; Jung, D.; Mayer-Popken, O.; Fuchs, J.; Gebhard, S.; et al. Occupational exposure to heavy metals: DNA damage induction and DNA repair inhibition prove co-exposures to cadmium, cobalt and lead as more dangerous than hitherto expected. Carcinogenesis 2003, 24, 63–73. [Google Scholar] [CrossRef]
- Adhikari, S.; Toretsky, J.A.; Yuan, L.; Roy, R. Magnesium, essential for base excision repair enzymes, inhibits substrate binding of N-methylpurine-DNA glycosylase. J. Biol. Chem. 2006, 281, 29525–29532. [Google Scholar] [CrossRef]
- Wang, P.; Guliaev, A.B.; Hang, B. Metal inhibition of human n-methylpurine-DNA glycosylase activity in base excision repair. Toxicol. Lett. 2006, 166, 237–247. [Google Scholar] [CrossRef]
- Whiteside, J.R.; Box, C.L.; McMillan, T.J.; Allinson, S.L. Cadmium and copper inhibit both DNA repair activities of polynucleotide kinase. DNA Repair 2010, 9, 83–89. [Google Scholar] [CrossRef]
- McNeill, D.R.; Narayana, A.; Wong, H.K.; Wilson, D.M., 3rd. Inhibition of APE1 nuclease activity by lead, iron, and cadmium. Environ. Health Perspect. 2004, 112, 799–804. [Google Scholar] [CrossRef]
- Hegde, M.L.; Jagannatha Rao, K.S. Challenges and complexities of alpha-synuclein toxicity: New postulates in unfolding the mystery associated with Parkinson’s disease. Arch. Biochem. Biophys. 2003, 418, 169–178. [Google Scholar] [CrossRef]
- Hegde, M.L. Molecular characterization of neuroprotective activities of plant based products could revive their utilization and lead discovery of new drug candidates for brain diseases. J. Pharm. Bioallied Sci. 2014, 6, 63–64. [Google Scholar]
- Romero, A.; Ramos, E.; de Los Rios, C.; Egea, J.; del Pino, J.; Reiter, R.J. A review of metal-catalyzed molecular damage: Protection by melatonin. J. Pineal Res. 2014, 56, 343–370. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mitra, J.; Guerrero, E.N.; Hegde, P.M.; Wang, H.; Boldogh, I.; Rao, K.S.; Mitra, S.; Hegde, M.L. New Perspectives on Oxidized Genome Damage and Repair Inhibition by Pro-Oxidant Metals in Neurological Diseases. Biomolecules 2014, 4, 678-703. https://doi.org/10.3390/biom4030678
Mitra J, Guerrero EN, Hegde PM, Wang H, Boldogh I, Rao KS, Mitra S, Hegde ML. New Perspectives on Oxidized Genome Damage and Repair Inhibition by Pro-Oxidant Metals in Neurological Diseases. Biomolecules. 2014; 4(3):678-703. https://doi.org/10.3390/biom4030678
Chicago/Turabian StyleMitra, Joy, Erika N. Guerrero, Pavana M. Hegde, Haibo Wang, Istvan Boldogh, Kosagi Sharaf Rao, Sankar Mitra, and Muralidhar L. Hegde. 2014. "New Perspectives on Oxidized Genome Damage and Repair Inhibition by Pro-Oxidant Metals in Neurological Diseases" Biomolecules 4, no. 3: 678-703. https://doi.org/10.3390/biom4030678