
Transcriptional Activation of Inflammatory Genes: Mechanistic Insight into Selectivity and Diversity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Inflammation and Cellular Homeostasis
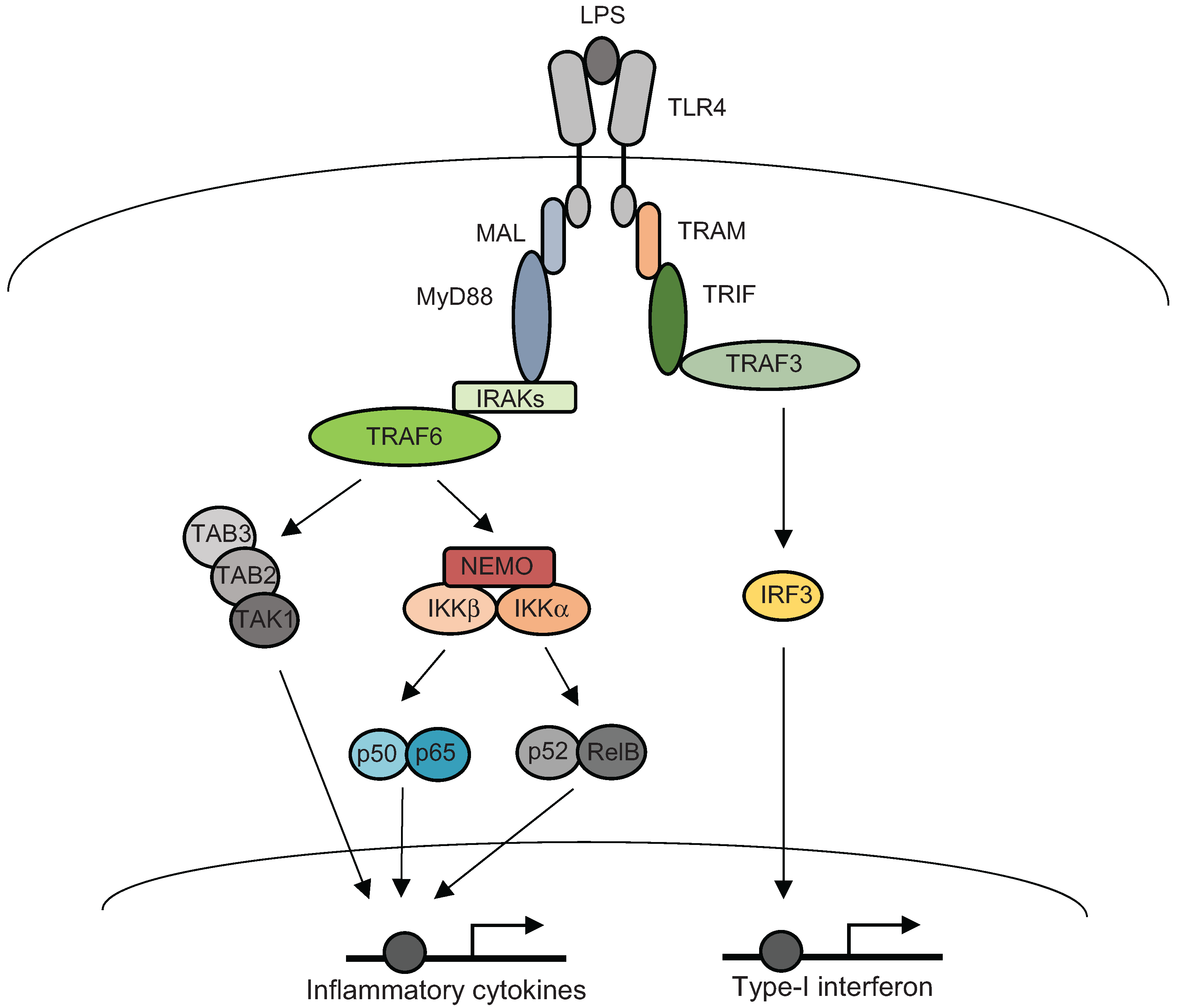
3. Inflammatory Signal Transductions

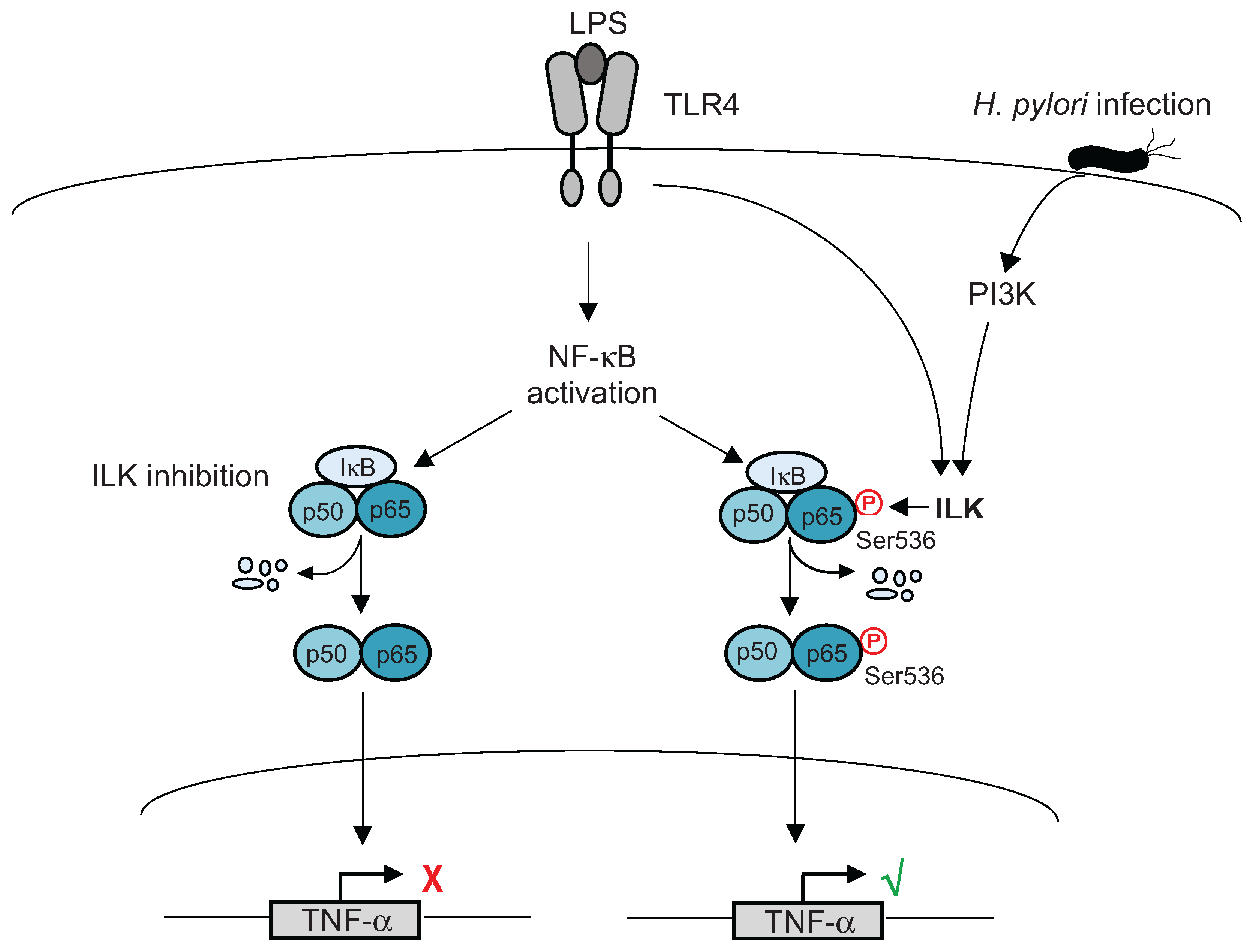
4. Post-Translational Modifications of NF-κB

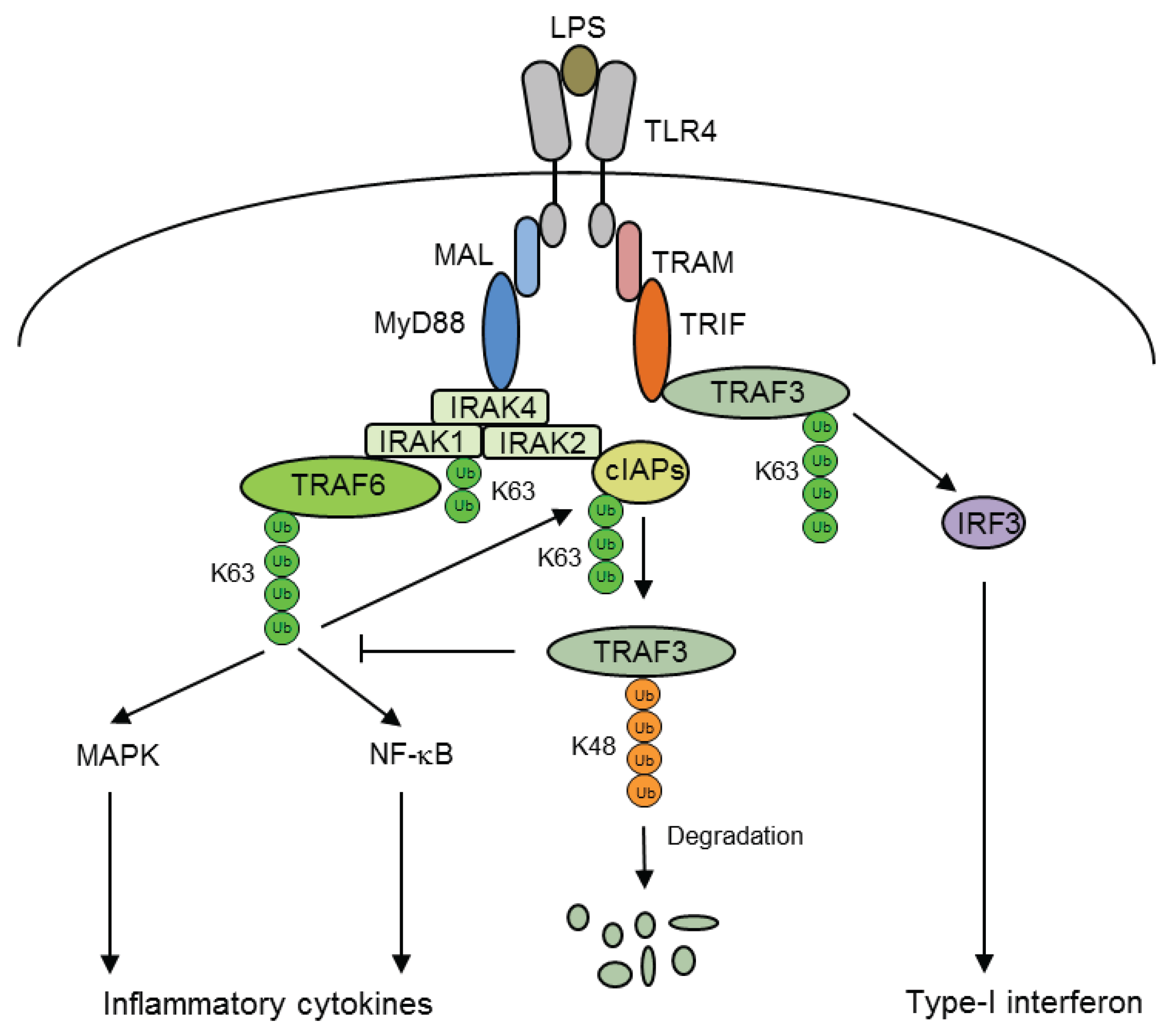
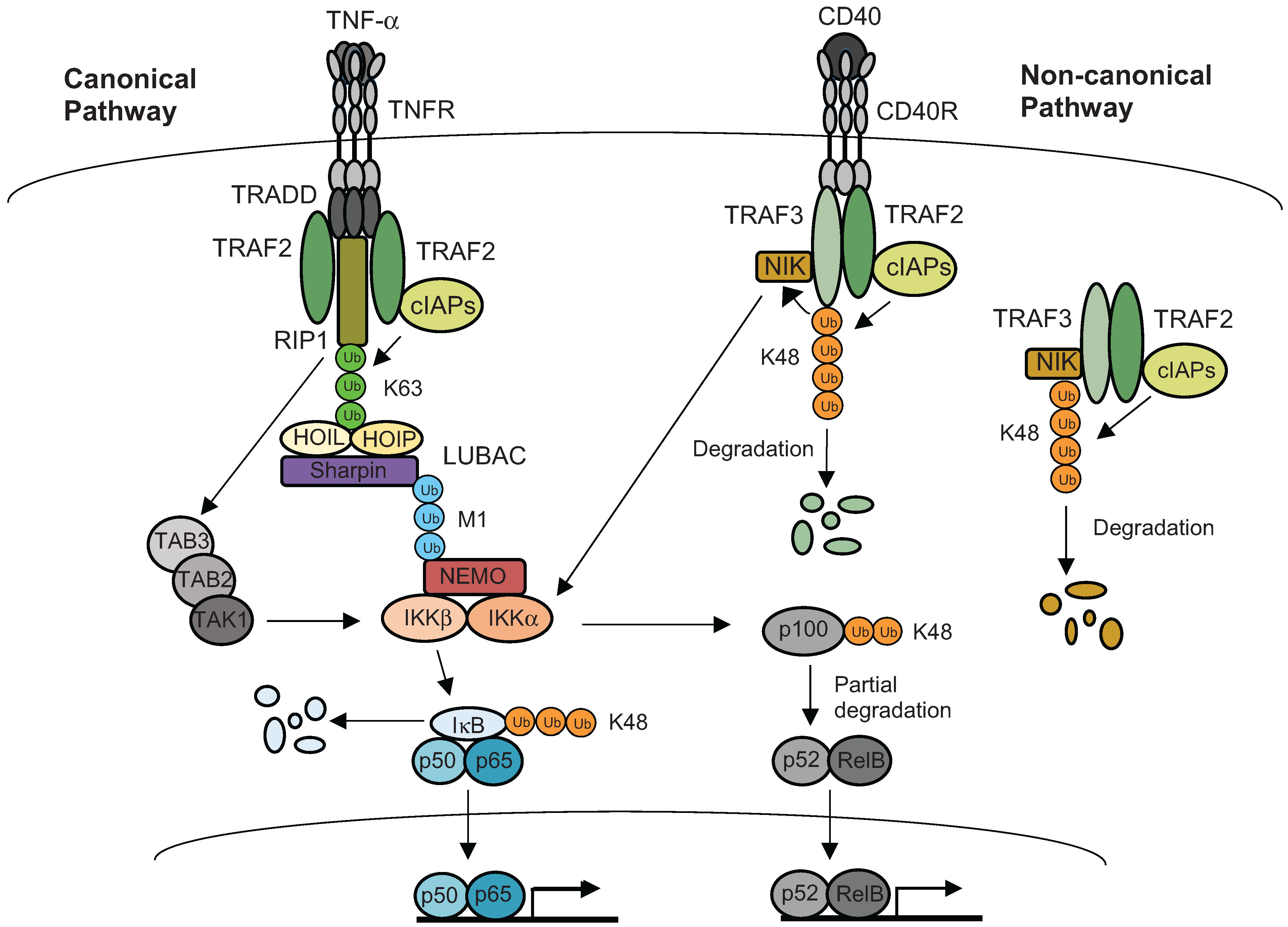
5. Ubiquitination of Inflammatory Signaling Complexes


6. Selective Regulation by Chromatin Structures

7. Additional Effects on Pro-Inflammatory Gene Transcription
7.1. Epigenetic Markers
7.2. Developmental Events
7.3. Physiological Relevance
7.4. Interplay between Signaling Pathways
8. Conclusions and Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Medzhitov, R. Inflammation 2010: New adventures of an old flame. Cell 2010, 140, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Carey, M.; Robbins, S.L. Robbins Basic Pathology, 7th ed.; Saunders: New York, NY, USA, 2003. [Google Scholar]
- Majno, G.; Joris, I. Cells, Tissues, and Diseases, 2nd ed.; Oxford University Press: Oxford, UK, 2004. [Google Scholar]
- Meroni, P.L.; Valentini, G.; Ayala, F.; Cattaneo, A.; Valesini, G. New strategies to address the pharmacodynamics and pharmacokinetics of tumor necrosis factor (TNF) inhibitors: A systematic analysis. Autoimmun. Rev. 2015, 14, 812–829. [Google Scholar] [CrossRef] [PubMed]
- Kodama, S.; Davis, M.; Faustman, D.L. The therapeutic potential of tumor necrosis factor for autoimmune disease: A mechanistically based hypothesis. Cell. Mol. Life Sci. 2005, 62, 1850–1862. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.R.; Alberts, B.M. Steroid receptors: Elements for modulation of eukaryotic transcription. Annu. Rev. Biochem. 1976, 45, 721–746. [Google Scholar] [CrossRef] [PubMed]
- Herschman, H.R. Primary response genes induced by growth factors and tumor promoters. Annu. Rev. Biochem. 1991, 60, 281–319. [Google Scholar] [CrossRef] [PubMed]
- Smale, S.T. Selective transcription in response to an inflammatory stimulus. Cell 2010, 140, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- McGettrick, A.F.; O’Neill, L.A. Regulators of TLR4 signaling by endotoxins. Subcell. Biochem. 2010, 53, 153–171. [Google Scholar] [PubMed]
- Newton, K.; Dixit, V.M. Signaling in innate immunity and inflammation. Cold Spring Harb. Perspect. Biol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Sen, R.; Baltimore, D. Inducibility of kappa immunoglobulin enhancer-binding protein NF-kappaB by a posttranslational mechanism. Cell 1986, 47, 921–928. [Google Scholar] [CrossRef]
- Ghosh, S.; May, M.J.; Kopp, E.B. NF-kappa B and Rel proteins: Evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol. 1998, 16, 225–260. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; Natoli, G.; Ghosh, G. Transcriptional regulation via the NF-kappaB signaling module. Oncogene 2006, 25, 6706–6716. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Karin, M. Missing pieces in the NF-kappaB puzzle. Cell 2002, 109, S81–S96. [Google Scholar] [CrossRef]
- Vallabhapurapu, S.; Karin, M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev. Immunol. 2009, 27, 693–733. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.U. An overview of inflammation: Mechanism and consequences. Front. Biol. 2011, 6, 274–281. [Google Scholar]
- Sun, S.C. The noncanonical NF-kappaB pathway. Immunol. Rev. 2012, 246, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.S.; Ley, S.C. Mitogen-activated protein kinases in innate immunity. Nat. Rev. Immunol. 2013, 13, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Sadler, A.J.; Rossello, F.J.; Yu, L.; Deane, J.A.; Yuan, X.; Wang, D.; Irving, A.T.; Kaparakis-Liaskos, M.; Gantier, M.P.; Ying, H.; et al. BTB-ZF transcriptional regulator PLZF modifies chromatin to restrain inflammatory signaling programs. Proc. Natl. Acad. Sci. USA 2015, 112, 1535–1540. [Google Scholar] [CrossRef] [PubMed]
- Sadler, A.J.; Suliman, B.A.; Yu, L.; Yuan, X.; Wang, D.; Irving, A.T.; Sarvestani, S.T.; Banerjee, A.; Mansell, A.S.; Liu, J.P.; et al. The acetyltransferase hat1 moderates the NF-kappaB response by regulating the transcription factor PLZF. Nat. Commun. 2015. [Google Scholar] [CrossRef] [PubMed]
- Carey, M.F.; Peterson, C.L.; Smale, S.T. Transcriptional Regulation in Eukaryotes: Concepts, Strategies, and Techniques; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2009. [Google Scholar]
- Thanos, D.; Maniatis, T. Virus induction of human IFN beta gene expression requires the assembly of an enhanceosome. Cell 1995, 83, 1091–1100. [Google Scholar] [CrossRef]
- Munshi, N.; Agalioti, T.; Lomvardas, S.; Merika, M.; Chen, G.; Thanos, D. Coordination of a transcriptional switch by HMGI(Y) acetylation. Science 2001, 293, 1133–1136. [Google Scholar] [CrossRef] [PubMed]
- Bosisio, D.; Marazzi, I.; Agresti, A.; Shimizu, N.; Bianchi, M.E.; Natoli, G. A hyper-dynamic equilibrium between promoter-bound and nucleoplasmic dimers controls NF-kappaB-dependent gene activity. EMBO J. 2006, 25, 798–810. [Google Scholar] [CrossRef] [PubMed]
- Hager, G.L.; McNally, J.G.; Misteli, T. Transcription dynamics. Mol. Cell 2009, 35, 741–753. [Google Scholar] [CrossRef] [PubMed]
- Panne, D.; Maniatis, T.; Harrison, S.C. Crystal structure of ATF-2/c-jun and IRF-3 bound to the interferon-beta enhancer. EMBO J. 2004, 23, 4384–4393. [Google Scholar] [CrossRef] [PubMed]
- Panne, D.; Maniatis, T.; Harrison, S.C. An atomic model of the interferon-beta enhanceosome. Cell 2007, 129, 1111–1123. [Google Scholar] [CrossRef] [PubMed]
- Wan, F.; Lenardo, M.J. Specification of DNA binding activity of NF-kappaB proteins. Cold Spring Harb. Perspect. Biol. 2009, 1, a000067. [Google Scholar] [CrossRef] [PubMed]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Voll, R.E.; Ghosh, S. Phosphorylation of NF-kappa B p65 by Pka stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator Cbp/p300. Mol. Cell 1998, 1, 661–671. [Google Scholar] [CrossRef]
- Zhong, H.; May, M.J.; Jimi, E.; Ghosh, S. The phosphorylation status of nuclear NF-kappaB determines its association with Cbp/p300 or Hdac-1. Mol. Cell 2002, 9, 625–636. [Google Scholar] [CrossRef]
- Dong, J.; Jimi, E.; Zhong, H.; Hayden, M.S.; Ghosh, S. Repression of gene expression by unphosphorylated NF-kappaB p65 through epigenetic mechanisms. Genes Dev. 2008, 22, 1159–1173. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, H.; Chiba, H.; Miyoshi, H.; Sugita, T.; Toriumi, W. Ikappab kinases phosphorylate NF-kappaB p65 subunit on serine 536 in the transactivation domain. J. Biol. Chem. 1999, 274, 30353–30356. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Tang, E.; Guan, K.; Wang, C.Y. IKK beta plays an essential role in the phosphorylation of RelA/p65 on serine 536 induced by lipopolysaccharide. J. Immunol. 2003, 170, 5630–5635. [Google Scholar] [CrossRef] [PubMed]
- Douillette, A.; Bibeau-Poirier, A.; Gravel, S.P.; Clement, J.F.; Chenard, V.; Moreau, P.; Servant, M.J. The proinflammatory actions of angiotensin II are dependent on p65 phosphorylation by the IkappaB kinase complex. J. Biol. Chem. 2006, 281, 13275–13284. [Google Scholar] [CrossRef] [PubMed]
- Mandrekar, P.; Jeliazkova, V.; Catalano, D.; Szabo, G. Acute alcohol exposure exerts anti-inflammatory effects by inhibiting IkappaB kinase activity and p65 phosphorylation in human monocytes. J. Immunol. 2007, 178, 7686–7693. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, C.; Batra, S.; Vargo, M.A.; Voss, O.H.; Gavrilin, M.A.; Wewers, M.D.; Guttridge, D.C.; Grotewold, E.; Doseff, A.I. Apigenin blocks lipopolysaccharide-induced lethality in vivo and proinflammatory cytokines expression by inactivating NF-kappaB through the suppression of p65 phosphorylation. J. Immunol. 2007, 179, 7121–7127. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, C.Y.; Barberi, T.J.; Ghosh, P.; Longo, D.L. Phosphorylation of RelA/p65 on serine 536 defines an I{kappa}B{alpha}-independent NF-{kappa}B pathway. J. Biol. Chem. 2005, 280, 34538–34547. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.U.; Sarvestani, S.T.; Gantier, M.P.; Williams, B.R.; Hannigan, G.E. Integrin-linked kinase modulates lipopolysaccharide- and helicobacter pylori-induced nuclear factor kappaB-activated tumor necrosis factor-alpha production via regulation of p65 serine 536 phosphorylation. J. Biol. Chem. 2014, 289, 27776–27793. [Google Scholar] [CrossRef] [PubMed]
- Hochrainer, K.; Racchumi, G.; Anrather, J. Site-specific phosphorylation of the p65 protein subunit mediates selective gene expression by differential NF-kappaB and RNA polymerase II promoter recruitment. J. Biol. Chem. 2013, 288, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Ea, C.K.; Baltimore, D. Regulation of NF-kappaB activity through lysine monomethylation of p65. Proc. Natl. Acad. Sci. USA 2009, 106, 18972–18977. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.F.; Greene, W.C. Regulation of distinct biological activities of the NF-kappaB transcription factor complex by acetylation. J. Mol. Med. 2003, 81, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Kiernan, R.; Bres, V.; Ng, R.W.; Coudart, M.P.; el Messaoudi, S.; Sardet, C.; Jin, D.Y.; Emiliani, S.; Benkirane, M. Post-activation turn-off of NF-kappa B-dependent transcription is regulated by acetylation of p65. J. Biol. Chem. 2003, 278, 2758–2766. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.F.; Mu, Y.; Greene, W.C. Acetylation of rela at discrete sites regulates distinct nuclear functions of NF-kappaB. EMBO J. 2002, 21, 6539–6548. [Google Scholar] [CrossRef] [PubMed]
- Smale, S.T. Dimer-specific regulatory mechanisms within the NF-kappaB family of transcription factors. Immunol. Rev. 2012, 246, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Saccani, S.; Pantano, S.; Natoli, G. Two waves of nuclear factor kappaB recruitment to target promoters. J. Exp. Med. 2001, 193, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Smale, S.T. Hierarchies of NF-kappaB target-gene regulation. Nat. Immunol. 2011, 12, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Corn, J.E.; Vucic, D. Ubiquitin in inflammation: The right linkage makes all the difference. Nat. Struct. Mol. Biol. 2014, 21, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Darding, M.; Meier, P. IAPs: Guardians of RIPK1. Cell Death Differ. 2012, 19, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Tseng, P.H.; Matsuzawa, A.; Zhang, W.; Mino, T.; Vignali, D.A.; Karin, M. Different modes of ubiquitination of the adaptor TRAF3 selectively activate the expression of type I interferons and proinflammatory cytokines. Nat. Immunol. 2010, 11, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Varfolomeev, E.; Goncharov, T.; Maecker, H.; Zobel, K.; Komuves, L.G.; Deshayes, K.; Vucic, D. Cellular inhibitors of apoptosis are global regulators of NF-kappaB and mapk activation by members of the TNF family of receptors. Sci. Signal. 2012. [Google Scholar] [CrossRef] [PubMed]
- De Almagro, M.C.; Vucic, D. The inhibitor of apoptosis (Iap) proteins are critical regulators of signaling pathways and targets for anti-cancer therapy. Exp. Oncol. 2012, 34, 200–211. [Google Scholar] [PubMed]
- Silke, J.; Vucic, D. IAP family of cell death and signaling regulators. Methods Enzymol. 2014, 545, 35–65. [Google Scholar] [PubMed]
- Luger, K.; Mader, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 1997, 389, 251–260. [Google Scholar] [PubMed]
- Weintraub, H.; Groudine, M. Chromosomal subunits in active genes have an altered conformation. Science 1976, 193, 848–856. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Bingham, P.M.; Livak, K.J.; Holmgren, R.; Elgin, S.C. The chromatin structure of specific genes: I. Evidence for higher order domains of defined DNA sequence. Cell 1979, 16, 797–806. [Google Scholar] [CrossRef]
- Clapier, C.R.; Cairns, B.R. The biology of chromatin remodeling complexes. Annu. Rev. Biochem. 2009, 78, 273–304. [Google Scholar] [CrossRef] [PubMed]
- Brownell, J.E.; Zhou, J.; Ranalli, T.; Kobayashi, R.; Edmondson, D.G.; Roth, S.Y.; Allis, C.D. Tetrahymena histone acetyltransferase A: A homolog to yeast Gcn5p linking histone acetylation to gene activation. Cell 1996, 84, 843–851. [Google Scholar] [CrossRef]
- Cote, J.; Quinn, J.; Workman, J.L.; Peterson, C.L. Stimulation of gal4 derivative binding to nucleosomal DNA by the yeast SWI/SNF complex. Science 1994, 265, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Imbalzano, A.N.; Kwon, H.; Green, M.R.; Kingston, R.E. Facilitated binding of TATA-binding protein to nucleosomal DNA. Nature 1994, 370, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.; Imbalzano, A.N.; Khavari, P.A.; Kingston, R.E.; Green, M.R. Nucleosome disruption and enhancement of activator binding by a human SW1/SNF complex. Nature 1994, 370, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Wang, W.; Rando, O.J.; Xue, Y.; Swiderek, K.; Kuo, A.; Crabtree, G.R. Rapid and phosphoinositol-dependent binding of the SWI/SNF-like BAF complex to chromatin after T lymphocyte receptor signaling. Cell 1998, 95, 625–636. [Google Scholar] [CrossRef]
- Ramirez-Carrozzi, V.R.; Nazarian, A.A.; Li, C.C.; Gore, S.L.; Sridharan, R.; Imbalzano, A.N.; Smale, S.T. Selective and antagonistic functions of SWI/SNF and MI-2beta nucleosome remodeling complexes during an inflammatory response. Genes Dev. 2006, 20, 282–296. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Carrozzi, V.R.; Braas, D.; Bhatt, D.M.; Cheng, C.S.; Hong, C.; Doty, K.R.; Black, J.C.; Hoffmann, A.; Carey, M.; Smale, S.T. A unifying model for the selective regulation of inducible transcription by CPG islands and nucleosome remodeling. Cell 2009, 138, 114–128. [Google Scholar] [CrossRef] [PubMed]
- Lomvardas, S.; Thanos, D. Modifying gene expression programs by altering core promoter chromatin architecture. Cell 2002, 110, 261–271. [Google Scholar] [CrossRef]
- Rogatsky, I.; Adelman, K. Preparing the first responders: Building the inflammatory transcriptome from the ground up. Mol. Cell 2014, 54, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Shibata, H.; Handa, H. Transcription elongation factors DSIF and NELF: Promoter-proximal pausing and beyond. Biochim. Biophys. Acta 2013, 1829, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Barboric, M.; Nissen, R.M.; Kanazawa, S.; Jabrane-Ferrat, N.; Peterlin, B.M. NF-kappaB binds p-tefb to stimulate transcriptional elongation by RNA polymerase II. Mol. Cell 2001, 8, 327–337. [Google Scholar] [CrossRef]
- Takahashi, H.; Parmely, T.J.; Sato, S.; Tomomori-Sato, C.; Banks, C.A.; Kong, S.E.; Szutorisz, H.; Swanson, S.K.; Martin-Brown, S.; Washburn, M.P.; et al. Human mediator subunit med26 functions as a docking site for transcription elongation factors. Cell 2011, 146, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Prinjha, R.; Tarakhovsky, A. Chromatin targeting drugs in cancer and immunity. Genes Dev. 2013, 27, 1731–1738. [Google Scholar] [CrossRef] [PubMed]
- Calo, E.; Wysocka, J. Modification of enhancer chromatin: What, how, and why? Mol. Cell 2013, 49, 825–837. [Google Scholar] [CrossRef] [PubMed]
- Spitz, F.; Furlong, E.E. Transcription factors: From enhancer binding to developmental control. Nat. Rev. Genet. 2012, 13, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Avni, O.; Rao, A. Cell-type-restricted binding of the transcription factor NFAT to a distal IL-4 enhancer in vivo. Immunity 2000, 12, 643–652. [Google Scholar] [CrossRef]
- Zhou, L.; Nazarian, A.A.; Xu, J.; Tantin, D.; Corcoran, L.M.; Smale, S.T. An inducible enhancer required for IL12b promoter activity in an insulated chromatin environment. Mol. Cell. Biol. 2007, 27, 2698–2712. [Google Scholar] [CrossRef] [PubMed]
- De Santa, F.; Barozzi, I.; Mietton, F.; Ghisletti, S.; Polletti, S.; Tusi, B.K.; Muller, H.; Ragoussis, J.; Wei, C.L.; Natoli, G. A large fraction of extragenic RNA pol II transcription sites overlap enhancers. PLoS Biol. 2010, 8, e1000384. [Google Scholar] [CrossRef] [PubMed]
- Lam, M.T.; Cho, H.; Lesch, H.P.; Gosselin, D.; Heinz, S.; Tanaka-Oishi, Y.; Benner, C.; Kaikkonen, M.U.; Kim, A.S.; Kosaka, M.; et al. Rev-erbs repress macrophage gene expression by inhibiting enhancer-directed transcription. Nature 2013, 498, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Melo, C.A.; Drost, J.; Wijchers, P.J.; van de Werken, H.; de Wit, E.; Oude Vrielink, J.A.; Elkon, R.; Melo, S.A.; Leveille, N.; Kalluri, R.; et al. Ernas are required for p53-dependent enhancer activity and gene transcription. Mol. Cell 2013, 49, 524–535. [Google Scholar] [CrossRef] [PubMed]
- Lai, D.; Wan, M.; Wu, J.; Preston-Hurlburt, P.; Kushwaha, R.; Grundstrom, T.; Imbalzano, A.N.; Chi, T. Induction of TLR4-target genes entails calcium/calmodulin-dependent regulation of chromatin remodeling. Proc. Natl. Acad. Sci. USA 2009, 106, 1169–1174. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, D.M.; Pandya-Jones, A.; Tong, A.J.; Barozzi, I.; Lissner, M.M.; Natoli, G.; Black, D.L.; Smale, S.T. Transcript dynamics of proinflammatory genes revealed by sequence analysis of subcellular RNA fractions. Cell 2012, 150, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Danko, C.G.; Hah, N.; Luo, X.; Martins, A.L.; Core, L.; Lis, J.T.; Siepel, A.; Kraus, W.L. Signaling pathways differentially affect RNA polymerase ii initiation, pausing, and elongation rate in cells. Mol. Cell 2013, 50, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Targeting the epigenome in the treatment of asthma and chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2009, 6, 693–696. [Google Scholar] [CrossRef] [PubMed]
- Bayarsaihan, D. Epigenetic mechanisms in inflammation. J. Dent. Res. 2011, 90, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Saccani, S.; Natoli, G. Dynamic changes in histone H3 LYS 9 methylation occurring at tightly regulated inducible inflammatory genes. Genes Dev. 2002, 16, 2219–2224. [Google Scholar] [CrossRef] [PubMed]
- De Santa, F.; Totaro, M.G.; Prosperini, E.; Notarbartolo, S.; Testa, G.; Natoli, G. The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell 2007, 130, 1083–1094. [Google Scholar] [CrossRef] [PubMed]
- De Santa, F.; Narang, V.; Yap, Z.H.; Tusi, B.K.; Burgold, T.; Austenaa, L.; Bucci, G.; Caganova, M.; Notarbartolo, S.; Casola, S.; et al. Jmjd3 contributes to the control of gene expression in lps-activated macrophages. EMBO J. 2009, 28, 3341–3352. [Google Scholar] [CrossRef] [PubMed]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Liang, G. Rethinking how DNA methylation patterns are maintained. Nat. Rev. Genet. 2009, 10, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Cheung, H.H.; Lee, T.L.; Rennert, O.M.; Chan, W.Y. DNA methylation of cancer genome. Birth Defects Res. C Embryo Today 2009, 87, 335–350. [Google Scholar] [CrossRef] [PubMed]
- Shuto, T.; Furuta, T.; Oba, M.; Xu, H.; Li, J.D.; Cheung, J.; Gruenert, D.C.; Uehara, A.; Suico, M.A.; Okiyoneda, T.; et al. Promoter hypomethylation of toll-like receptor-2 gene is associated with increased proinflammatory response toward bacterial peptidoglycan in cystic fibrosis bronchial epithelial cells. FASEB J. 2006, 20, 782–784. [Google Scholar] [CrossRef] [PubMed]
- Heintzman, N.D.; Stuart, R.K.; Hon, G.; Fu, Y.; Ching, C.W.; Hawkins, R.D.; Barrera, L.O.; van Calcar, S.; Qu, C.; Ching, K.A.; et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007, 39, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Visel, A.; Blow, M.J.; Li, Z.; Zhang, T.; Akiyama, J.A.; Holt, A.; Plajzer-Frick, I.; Shoukry, M.; Wright, C.; Chen, F.; et al. Chip-seq accurately predicts tissue-specific activity of enhancers. Nature 2009, 457, 854–858. [Google Scholar] [CrossRef] [PubMed]
- Lefevre, P.; Witham, J.; Lacroix, C.E.; Cockerill, P.N.; Bonifer, C. The LPS-induced transcriptional upregulation of the chicken lysozyme locus involves CTCF eviction and noncoding RNA transcription. Mol. Cell 2008, 32, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Zaret, K.S.; Watts, J.; Xu, J.; Wandzioch, E.; Smale, S.T.; Sekiya, T. Pioneer factors, genetic competence, and inductive signaling: Programming liver and pancreas progenitors from the endoderm. Cold Spring Harb. Symp. Quant. Biol. 2008, 73, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Watts, J.A.; Pope, S.D.; Gadue, P.; Kamps, M.; Plath, K.; Zaret, K.S.; Smale, S.T. Transcriptional competence and the active marking of tissue-specific enhancers by defined transcription factors in embryonic and induced pluripotent stem cells. Genes Dev. 2009, 23, 2824–2838. [Google Scholar] [CrossRef] [PubMed]
- Foster, S.L.; Hargreaves, D.C.; Medzhitov, R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature 2007, 447, 972–978. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.; Levine, A. P53 research: The past thirty years and the next thirty years. Cold Spring Harb. Perspect. Biol. 2010. [Google Scholar] [CrossRef] [PubMed]
- Menendez, D.; Shatz, M.; Azzam, K.; Garantziotis, S.; Fessler, M.B.; Resnick, M.A. The toll-like receptor gene family is integrated into human DNA damage and p53 networks. PLoS Genet. 2011, 7, e1001360. [Google Scholar] [CrossRef] [PubMed]
- Lowe, J.M.; Menendez, D.; Bushel, P.R.; Shatz, M.; Kirk, E.L.; Troester, M.A.; Garantziotis, S.; Fessler, M.B.; Resnick, M.A. P53 and NF-kappaB coregulate proinflammatory gene responses in human macrophages. Cancer Res. 2014, 74, 2182–2192. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.; Mui, A.; Dedhar, S. Integrin-linked kinase regulates inducible nitric oxide synthase and cyclooxygenase-2 expression in an NF-kappa B-dependent manner. J. Biol. Chem. 2002, 277, 3109–3116. [Google Scholar] [CrossRef] [PubMed]
- Makino, K.; Day, C.P.; Wang, S.C.; Li, Y.M.; Hung, M.C. Upregulation of IKKalpha/IKKbeta by integrin-linked kinase is required for Her2/Neu-induced NF-kappaB antiapoptotic pathway. Oncogene 2004, 23, 3883–3887. [Google Scholar] [CrossRef] [PubMed]
- Agouni, A.; Sourbier, C.; Danilin, S.; Rothhut, S.; Lindner, V.; Jacqmin, D.; Helwig, J.J.; Lang, H.; Massfelder, T. Parathyroid hormone-related protein induces cell survival in human renal cell carcinoma through the pi3k AKT pathway: Evidence for a critical role for integrin-linked kinase and nuclear factor kappa B. Carcinogenesis 2007, 28, 1893–1901. [Google Scholar] [CrossRef] [PubMed]
- Medici, D.; Nawshad, A. Type I collagen promotes epithelial-mesenchymal transition through ILK-dependent activation of NF-kappab and LEF-1. Matrix Biol. 2010, 29, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Del Nogal, M.; Luengo, A.; Olmos, G.; Lasa, M.; Rodriguez-Puyol, D.; Rodriguez-Puyol, M.; Calleros, L. Balance between apoptosis or survival induced by changes in extracellular-matrix composition in human mesangial cells: A key role for ILK-NFkappab pathway. Apoptosis 2012, 17, 1261–1274. [Google Scholar] [CrossRef] [PubMed]
- Wani, A.A.; Jafarnejad, S.M.; Zhou, J.; Li, G. Integrin-linked kinase regulates melanoma angiogenesis by activating NF-kappaB/interleukin-6 signaling pathway. Oncogene 2011, 30, 2778–2788. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Jin, J.; Xu, S.; Liu, H.; Li, N.; Cao, X. Integrin CD11b negatively regulates TLR-triggered inflammatory responses by activating Syk and promoting degradation of MyD88 and TRIF via Cbl-b. Nat. Immunol. 2010, 11, 734–742. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Gordon, R.A.; Huynh, L.; Su, X.; Park Min, K.H.; Han, J.; Arthur, J.S.; Kalliolias, G.D.; Ivashkiv, L.B. Indirect inhibition of toll-like receptor and type I interferon responses by itam-coupled receptors and integrins. Immunity 2010, 32, 518–530. [Google Scholar] [CrossRef] [PubMed]
- Yee, N.K.; Hamerman, J.A. Beta2 integrins inhibit TLR responses by regulating NF-kappaB pathway and p38 MAPK activation. Eur. J. Immunol. 2013, 43, 779–792. [Google Scholar] [CrossRef] [PubMed]
- Kagan, J.C.; Medzhitov, R. Phosphoinositide-mediated adaptor recruitment controls toll-like receptor signaling. Cell 2006, 125, 943–955. [Google Scholar] [CrossRef] [PubMed]
- Medvedev, A.E.; Flo, T.; Ingalls, R.R.; Golenbock, D.T.; Teti, G.; Vogel, S.N.; Espevik, T. Involvement of CD14 and complement receptors CR3 and CR4 in nuclear factor-kappaB activation and TNF production induced by lipopolysaccharide and group B streptococcal cell walls. J. Immunol. 1998, 160, 4535–4542. [Google Scholar] [PubMed]
- Perera, P.Y.; Mayadas, T.N.; Takeuchi, O.; Akira, S.; Zaks-Zilberman, M.; Goyert, S.M.; Vogel, S.N. CD11b/CD18 acts in concert with CD14 and toll-like receptor (TLR) 4 to elicit full lipopolysaccharide and taxol-inducible gene expression. J. Immunol. 2001, 166, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Petzold, T.; Orr, A.W.; Hahn, C.; Jhaveri, K.A.; Parsons, J.T.; Schwartz, M.A. Focal adhesion kinase modulates activation of NF-kappaB by flow in endothelial cells. Am. J. Physiol. Cell Physiol. 2009, 297, C814–C822. [Google Scholar] [CrossRef] [PubMed]
- Kwok, T.; Zabler, D.; Urman, S.; Rohde, M.; Hartig, R.; Wessler, S.; Misselwitz, R.; Berger, J.; Sewald, N.; Konig, W.; et al. Helicobacter exploits integrin for type IV secretion and kinase activation. Nature 2007, 449, 862–866. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Zhao, S.; Wu, J.; Zheng, F.; Yang, L.; Hu, J.; Hann, S.S. Inhibition of integrin-linked kinase expression by emodin through crosstalk of ampkalpha and Erk1/2 signaling and reciprocal interplay of sp1 and C-jun. Cell. Signal. 2015, 27, 1469–1477. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Liu, Q.; Zhang, Y.; Zhao, L. Involvement of Ilk/erk1/2 and Ilk/p38 pathways in mediating the enhanced osteoblast differentiation by micro/nanotopography. Acta Biomater. 2014, 10, 3705–3715. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Yuan, X.; Wang, D.; Barakat, B.; Williams, E.D.; Hannigan, G.E. Selective regulation of p38beta protein and signaling by integrin-linked kinase mediates bladder cancer cell migration. Oncogene 2014, 33, 690–701. [Google Scholar] [CrossRef] [PubMed]
- Hashiramoto, A.; Murata, M.; Kawazoe, T.; Yoshida, K.; Akiyama, C.; Shiozawa, K.; Shiozawa, S. Heat shock protein 90 maintains the tumour-like character of rheumatoid synovial cells by stabilizing integrin-linked kinase, extracellular signal-regulated kinase and protein kinase B. Rheumatology 2011, 50, 852–861. [Google Scholar] [CrossRef] [PubMed]
- Noh, K.T.; Park, Y.M.; Cho, S.G.; Choi, E.J. Gsk-3beta-induced ask1 stabilization is crucial in Lps-induced endotoxin shock. Exp. Cell Res. 2011, 317, 1663–1668. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmed, A.U.; Williams, B.R.G.; Hannigan, G.E. Transcriptional Activation of Inflammatory Genes: Mechanistic Insight into Selectivity and Diversity. Biomolecules 2015, 5, 3087-3111. https://doi.org/10.3390/biom5043087
Ahmed AU, Williams BRG, Hannigan GE. Transcriptional Activation of Inflammatory Genes: Mechanistic Insight into Selectivity and Diversity. Biomolecules. 2015; 5(4):3087-3111. https://doi.org/10.3390/biom5043087
Chicago/Turabian StyleAhmed, Afsar U., Bryan R. G. Williams, and Gregory E. Hannigan. 2015. "Transcriptional Activation of Inflammatory Genes: Mechanistic Insight into Selectivity and Diversity" Biomolecules 5, no. 4: 3087-3111. https://doi.org/10.3390/biom5043087