
The Role of miRNAs in Common Inflammatory Arthropathies: Osteoarthritis and Gouty Arthritis

{kind=link}
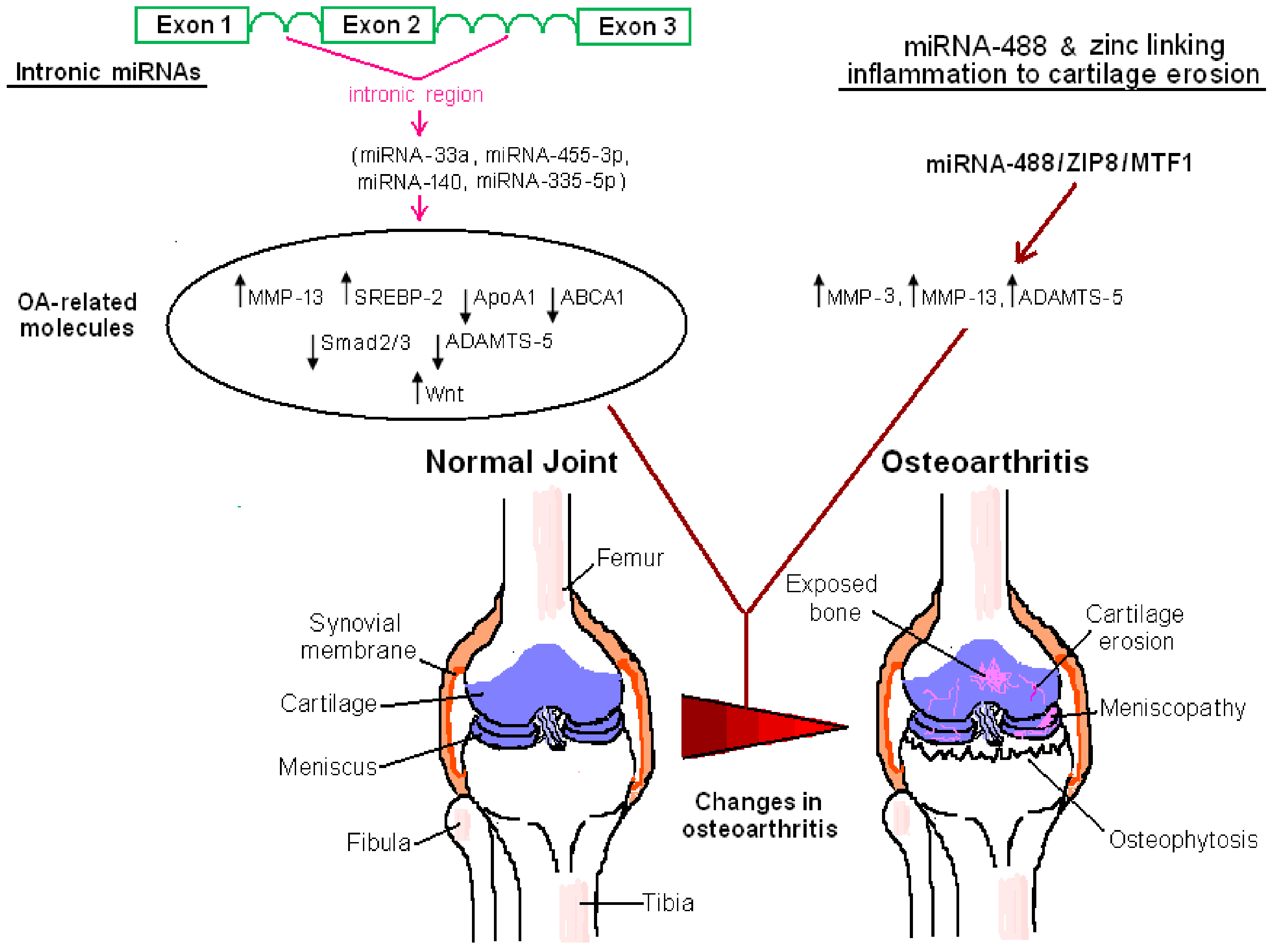
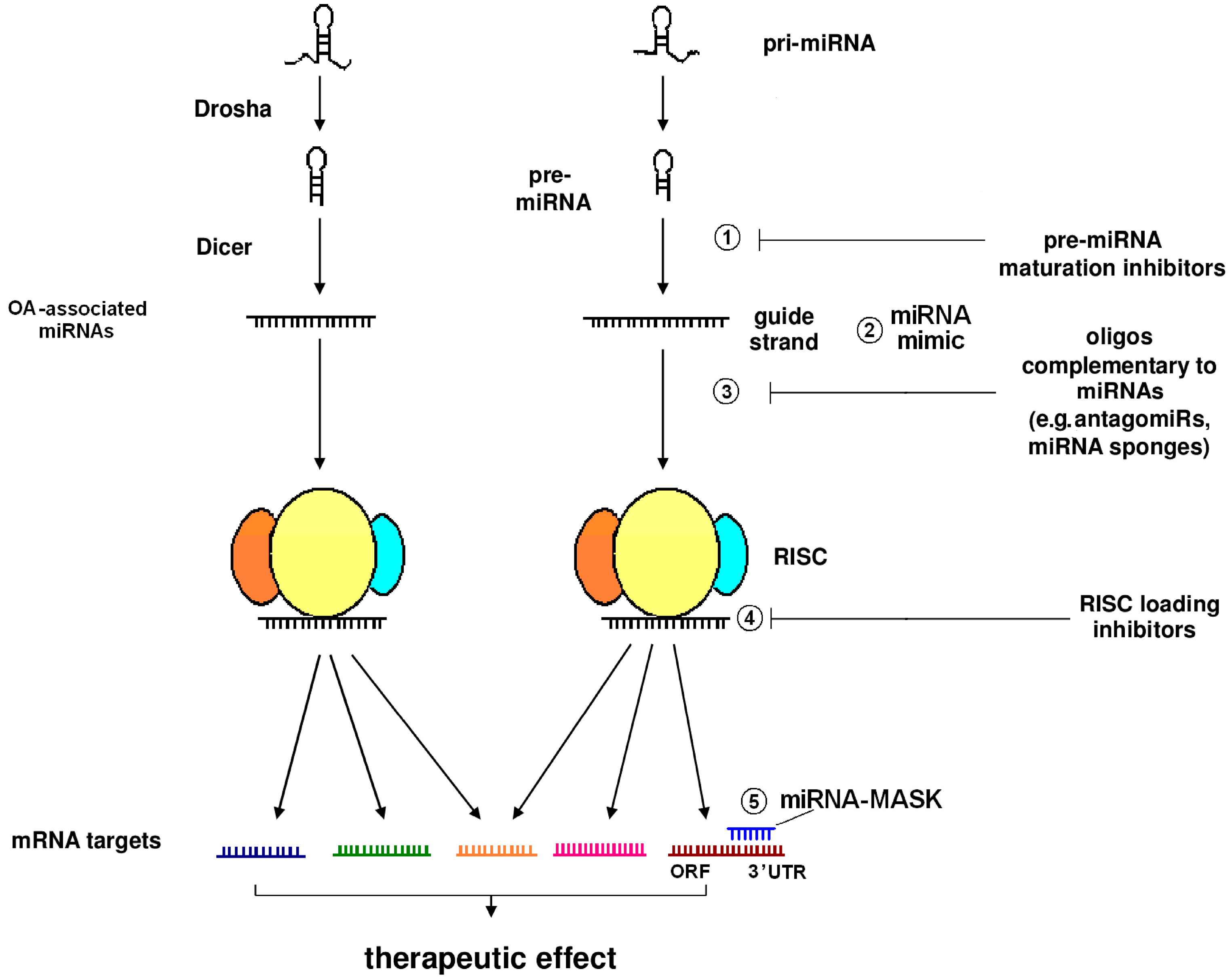{kind=link}
Abstract
:1. Introduction
2. OA and Gouty Arthritis: Overview of Pathophysiological Features
3. OA and Overweight: A Perplexed Interplay
4. miRNA-Associated Pathways in OA
4.1. Metabolic Pathways
4.2. Inflammatory Networks, Degenerative Enzymes, and Zinc as the Molecular Link among Them
4.2.1. Inflammatory Signaling Pathways, MMPs, and A Disintegrin and Metalloproteinase with Thrombospondin Motifs Enzymes
4.2.2. Zinc at the Crossroads of Inflammation and Cartilage Erosion
4.3. TGF-β/Smad, Wingless , and Vascular Endothelial Growth Factor Pathway
4.4. Autophagy
4.5. Nociception
4.6. Mesenchymal Stem Cell-Associated Pathways
5. miRNA-Associated Pathways in Gouty Arthritis
6. miRNA-Dependent Pathways as Therapeutic Targets in OA and Gouty Arthritis: At the Juncture of Biotechnology and Traditional Medicine
7. Conclusions and Future Perspectives
Acknowledgments
Conflicts of interest
References
- Saetrom, P.; Snove, O., Jr.; Rossi, J.J. Epigenetics and microRNAs. Pediatr. Res. 2007, 61, 17R–23R. [Google Scholar] [CrossRef] [PubMed]
- Selbach, M.; Schwanhausser, B.; Thierfelder, N.; Fang, Z.; Khanin, R.; Rajewsky, N. Widespread changes in protein synthesis induced by microRNAs. Nature 2008, 455, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Baek, D.; Villen, J.; Shin, C.; Camargo, F.D.; Gygi, S.P.; Bartel, D.P. The impact of microRNAs on protein output. Nature 2008, 455, 64–71. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, R.M.; Rao, D.S.; Chaudhuri, A.A.; Baltimore, D. Physiological and pathological roles for microRNAs in the immune system. Nat. Rev. Immunol. 2010, 10, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Gregory, R.I. MicroRNA biogenesis pathways in cancer. Nat. Rev. Cancer 2015, 15, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Pasquinelli, A.E.; Reinhart, B.J.; Slack, F.; Martindale, M.Q.; Kuroda, M.I.; Maller, B.; Hayward, D.C.; Ball, E.E.; Degnan, B.; Muller, P.; et al. Conservation of the sequence and temporal expression of let-7 heterochronic regulatory RNA. Nature 2000, 408, 86–89. [Google Scholar] [PubMed]
- Chen, C.Z.; Li, L.; Lodish, H.F.; Bartel, D.P. MicroRNAs modulate hematopoietic lineage differentiation. Science 2004, 303, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Nicolas, E.; Marks, D.; Sander, C.; Lerro, A.; Buendia, M.A.; Xu, C.; Mason, W.S.; Moloshok, T.; Bort, R.; et al. miR-122, a mammalian liver-specific microRNA, is processed from hcr mrna and may downregulate the high affinity cationic amino acid transporter CAT-1. RNA Biol. 2004, 1, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.F.; Mandel, E.M.; Thomson, J.M.; Wu, Q.; Callis, T.E.; Hammond, S.M.; Conlon, F.L.; Wang, D.Z. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat. Genet. 2006, 38, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Saini, H.K.; Enright, A.J.; Griffiths-Jones, S. Annotation of mammalian primary microRNAs. BMC Genom. 2008, 9, 564. [Google Scholar] [CrossRef] [PubMed]
- Baulina, N.M.; Kulakova, O.G.; Favorova, O.O. MicroRNAs: The role in autoimmune inflammation. Acta Nat. 2016, 8, 21–33. [Google Scholar]
- Hutvagner, G.; McLachlan, J.; Pasquinelli, A.E.; Balint, E.; Tuschl, T.; Zamore, P.D. A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science 2001, 293, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, E.; Caudy, A.A.; Hammond, S.M.; Hannon, G.J. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 2001, 409, 363–366. [Google Scholar] [CrossRef] [PubMed]
- Grishok, A.; Pasquinelli, A.E.; Conte, D.; Li, N.; Parrish, S.; Ha, I.; Baillie, D.L.; Fire, A.; Ruvkun, G.; Mello, C.C. Genes and mechanisms related to RNA interference regulate expression of the small temporal RNAs that control C. elegans developmental timing. Cell 2001, 106, 23–34. [Google Scholar] [CrossRef]
- Lee, Y.; Ahn, C.; Han, J.; Choi, H.; Kim, J.; Yim, J.; Lee, J.; Provost, P.; Radmark, O.; Kim, S.; et al. The nuclear RNase III Drosha initiates microRNA processing. Nature 2003, 425, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Kay, M.A. How do miRNAs mediate translational repression? Silence 2010, 1, 11. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.H.; Ghoshal, K. MicroRNAs in liver health and disease. Curr. Pathobiol. Rep. 2013, 1, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.P.; Massachi, I.; Manickavel, S.; Singh, S.; Rao, N.P.; Hasan, S.; Mc Curdy, D.K.; Sharma, S.; Wong, D.; Hahn, B.H.; et al. The role of miRNA in inflammation and autoimmunity. Autoimmun. Rev. 2013, 12, 1160–1165. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Santos, M.C.; Aragon-Raygoza, A.; Espinal-Centeno, A.; Arteaga-Vazquez, M.; Cruz-Hernandez, A.; Bako, L.; Cruz-Ramirez, A. The role of microRNAs in animal cell reprogramming. Stem Cells Dev. 2016, 25, 1035–1049. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, M.; Kishikawa, T.; Yoshikawa, T.; Yamagami, M.; Ohno, M.; Takata, A.; Shibata, C.; Ishibashi, R.; Koike, K. MicroRNAs and liver disease. J. Hum. Genet. 2016. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.D.; Li, L.; Chan, W.Y. Micrornas: Key regulators in the central nervous system and their implication in neurological diseases. Int. J. Mol. Sci. 2016, 17, 842. [Google Scholar] [CrossRef] [PubMed]
- Asghari, F.; Haghnavaz, N.; Baradaran, B.; Hemmatzadeh, M.; Kazemi, T. Tumor suppressor microRNAs: Targeted molecules and signaling pathways in breast cancer. Biomed. Pharmacother. 2016, 81, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Sanmarti, R.; Canete, J.D.; Salvador, G. Palindromic rheumatism and other relapsing arthritis. Best Pract. Res. Clin. Rheumatol. 2004, 18, 647–661. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Feng, M.; Wu, F.; Ma, X.; Lu, J.; Kang, M.; Liu, Z. Plasma miR-26a as a diagnostic biomarker regulates cytokine expression in systemic juvenile idiopathic arthritis. J. Rheumatol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Murphy, L.; Helmick, C.G. The impact of osteoarthritis in the united states: A population-health perspective. Am. J. Nurs. 2012, 112, S13–S19. [Google Scholar] [CrossRef] [PubMed]
- Nakasa, T.; Shibuya, H.; Nagata, Y.; Niimoto, T.; Ochi, M. The inhibitory effect of microRNA-146a expression on bone destruction in collagen-induced arthritis. Arthritis Rheum. 2011, 63, 1582–1590. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.T.; Chen, S.Y.; Wang, C.R.; Liu, M.F.; Lin, C.C.; Jou, I.M.; Shiau, A.L.; Wu, C.L. Brief report: Amelioration of collagen-induced arthritis in mice by lentivirus-mediated silencing of microRNA-223. Arthritis Rheum. 2012, 64, 3240–3245. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.S.; Chen, S.Y.; Wu, C.L.; Chong, H.E.; Ding, Y.C.; Shiau, A.L.; Wang, C.R. Amelioration of experimental autoimmune arthritis through targeting of synovial fibroblasts by intraarticular delivery of microRNAs 140-3p and 140-5p. Arthritis Rheumatol. 2016, 68, 370–381. [Google Scholar] [CrossRef] [PubMed]
- Ceribelli, A.; Nahid, M.A.; Satoh, M.; Chan, E.K. MicroRNAs in rheumatoid arthritis. FEBS Lett. 2011, 585, 3667–3674. [Google Scholar] [CrossRef] [PubMed]
- Churov, A.V.; Oleinik, E.K.; Knip, M. MicroRNAs in rheumatoid arthritis: Altered expression and diagnostic potential. Autoimmun. Rev. 2015, 14, 1029–1037. [Google Scholar] [CrossRef] [PubMed]
- Garo, L.P.; Murugaiyan, G. Contribution of microRNAs to autoimmune diseases. Cell. Mol. Life Sci. 2016, 73, 2041–2051. [Google Scholar] [CrossRef] [PubMed]
- Riches, P.L.; Wright, A.F.; Ralston, S.H. Recent insights into the pathogenesis of hyperuricaemia and gout. Hum. Mol. Genet. 2009, 18, R177–R184. [Google Scholar] [CrossRef] [PubMed]
- Kostopoulou, F.; Malizos, K.N.; Papathanasiou, I.; Tsezou, A. MicroRNA-33a regulates cholesterol synthesis and cholesterol efflux-related genes in osteoarthritic chondrocytes. Arthritis Res. Ther. 2015, 17, 42. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.Y.; Zhu, W.H.; Chen, Z.W.; Dai, H.L.; Ren, J.J.; Chen, J.H.; Chen, L.Q.; Fang, L.Z. Relationship between hyperuricemia and metabolic syndrome. J. Zhejiang Univ. Sci. B 2007, 8, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.K.; Ford, E.S. Prevalence of the metabolic syndrome in individuals with hyperuricemia. Am. J. Med. 2007, 120, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Chu, C.H.; Bai, C.H.; You, S.L.; Chou, Y.C.; Chou, W.Y.; Chien, K.L.; Hwang, L.C.; Su, T.C.; Tseng, C.H.; et al. Uric acid level as a risk marker for metabolic syndrome: A chinese cohort study. Atherosclerosis 2012, 220, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Le, L.T.; Swingler, T.E.; Clark, I.M. Review: The role of microRNAs in osteoarthritis and chondrogenesis. Arthritis Rheum. 2013, 65, 1963–1974. [Google Scholar] [CrossRef] [PubMed]
- Sondag, G.R.; Haqqi, T.M. The role of microRNAs and their targets in osteoarthritis. Curr. Rheumatol. Rep. 2016, 18, 56. [Google Scholar] [CrossRef] [PubMed]
- Plummer, J.; Park, M.; Perodin, F.; Horowitz, M.C.; Hens, J.R. Methionine-restricted diet increases miRNAs that can target RUNX2 expression and alters bone structure in young mice. J. Cell. Biochem. 2016. [Google Scholar] [CrossRef] [PubMed]
- Vinod, M.; Patankar, J.V.; Sachdev, V.; Frank, S.; Graier, W.F.; Kratky, D.; Kostner, G.M. miR-206 is expressed in pancreatic islets and regulates glucokinase activity. Am. J. Physiol. Endocrinol. Metab. 2016, 311, E175–E185. [Google Scholar] [CrossRef] [PubMed]
- Massart, J.; Katayama, M.; Krook, A. Micromanaging glucose and lipid metabolism in skeletal muscle: Role of microRNAs. Biochim. Biophys. Acta 2016. [Google Scholar] [CrossRef]
- Reyes, C.; Leyland, K.M.; Peat, G.; Cooper, C.; Arden, N.K.; Prieto-Alhambra, D. Association between overweight and obesity and risk of clinically diagnosed knee, hip, and hand osteoarthritis: A population-based cohort study. Arthritis Rheumatol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Sellam, J.; Berenbaum, F. The role of synovitis in pathophysiology and clinical symptoms of osteoarthritis. Nat. Rev. Rheumatol. 2010, 6, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Yin, J.; Gao, J.; Cheng, T.S.; Pavlos, N.J.; Zhang, C.; Zheng, M.H. Subchondral bone in osteoarthritis: Insight into risk factors and microstructural changes. Arthritis Res. Ther. 2013, 15, 223. [Google Scholar] [CrossRef] [PubMed]
- Mancarella, L.; Addimanda, O.; Cavallari, C.; Meliconi, R. Synovial inflammation drives structural damage in hand osteoarthritis: A narrative literature review. Curr. Rheumatol. Rev. 2016, in press. [Google Scholar] [CrossRef]
- Aspden, R.M.; Scheven, B.A.; Hutchison, J.D. Osteoarthritis as a systemic disorder including stromal cell differentiation and lipid metabolism. Lancet 2001, 357, 1118–1120. [Google Scholar] [CrossRef]
- Lieben, L. Osteoarthritis: Osteophyte formation involves PAR2. Nat. Rev. Rheumatol. 2016, 12, 70–71. [Google Scholar] [CrossRef] [PubMed]
- Hosaka, Y.; Saito, T.; Sugita, S.; Hikata, T.; Kobayashi, H.; Fukai, A.; Taniguchi, Y.; Hirata, M.; Akiyama, H.; Chung, U.I.; et al. Notch signaling in chondrocytes modulates endochondral ossification and osteoarthritis development. Proc. Natl. Acad. Sci. USA 2013, 110, 1875–1880. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Jeon, J.; Shin, M.; Won, Y.; Lee, M.; Kwak, J.S.; Lee, G.; Rhee, J.; Ryu, J.H.; Chun, C.H.; et al. Regulation of the catabolic cascade in osteoarthritis by the zinc-ZIP8-MTF1 axis. Cell 2014, 156, 730–743. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Won, Y.; Shin, Y.; Kim, J.H.; Chun, J.S. Reciprocal activation of hypoxia-inducible factor (HIF)-2alpha and the zinc-ZIP8-MTF1 axis amplifies catabolic signaling in osteoarthritis. Osteoarthr. Res. Soc. 2016, 24, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Kostopoulou, F.; Gkretsi, V.; Malizos, K.N.; Iliopoulos, D.; Oikonomou, P.; Poultsides, L.; Tsezou, A. Central role of SREBP-2 in the pathogenesis of osteoarthritis. PLoS ONE 2012, 7, e35753. [Google Scholar] [CrossRef] [PubMed]
- Portal-Nunez, S.; Esbrit, P.; Alcaraz, M.J.; Largo, R. Oxidative stress, autophagy, epigenetic changes and regulation by miRNAs as potential therapeutic targets in osteoarthritis. Biochem. Pharmacol. 2016, 108, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Tsezou, A.; Iliopoulos, D.; Malizos, K.N.; Simopoulou, T. Impaired expression of genes regulating cholesterol efflux in human osteoarthritic chondrocytes. J. Orthop. Res. 2010, 28, 1033–1039. [Google Scholar] [CrossRef] [PubMed]
- Prasadam, I.; Batra, J.; Perry, S.; Gu, W.; Crawford, R.; Xiao, Y. Systematic identification, characterization and target gene analysis of microRNAs involved in osteoarthritis subchondral bone pathogenesis. Calcif. Tissue Int. 2016, 99, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Rozelle, A.L.; Lepus, C.M.; Scanzello, C.R.; Song, J.J.; Larsen, D.M.; Crish, J.F.; Bebek, G.; Ritter, S.Y.; Lindstrom, T.M.; et al. Identification of a central role for complement in osteoarthritis. Nat. Med. 2011, 17, 1674–1679. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, N.; Rasheed, Z.; Ramamurthy, S.; Anbazhagan, A.N.; Voss, F.R.; Haqqi, T.M. MicroRNA-27b regulates the expression of matrix metalloproteinase 13 in human osteoarthritis chondrocytes. Arthritis Rheum. 2010, 62, 1361–1371. [Google Scholar] [CrossRef] [PubMed]
- Katz, J.D.; Agrawal, S.; Velasquez, M. Getting to the heart of the matter: Osteoarthritis takes its place as part of the metabolic syndrome. Curr. Opin. Rheumatol. 2010, 22, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Ahn, C.; Chun, C.H.; Jin, E.J. A long non-coding RNA, GAS5, plays a critical role in the regulation of miR-21 during osteoarthritis. J. Orthop. Res. 2014, 32, 1628–1635. [Google Scholar] [CrossRef] [PubMed]
- Nugent, M. MicroRNAs: Exploring new horizons in osteoarthritis. Osteoarthr. Res. Soc. 2016, 24, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Crowe, N.; Swingler, T.E.; Le, L.T.; Barter, M.J.; Wheeler, G.; Pais, H.; Donell, S.T.; Young, D.A.; Dalmay, T.; Clark, I.M. Detecting new microRNAs in human osteoarthritic chondrocytes identifies miR-3085 as a human, chondrocyte-selective, microRNA. Osteoarthr. Res. Soc. 2016, 24, 534–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jingsheng, S.; Yibing, W.; Jun, X.; Siqun, W.; Jianguo, W.; Feiyan, C.; Gangyong, H.; Jie, C. MicroRNAs are potential prognostic and therapeutic targets in diabetic osteoarthritis. J. Bone Miner. Metab. 2015, 33, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Huang, Y.; Cai, D.; Liu, J.; Cao, X. Analysis of differences in the molecular mechanism of rheumatoid arthritis and osteoarthritis based on integration of gene expression profiles. Immunol. Lett. 2015, 168, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Lawry, G.V.; Fan, P.T.; Bluestone, R. Polyarticular versus monoarticular gout: A prospective, comparative analysis of clinical features. Medicine 1988, 67, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Burns, C.M.; Wortmann, R.L. Latest evidence on gout management: What the clinician needs to know. Ther. Adv. Chronic Dis. 2012, 3, 271–286. [Google Scholar] [CrossRef] [PubMed]
- Sewell, K.L.; Petrucci, R.; Keiser, H.D. Misdiagnosis of rheumatoid arthritis in an elderly woman with gout. J. Am. Geriatr. Soc. 1991, 39, 403–406. [Google Scholar] [CrossRef] [PubMed]
- Scott, P.; Ma, H.; Viriyakosol, S.; Terkeltaub, R.; Liu-Bryan, R. Engagement of CD14 mediates the inflammatory potential of monosodium urate crystals. J. Immunol. 2006, 177, 6370–6378. [Google Scholar] [CrossRef] [PubMed]
- Busso, N.; So, A. Mechanisms of inflammation in gout. Arthritis Res. Ther. 2010, 12, 206. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, D.; Wang, B.; Hou, X. Could microRNAs be regulators of gout pathogenesis? Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2015, 36, 2085–2092. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, D.; Malizos, K.N.; Oikonomou, P.; Tsezou, A. Integrative microRNA and proteomic approaches identify novel osteoarthritis genes and their collaborative metabolic and inflammatory networks. PLoS ONE 2008, 3, e3740. [Google Scholar] [CrossRef]
- Dunn, W.; DuRaine, G.; Reddi, A.H. Profiling microRNA expression in bovine articular cartilage and implications for mechanotransduction. Arthritis Rheum. 2009, 60, 2333–2339. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.J.; Yang, X.; Wei, L.; Chen, Q. miR-365: A mechanosensitive microRNA stimulates chondrocyte differentiation through targeting histone deacetylase 4. FASEB J. 2011, 25, 4457–4466. [Google Scholar] [CrossRef] [PubMed]
- Ge, Q.; Brichard, S.; Yi, X.; Li, Q. MicroRNAs as a new mechanism regulating adipose tissue inflammation in obesity and as a novel therapeutic strategy in the metabolic syndrome. J. Immunol. Res. 2014, 2014, 987285. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M. The role of PPARalpha in lipid metabolism and obesity: Focusing on the effects of estrogen on PPARalpha actions. Pharmacol. Res. 2009, 60, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Wei, M.; Kang, X.; Liu, D.; Quan, Y.; Pan, X.; Liu, X.; Liao, D.; Liu, J.; Zhang, B. Reciprocal inhibition between miR-26a and NF-kappaB regulates obesity-related chronic inflammation in chondrocytes. Biosci. Rep. 2015, 35, e00204. [Google Scholar] [PubMed]
- Tardif, G.; Hum, D.; Pelletier, J.P.; Duval, N.; Martel-Pelletier, J. Regulation of the IGFBP-5 and MMP-13 genes by the microRNAs miR-140 and miR-27a in human osteoarthritic chondrocytes. BMC Musculoskelet. Dis. 2009, 10, 148. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Kim, A.Y.; Lee, H.W.; Son, Y.H.; Lee, G.Y.; Lee, J.W.; Lee, Y.S.; Kim, J.B. miR-27a is a negative regulator of adipocyte differentiation via suppressing PPARgamma expression. Biochem. Biophys. Res. Commun. 2010, 392, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Marquart, T.J.; Allen, R.M.; Ory, D.S.; Baldan, A. miR-33 links SREBP-2 induction to repression of sterol transporters. Proc. Natl. Acad. Sci. USA 2010, 107, 12228–12232. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, C.M.; Goedeke, L.; Fernandez-Hernando, C. “Micromanaging” metabolic syndrome. Cell Cycle 2011, 10, 3249–3252. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.H.; Elvington, A.; Randolph, G.J. The role of the lymphatic system in cholesterol transport. Front. Pharmacol. 2015, 6, 182. [Google Scholar] [CrossRef] [PubMed]
- Blaney Davidson, E.N.; Remst, D.F.; Vitters, E.L.; van Beuningen, H.M.; Blom, A.B.; Goumans, M.J.; van den Berg, W.B.; van der Kraan, P.M. Increase in ALK1/ALK5 ratio as a cause for elevated MMP-13 expression in osteoarthritis in humans and mice. J. Immunol. 2009, 182, 7937–7945. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.W.; Watkins, G.; Le Good, N.; Roberts, S.; Murphy, C.L.; Brockbank, S.M.; Needham, M.R.; Read, S.J.; Newham, P. The identification of differentially expressed microRNA in osteoarthritic tissue that modulate the production of TNF-alpha and MMP13. Osteoarthr. Res. Soc. 2009, 17, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Cao, X.; Li, J.; Zhao, G. miR-210 inhibits NF-kappaB signaling pathway by targeting DR6 in osteoarthritis. Sci. Rep. 2015, 5, 12775. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Sun, Y.; Wang, Y.; Liu, R.; Bao, Y.; Li, Q. miR-502–5p inhibits IL-1beta-induced chondrocyte injury by targeting TRAF2. Cell. Immunol. 2016, 302, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Le, L.T.; Swingler, T.E.; Crowe, N.; Vincent, T.L.; Barter, M.J.; Donell, S.T.; Delany, A.M.; Dalmay, T.; Young, D.A.; Clark, I.M. The microRNA-29 family in cartilage homeostasis and osteoarthritis. J. Mol. Med. 2016, 94, 583–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santini, P.; Politi, L.; Dalla Vedova, P.; Scandurra, R.; d’Abusco, A.S. The inflammatory circuitry of miR-149 as a pathological mechanism in osteoarthritis. Rheumatol. Int. 2014, 34, 711–716. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, Z.; Al-Shobaili, H.A.; Rasheed, N.; Al Salloom, A.A.; Al-Shaya, O.; Mahmood, A.; Alajez, N.M.; Alghamdi, A.S.; Mehana el, S.E. Integrated study of globally expressed microRNAs in IL-1beta-stimulated human osteoarthritis chondrocytes and osteoarthritis relevant genes: A microarray and bioinformatics analysis. Nucleosides Nucleotides Nucl. Acids 2016, 35, 335–355. [Google Scholar] [CrossRef] [PubMed]
- Ji, Q.; Xu, X.; Xu, Y.; Fan, Z.; Kang, L.; Li, L.; Liang, Y.; Guo, J.; Hong, T.; Li, Z.; et al. miR-105/Runx2 axis mediates FGF2-induced ADAMTs expression in osteoarthritis cartilage. J. Mol. Med. 2016, 94, 681–694. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Jia, J.; Liu, X.; Yang, S.; Ye, S.; Yang, W.; Zhang, Y. MicroRNA-16–5p controls development of osteoarthritis by targeting SMAD3 in chondrocytes. Curr. Pharm. Des. 2015, 21, 5160–5167. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Liu, Y.; Deng, M.; Li, J.; Cai, H.; Meng, Q.; Fang, W.; Long, X.; Ke, J. MicroRNA221–3p modulates Ets-1 expression in synovial fibroblasts from patients with osteoarthritis of temporomandibular joint. Osteoarthr. Res. Soc. 2016, 24, 2003–2011. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Jin, E.H.; Kim, D.; Kim, K.Y.; Chun, C.H.; Jin, E.J. MicroRNA-222 regulates MMP-13 via targeting HDAC-4 during osteoarthritis pathogenesis. BBA Clin. 2015, 3, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, N.; Makki, M.S.; Haqqi, T.M. MicroRNA-602 and microRNA-608 regulate sonic hedgehog expression via target sites in the coding region in human chondrocytes. Arthritis Rheumatol. 2015, 67, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Miyaki, S.; Sato, T.; Inoue, A.; Otsuki, S.; Ito, Y.; Yokoyama, S.; Kato, Y.; Takemoto, F.; Nakasa, T.; Yamashita, S.; et al. MicroRNA-140 plays dual roles in both cartilage development and homeostasis. Genes Dev. 2010, 24, 1173–1185. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.J.; Zhuang, H.; Wang, G.X.; Li, Z.; Zhang, H.T.; Yu, T.Q.; Zhang, B.D. MiRNA-140 is a negative feedback regulator of MMP-13 in IL-1beta-stimulated human articular chondrocyte C28/I2 cells. Inflamm. Res. 2012, 61, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Makki, M.S.; Haqqi, T.M. niR-139 modulates MCPIP1/IL-6 expression and induces apoptosis in human OA chondrocytes. Exp. Mol. Med. 2015, 47, e189. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Zhang, X.; Hu, X.; Liu, Q.; Man, Z.; Huang, H.; Meng, Q.; Zhou, C.; Ao, Y. Silencing of miR-101 prevents cartilage degradation by regulating extracellular matrix-related genes in a rat model of osteoarthritis. Mol. Ther. 2015, 23, 1331–1340. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Guo, Y.; Wang, C.; Yu, H.; Yu, X.; Yu, H. MicroRNA-142-3p inhibits chondrocyte apoptosis and inflammation in osteoarthritis by targeting HMGB1. Inflammation 2016, 39, 1718–1728. [Google Scholar] [CrossRef] [PubMed]
- Katiyar, S.K.; Raman, C. Green tea: A new option for the prevention or control of osteoarthritis. Arthritis Res. Ther. 2011, 13, 121. [Google Scholar] [CrossRef] [PubMed]
- Leong, D.J.; Choudhury, M.; Hanstein, R.; Hirsh, D.M.; Kim, S.J.; Majeska, R.J.; Schaffler, M.B.; Hardin, J.A.; Spray, D.C.; Goldring, M.B.; et al. Green tea polyphenol treatment is chondroprotective, anti-inflammatory and palliative in a mouse post-traumatic osteoarthritis model. Arthritis Res. Ther. 2014, 16, 508. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, Z.; Rasheed, N.; Al-Shobaili, H.A. Epigallocatechin-3-O-gallate up-regulates microRNA-199a-3p expression by down-regulating the expression of cyclooxygenase-2 in stimulated human osteoarthritis chondrocytes. J. Cell. Mol. Med. 2016. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, K.; Nakasa, T.; Miyaki, S.; Ishikawa, M.; Deie, M.; Adachi, N.; Yasunaga, Y.; Asahara, H.; Ochi, M. Expression of microRNA-146a in osteoarthritis cartilage. Arthritis Rheum. 2009, 60, 1035–1041. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Gibson, G.; Kim, J.S.; Kroin, J.; Xu, S.; van Wijnen, A.J.; Im, H.J. MicroRNA-146a is linked to pain-related pathophysiology of osteoarthritis. Gene 2011, 480, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Ma, N.; Yan, F.; Yu, Z.; Wu, G.; Qiao, Y.; Han, D.; Xiang, Y.; Li, F.; Wang, W.; et al. The expression of intronic miRNAs, miR-483 and miR-483*, and their host gene, IGF2, in murine osteoarthritis cartilage. Int. J. Biol. Macromol. 2013, 61, 43–49. [Google Scholar] [CrossRef]
- Song, J.; Kim, D.; Lee, C.H.; Lee, M.S.; Chun, C.H.; Jin, E.J. MicroRNA-488 regulates zinc transporter SLC39A8/ZIP8 during pathogenesis of osteoarthritis. J. Biomed. Sci. 2013, 20, 31. [Google Scholar] [CrossRef]
- Kraus, V.B. Osteoarthritis: The zinc link. Nature 2014, 507, 441–442. [Google Scholar] [CrossRef] [PubMed]
- Rushton, M.D.; Young, D.A.; Loughlin, J.; Reynard, L.N. Differential DNA methylation and expression of inflammatory and zinc transporter genes defines subgroups of osteoarthritic hip patients. Ann. Rheum. Dis. 2015, 74, 1778–1782. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Prado, S.; Cicione, C.; Muinos-Lopez, E.; Hermida-Gomez, T.; Oreiro, N.; Fernandez-Lopez, C.; Blanco, F.J. Characterization of microRNA expression profiles in normal and osteoarthritic human chondrocytes. BMC Musculoskelet. Dis. 2012, 13, 144. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Yang, C.; Song, Y.; Liu, W.; Wang, K.; Li, S.; Zhang, Y. MicroRNA-23a-3p promotes the development of osteoarthritis by directly targeting SMAD3 in chondrocytes. Biochem. Biophys. Res. Commun. 2016, 478, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Swingler, T.E.; Wheeler, G.; Carmont, V.; Elliott, H.R.; Barter, M.J.; Abu-Elmagd, M.; Donell, S.T.; Boot-Handford, R.P.; Hajihosseini, M.K.; Munsterberg, A.; et al. The expression and function of microRNAs in chondrogenesis and osteoarthritis. Arthritis Rheum. 2012, 64, 1909–1919. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Inloes, J.B.; Katagiri, T.; Kobayashi, T. Chondrocyte-specific microRNA-140 regulates endochondral bone development and targets Dnpep to modulate bone morphogenetic protein signaling. Mol. Cell. Biol. 2011, 31, 3019–3028. [Google Scholar] [CrossRef] [PubMed]
- Hjorten, R.; Hansen, U.; Underwood, R.A.; Telfer, H.E.; Fernandes, R.J.; Krakow, D.; Sebald, E.; Wachsmann-Hogiu, S.; Bruckner, P.; Jacquet, R.; et al. Type XXVII collagen at the transition of cartilage to bone during skeletogenesis. Bone 2007, 41, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Chen, X.; Shim, J.H.; Huang, Z.; Brady, N.; Hu, D.; Drapp, R.; Sigrist, K.; Glimcher, L.H.; Jones, D. The E3 ubiquitin ligase WWP2 regulates craniofacial development through mono-ubiquitylation of goosecoid. Nat. Cell Biol. 2011, 13, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Huang, J.; Dai, L.; Yu, D.; Chen, Q.; Zhang, X.; Dai, K. miR-146a, an IL-1beta responsive miRNA, induces vascular endothelial growth factor and chondrocyte apoptosis by targeting Smad4. Arthritis Res. Ther. 2012, 14, R75. [Google Scholar] [CrossRef] [PubMed]
- Carames, B.; Taniguchi, N.; Otsuki, S.; Blanco, F.J.; Lotz, M. Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. Arthritis Rheum. 2010, 62, 791–801. [Google Scholar] [CrossRef] [PubMed]
- D’Adamo, S.; Alvarez-Garcia, O.; Muramatsu, Y.; Flamigni, F.; Lotz, M.K. MicroRNA-155 suppresses autophagy in chondrocytes by modulating expression of autophagy proteins. Osteoarthr. Res. Soc. 2016, 24, 1082–1091. [Google Scholar] [CrossRef] [PubMed]
- Premkumar, L.S.; Sikand, P. TRPV1: A target for next generation analgesics. Curr. Neuropharmacol. 2008, 6, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Tornero-Esteban, P.; Rodriguez-Rodriguez, L.; Abasolo, L.; Tome, M.; Lopez-Romero, P.; Herranz, E.; Gonzalez, M.A.; Marco, F.; Moro, E.; Fernandez-Gutierrez, B.; et al. Signature of microRNA expression during osteogenic differentiation of bone marrow MSCs reveals a putative role of miR-335-5p in osteoarthritis. BMC Musculoskelet. Dis. 2015, 16, 182. [Google Scholar] [CrossRef] [PubMed]
- Wen, C.; Lu, W.W.; Chiu, K.Y. Importance of subchondral bone in the pathogenesis and management of osteoarthritis from bench to bed. J. Orthop. Trans. 2014, 2, 16–25. [Google Scholar] [CrossRef]
- Tian, Y.; Guo, R.; Shi, B.; Chen, L.; Yang, L.; Fu, Q. MicroRNA-30a promotes chondrogenic differentiation of mesenchymal stem cells through inhibiting delta-like 4 expression. Life Sci. 2016, 148, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Ham, O.; Lee, C.Y.; Kim, R.; Lee, J.; Oh, S.; Lee, M.Y.; Kim, J.; Hwang, K.C.; Maeng, L.S.; Chang, W. Therapeutic potential of differentiated mesenchymal stem cells for treatment of osteoarthritis. Int. J. Mol. Sci. 2015, 16, 14961–14978. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.M.; Kim, T.J.; Choi, J.H.; Kim, M.J.; Cho, Y.N.; Nam, K.I.; Kee, S.J.; Moon, J.B.; Choi, S.Y.; Park, D.J.; et al. MicroRNA-155 as a proinflammatory regulator via SHIP-1 down-regulation in acute gouty arthritis. Arthritis Res. Ther. 2014, 16, R88. [Google Scholar] [CrossRef] [PubMed]
- Dalbeth, N.; Pool, B.; Shaw, O.M.; Harper, J.L.; Tan, P.; Franklin, C.; House, M.E.; Cornish, J.; Naot, D. Role of miR-146a in regulation of the acute inflammatory response to monosodium urate crystals. Ann. Rheum. Dis. 2015, 74, 786–790. [Google Scholar] [CrossRef] [PubMed]
- Marsland, D.; Mumith, A.; Barlow, I.W. Systematic review: The safety of intra-articular corticosteroid injection prior to total knee arthroplasty. Knee 2014, 21, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Lapi, F.; Piccinni, C.; Simonetti, M.; Levi, M.; Lora Aprile, P.; Cricelli, I.; Cricelli, C.; Fanelli, A. Non-steroidal anti-inflammatory drugs and risk of cerebrovascular events in patients with osteoarthritis: A nested case-control study. Intern. Emerg. Med. 2016, 11, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, J.P.; Martel-Pelletier, J.; Rannou, F.; Cooper, C. Efficacy and safety of oral NSAIDs and analgesics in the management of osteoarthritis: Evidence from real-life setting trials and surveys. Semin. Arthritis Rheum. 2016, 45, S22–S27. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.M.; Meng, H.Y.; Yuan, X.L.; Wang, Y.; Guo, Q.Y.; Peng, J.; Wang, A.Y.; Lu, S.B. MicroRNAs’ involvement in osteoarthritis and the prospects for treatments. Evid. Based Complement. Altern. Med. 2015, 2015, 236179. [Google Scholar] [CrossRef] [PubMed]
- Jopling, C.L.; Yi, M.; Lancaster, A.M.; Lemon, S.M.; Sarnow, P. Modulation of hepatitis C virus RNA abundance by a liver-specific microRNA. Science 2005, 309, 1577–1581. [Google Scholar] [CrossRef] [PubMed]
- Krutzfeldt, J.; Rajewsky, N.; Braich, R.; Rajeev, K.G.; Tuschl, T.; Manoharan, M.; Stoffel, M. Silencing of microRNAs in vivo with ‘antagomirs’. Nature 2005, 438, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Stenvang, J.; Petri, A.; Lindow, M.; Obad, S.; Kauppinen, S. Inhibition of microRNA function by antimiR oligonucleotides. Silence 2012, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Sastre, A.; Evans, M.J. miR-122 is more than a shield for the hepatitis C virus genome. Proc. Natl. Acad. Sci. USA 2013, 110, 1571–1572. [Google Scholar] [CrossRef] [PubMed]
- Janssen, H.L.; Reesink, H.W.; Lawitz, E.J.; Zeuzem, S.; Rodriguez-Torres, M.; Patel, K.; van der Meer, A.J.; Patick, A.K.; Chen, A.; Zhou, Y.; et al. Treatment of HCV infection by targeting microRNA. N. Engl. J. Med. 2013, 368, 1685–1694. [Google Scholar] [CrossRef] [PubMed]
- Obad, S.; dos Santos, C.O.; Petri, A.; Heidenblad, M.; Broom, O.; Ruse, C.; Fu, C.; Lindow, M.; Stenvang, J.; Straarup, E.M.; et al. Silencing of microRNA families by seed-targeting tiny LNAs. Nat. Genet. 2011, 43, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.J.; Saltzman, W.M.; Slack, F.J. Canonical and non-canonical barriers facing antimiR cancer therapeutics. Curr. Med. Chem. 2013, 20, 3582–3593. [Google Scholar] [CrossRef] [PubMed]
- Ebert, M.S.; Neilson, J.R.; Sharp, P.A. MicroRNA sponges: Competitive inhibitors of small RNAs in mammalian cells. Nat. Methods 2007, 4, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Ebert, M.S.; Sharp, P.A. MicroRNA sponges: Progress and possibilities. RNA 2010, 16, 2043–2050. [Google Scholar] [CrossRef] [PubMed]
- Garzon, R.; Marcucci, G.; Croce, C.M. Targeting microRNAs in cancer: Rationale, strategies and challenges. Nat. Rev. Drug Dis. 2010, 9, 775–789. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z. The guideline of the design and validation of miRNA mimics. Methods Mol. Biol. 2011, 676, 211–223. [Google Scholar] [PubMed]
- Van den Berg, F.T.; Rossi, J.J.; Arbuthnot, P.; Weinberg, M.S. Design of effective primary microRNA mimics with different basal stem conformations. Mol. Ther. Nucl. Acids 2016, 5, e278. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Yang, C.; Liu, X.; Yang, S.; Ye, S.; Jia, J.; Liu, W.; Zhang, Y. Elevated expression of microRNA-30b in osteoarthritis and its role in ERG regulation of chondrocyte. Biomed. Pharmacother. 2015, 76, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Wang, S.; Cai, H.; Lin, Y.; Zheng, X.; Zhang, B.; Xia, C. Overexpression of microRNA-634 suppresses survival and matrix synthesis of human osteoarthritis chondrocytes by targeting PIK3R1. Sci. Rep. 2016, 6, 23117. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jia, J.; Yang, S.; Liu, X.; Ye, S.; Tian, H. MicroRNA-21 controls the development of osteoarthritis by targeting GDF-5 in chondrocytes. Exp. Mol. Med. 2014, 46, e79. [Google Scholar] [CrossRef] [PubMed]
- Tan, G.S.; Chiu, C.H.; Garchow, B.G.; Metzler, D.; Diamond, S.L.; Kiriakidou, M. Small molecule inhibition of RISC loading. ACS Chem. Biol. 2012, 7, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Vigorito, E.; Perks, K.L.; Abreu-Goodger, C.; Bunting, S.; Xiang, Z.; Kohlhaas, S.; Das, P.P.; Miska, E.A.; Rodriguez, A.; Bradley, A.; et al. MicroRNA-155 regulates the generation of immunoglobulin class-switched plasma cells. Immunity 2007, 27, 847–859. [Google Scholar] [CrossRef] [PubMed]
- Asahara, H. Current status and strategy of microRNA research for cartilage development and osteoarthritis pathogenesis. J. Bone Metab. 2016, 23, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, Z.; Gemeinhart, R.A. Progress in microRNA delivery. J. Control. Release 2013, 172, 962–974. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.F.; Zhang, X.X.; Sun, F.Y.; Xu, W.; Liang, J.; Feng, S.M.; Wang, T. MicroRNA expression patterns of the kidney in hyperuricemia mice treated with Xiezhuo Chubi decoction. Chin. J. Integr. Med. 2011, 17, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.F.; Zhu, M.M.; Li, J.; Zhang, X.X.; Liu, Y.W.; Wu, X.R.; Liu, Z.G. Effects of Xie-Zhuo-Chu-Bi-Fang on miR-34a and URAT1 and their relationship in hyperuricemic mice. J. Ethnopharmacol. 2015, 161, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Knake, C.; Stamp, L.; Bahn, A. Molecular mechanism of an adverse drug-drug interaction of allopurinol and furosemide in gout treatment. Biochem. Biophys. Res. Commun. 2014, 452, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Sakiyama, M.; Matsuo, H.; Shimizu, S.; Nakashima, H.; Nakamura, T.; Nakayama, A.; Higashino, T.; Naito, M.; Suma, S.; Hishida, A.; et al. The effects of URAT1/SLC22A12 nonfunctional variants, R90H and W258X, on serum uric acid levels and gout/hyperuricemia progression. Sci. Rep. 2016, 6, 20148. [Google Scholar] [CrossRef] [PubMed]
- Dalbeth, N.; Stamp, L. Allopurinol dosing in renal impairment: Walking the tightrope between adequate urate lowering and adverse events. Semin. Dial. 2007, 20, 391–395. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.; Griffiths-Jones, S.; Ashurst, J.L.; Bradley, A. Identification of mammalian microRNA host genes and transcription units. Genome Res. 2004, 14, 1902–1910. [Google Scholar] [CrossRef] [PubMed]
- Franca, G.S.; Vibranovski, M.D.; Galante, P.A. Host gene constraints and genomic context impact the expression and evolution of human micrornas. Nat. Commun. 2016, 7, 11438. [Google Scholar] [CrossRef] [PubMed]
- Bali, K.K.; Kuner, R. Noncoding RNAs: Key molecules in understanding and treating pain. Trends Mol. Med. 2014, 20, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Beyer, C.; Zampetaki, A.; Lin, N.Y.; Kleyer, A.; Perricone, C.; Iagnocco, A.; Distler, A.; Langley, S.R.; Gelse, K.; Sesselmann, S.; et al. Signature of circulating microRNAs in osteoarthritis. Ann. Rheum. Dis. 2015, 74, e18. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Cheng, Q.; Zhang, B.H.; Zhang, M.Z. Circulating microRNAs as biomarkers in hepatocellular carcinoma screening: A validation set from China. Medicine 2015, 94, e603. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.T.; Liu, S.M.; Ma, H.; Yang, Y.; Zhang, X.; Sun, H.; Zhang, X.; Xu, J.; Wang, J. Systematic review and meta-analysis: Circulating miRNAs for diagnosis of hepatocellular carcinoma. J. Cell. Physiol. 2016, 231, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Kung, L.H.; Zaki, S.; Ravi, V.; Rowley, L.; Smith, M.M.; Bell, K.M.; Bateman, J.F.; Little, C.B. Utility of circulating serum miRNAs as biomarkers of early cartilage degeneration in animal models of post-traumatic osteoarthritis and inflammatory arthritis. Osteoarthr. Res. Soc. 2016, in press. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papanagnou, P.; Stivarou, T.; Tsironi, M. The Role of miRNAs in Common Inflammatory Arthropathies: Osteoarthritis and Gouty Arthritis. Biomolecules 2016, 6, 44. https://doi.org/10.3390/biom6040044
Papanagnou P, Stivarou T, Tsironi M. The Role of miRNAs in Common Inflammatory Arthropathies: Osteoarthritis and Gouty Arthritis. Biomolecules. 2016; 6(4):44. https://doi.org/10.3390/biom6040044
Chicago/Turabian StylePapanagnou, Panagiota, Theodora Stivarou, and Maria Tsironi. 2016. "The Role of miRNAs in Common Inflammatory Arthropathies: Osteoarthritis and Gouty Arthritis" Biomolecules 6, no. 4: 44. https://doi.org/10.3390/biom6040044