
The Emergence of Pan-Cancer CIMP and Its Elusive Interpretation

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Identification of CIMP in Colorectal Cancer
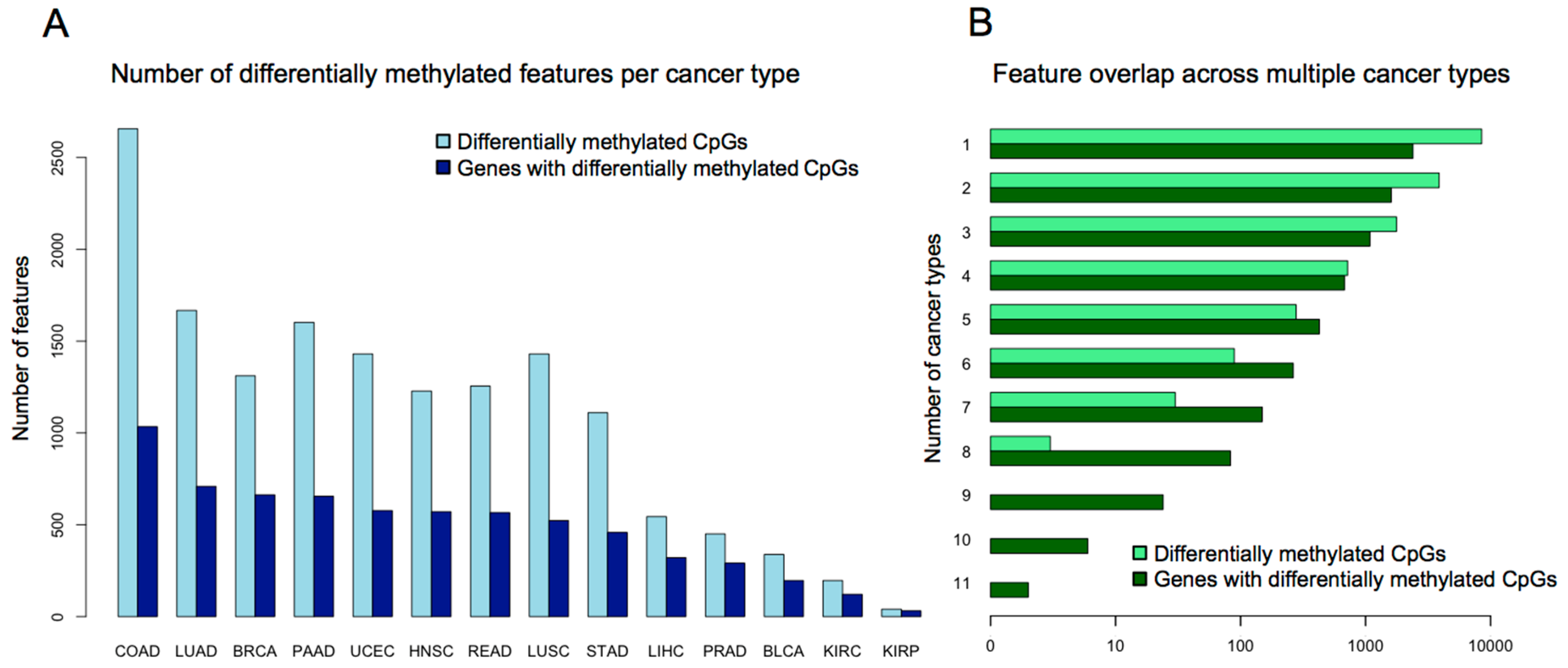
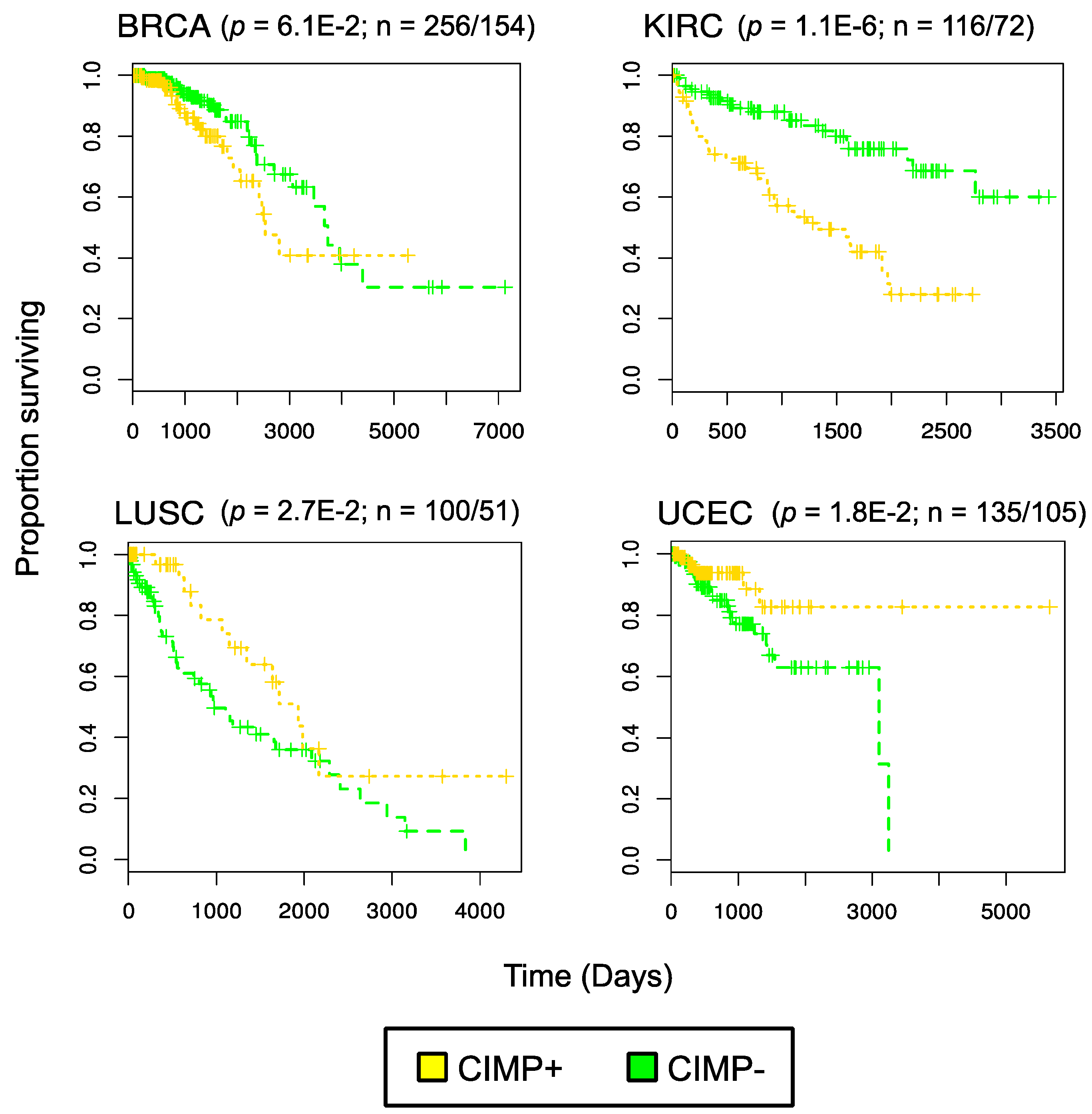
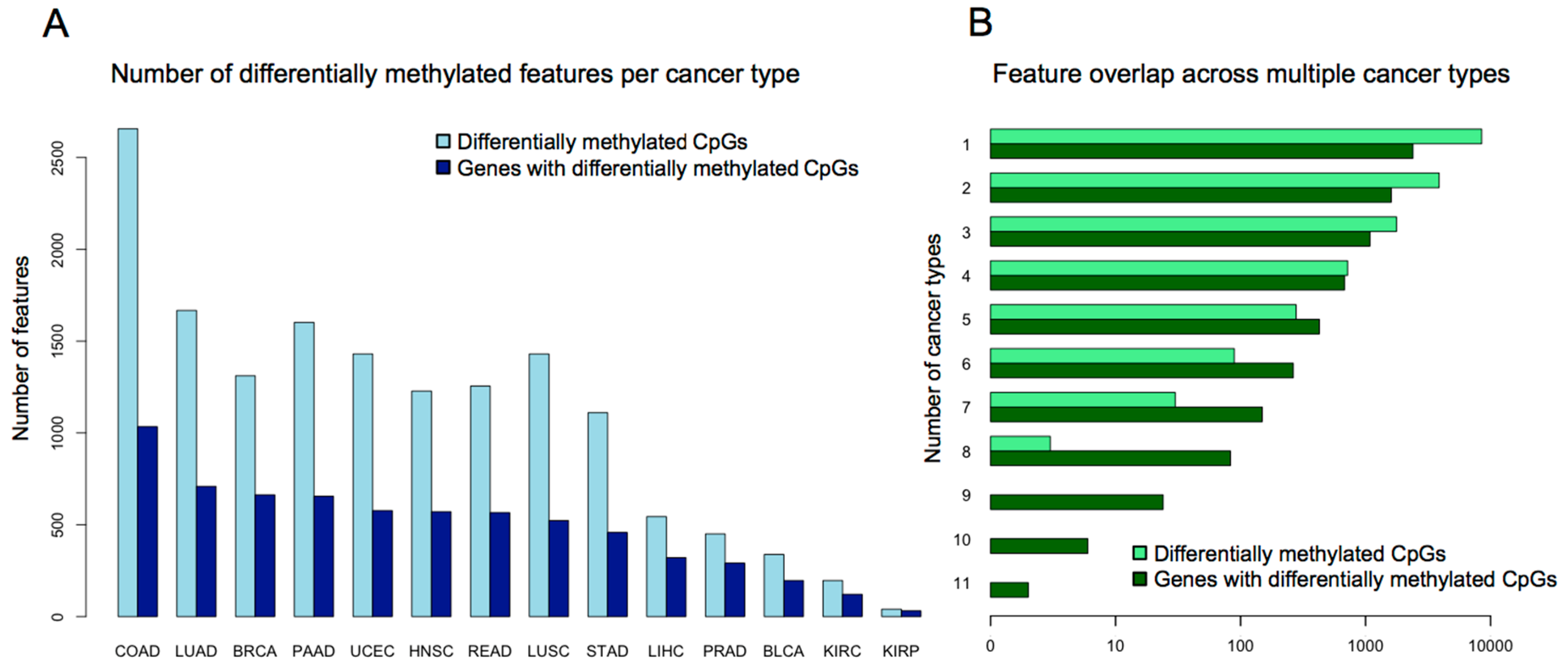
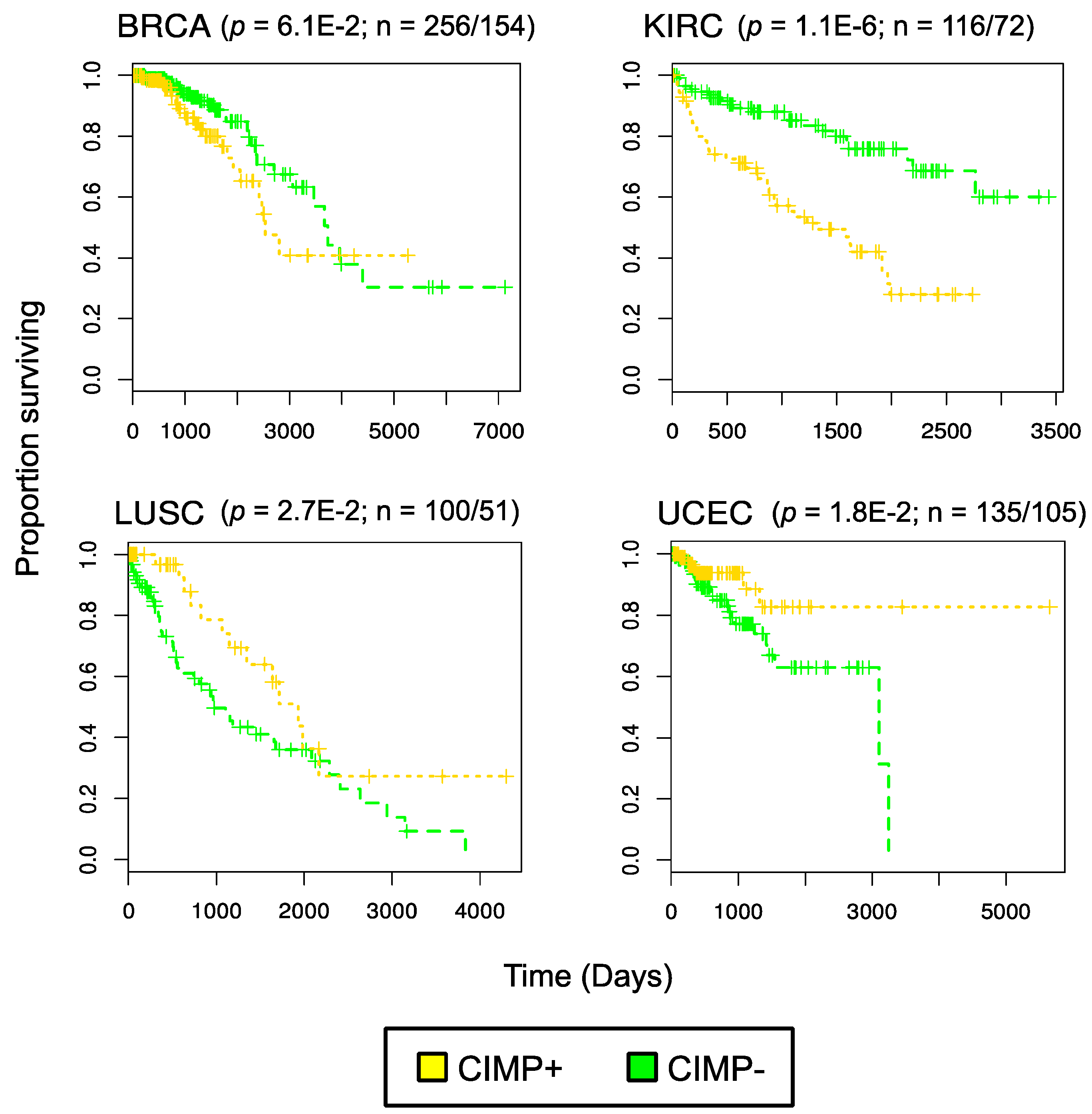
3. Evidence for Pan-Cancer CIMP
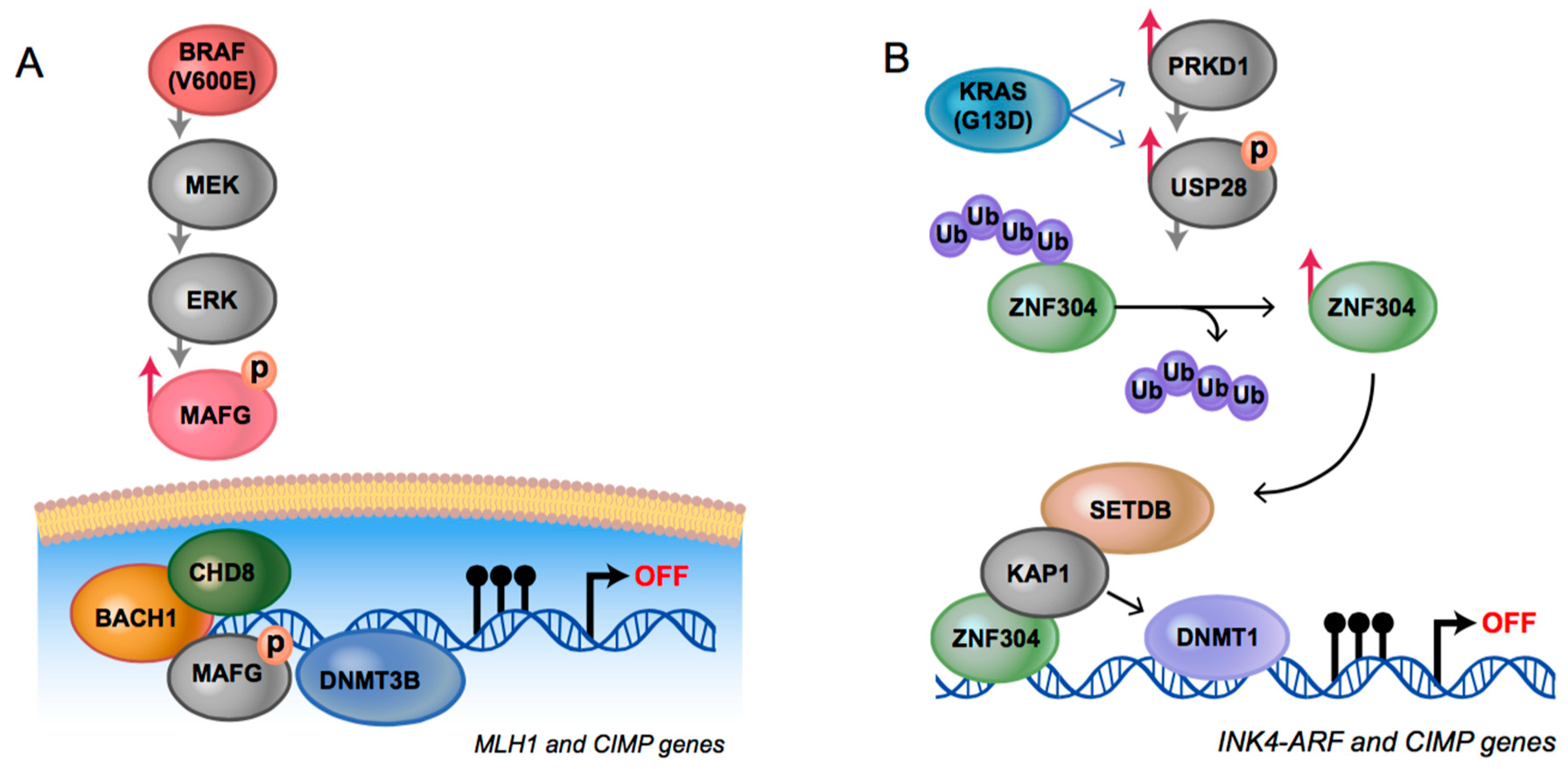
4. Shared Pathways and Common Mechanisms for CIMP
5. Passenger and Driver Events in DNA Methylation
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- You, J.S.; Jones, P.A. Cancer genetics and epigenetics: Two sides of the same coin? Cancer Cell 2012, 22, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Laird, P.W. Interplay between the cancer genome and epigenome. Cell 2013, 153, 38–55. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Timp, W.; Feinberg, A.P. Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host. Nat. Rev. Cancer 2013, 13, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Plass, C.; Pfister, S.M.; Lindroth, A.M.; Bogatyrova, O.; Claus, R.; Lichter, P. Mutations in regulators of the epigenome and their connections to global chromatin patterns in cancer. Nat. Rev. Genet. 2013, 14, 765–780. [Google Scholar] [CrossRef] [PubMed]
- Church, T.R.; Wandell, M.; Lofton-Day, C.; Mongin, S.J.; Burger, M.; Payne, S.R.; Castanos-Velez, E.; Blumenstein, B.A.; Rosch, T.; Osborn, N.; et al. Prospective evaluation of methylated SEPT9 in plasma for detection of asymptomatic colorectal cancer. Gut 2014, 63, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Laird, P.W. The power and the promise of DNA methylation markers. Nat. Rev. Cancer 2003, 3, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome—Biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Mikeska, T.; Craig, J.M. DNA methylation biomarkers: Cancer and beyond. Genes 2014, 5, 821–864. [Google Scholar] [CrossRef] [PubMed]
- Heyn, H.; Esteller, M. DNA methylation profiling in the clinic: Applications and challenges. Nat. Rev. Genet. 2012, 13, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Margolin, G.; Petrykowska, H.M.; Jameel, N.; Bell, D.W.; Young, A.C.; Elnitski, L. Robust detection of DNA hypermethylation of ZNF154 as a pan-cancer locus with in silico modeling for blood-based diagnostic development. J. Mol. Diagn. 2016, 18, 283–298. [Google Scholar] [CrossRef] [PubMed]
- Minucci, S.; Pelicci, P.G. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer 2006, 6, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Graham, J.S.; Kaye, S.B.; Brown, R. The promises and pitfalls of epigenetic therapies in solid tumours. Eur. J. Cancer 2009, 45, 1129–1136. [Google Scholar] [CrossRef] [PubMed]
- Hughes, L.A.; Melotte, V.; de Schrijver, J.; de Maat, M.; Smit, V.T.; Bovee, J.V.; French, P.J.; van den Brandt, P.A.; Schouten, L.J.; de Meyer, T.; et al. The CpG island methylator phenotype: What’s in a name? Cancer Res. 2013, 73, 5858–5868. [Google Scholar] [CrossRef] [PubMed]
- Toyota, M.; Ahuja, N.; Ohe-Toyota, M.; Herman, J.G.; Baylin, S.B.; Issa, J.P. CpG island methylator phenotype in colorectal cancer. Proc. Natl. Acad. Sci. USA 1999, 96, 8681–8686. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Herman, J.G.; Graff, J.R.; Vertino, P.M.; Issa, J.P. Alterations in DNA methylation: A fundamental aspect of neoplasia. Adv. Cancer Res. 1998, 72, 141–196. [Google Scholar] [PubMed]
- Yamashita, K.; Dai, T.; Dai, Y.; Yamamoto, F.; Perucho, M. Genetics supersedes epigenetics in colon cancer phenotype. Cancer Cell 2003, 4, 121–131. [Google Scholar] [CrossRef]
- Issa, J.P. CpG island methylator phenotype in cancer. Nat. Rev. Cancer 2004, 4, 988–993. [Google Scholar] [CrossRef] [PubMed]
- Weisenberger, D.J.; Siegmund, K.D.; Campan, M.; Young, J.; Long, T.I.; Faasse, M.A.; Kang, G.H.; Widschwendter, M.; Weener, D.; Buchanan, D.; et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat. Genet. 2006, 38, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Kawasaki, T.; Kirkner, G.J.; Kraft, P.; Loda, M.; Fuchs, C.S. Evaluation of markers for CpG island methylator phenotype (CIMP) in colorectal cancer by a large population-based sample. J. Mol. Diagn. 2007, 9, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Toyota, M.; Kondo, Y.; Lin, E.; Zhang, L.; Guo, Y.; Hernandez, N.S.; Chen, X.; Ahmed, S.; Konishi, K.; et al. Integrated genetic and epigenetic analysis identifies three different subclasses of colon cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 18654–18659. [Google Scholar] [CrossRef] [PubMed]
- Hinoue, T.; Weisenberger, D.J.; Lange, C.P.; Shen, H.; Byun, H.M.; Van Den Berg, D.; Malik, S.; Pan, F.; Noushmehr, H.; van Dijk, C.M.; et al. Genome-scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res. 2012, 22, 271–282. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar]
- Fang, M.; Ou, J.; Hutchinson, L.; Green, M.R. The BRAF oncoprotein functions through the transcriptional repressor MAFG to mediate the CpG Island Methylator phenotype. Mol. Cell 2014, 55, 904–915. [Google Scholar] [CrossRef] [PubMed]
- Serra, R.W.; Fang, M.; Park, S.M.; Hutchinson, L.; Green, M.R. A KRAS-directed transcriptional silencing pathway that mediates the CpG island methylator phenotype. eLife 2014, 3, e02313. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, R.; Toyooka, S.; Toyooka, K.O.; Harada, K.; Virmani, A.K.; Zochbauer-Muller, S.; Farinas, A.J.; Vakar-Lopez, F.; Minna, J.D.; Sagalowsky, A.; et al. Aberrant promoter methylation profile of bladder cancer and its relationship to clinicopathological features. Cancer Res. 2001, 61, 8659–8663. [Google Scholar] [PubMed]
- Bae, Y.K.; Brown, A.; Garrett, E.; Bornman, D.; Fackler, M.J.; Sukumar, S.; Herman, J.G.; Gabrielson, E. Hypermethylation in histologically distinct classes of breast cancer. Clin. Cancer Res. 2004, 10, 5998–6005. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Turcan, S.; Rimner, A.; Kaufman, A.; Giri, D.; Morris, L.G.; Shen, R.; Seshan, V.; Mo, Q.; Heguy, A.; et al. Breast cancer methylomes establish an epigenomic foundation for metastasis. Sci. Transl. Med. 2011, 3, 75ra25. [Google Scholar] [CrossRef] [PubMed]
- Jing, F.; Yuping, W.; Yong, C.; Jie, L.; Jun, L.; Xuanbing, T.; Lihua, H. CpG island methylator phenotype of multigene in serum of sporadic breast carcinoma. Tumour Biol. 2010, 31, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Whitcomb, B.P.; Mutch, D.G.; Herzog, T.J.; Rader, J.S.; Gibb, R.K.; Goodfellow, P.J. Frequent HOXA11 and THBS2 promoter methylation, and a methylator phenotype in endometrial adenocarcinoma. Clin. Cancer Res. 2003, 9, 2277–2287. [Google Scholar] [PubMed]
- Zhang, Q.Y.; Yi, D.Q.; Zhou, L.; Zhang, D.H.; Zhou, T.M. Status and significance of CpG island methylator phenotype in endometrial cancer. Gynecol. Obstet. Investig. 2011, 72, 183–191. [Google Scholar] [CrossRef] [PubMed]
- An, C.; Choi, I.S.; Yao, J.C.; Worah, S.; Xie, K.; Mansfield, P.F.; Ajani, J.A.; Rashid, A.; Hamilton, S.R.; Wu, T.T. Prognostic significance of CpG island methylator phenotype and microsatellite instability in gastric carcinoma. Clin. Cancer Res. 2005, 11, 656–663. [Google Scholar] [PubMed]
- Etoh, T.; Kanai, Y.; Ushijima, S.; Nakagawa, T.; Nakanishi, Y.; Sasako, M.; Kitano, S.; Hirohashi, S. Increased DNA methyltransferase 1 (DNMT1) protein expression correlates significantly with poorer tumor differentiation and frequent DNA hypermethylation of multiple CpG islands in gastric cancers. Am. J. Pathol. 2004, 164, 689–699. [Google Scholar] [CrossRef]
- Kim, H.; Kim, Y.H.; Kim, S.E.; Kim, N.G.; Noh, S.H.; Kim, H. Concerted promoter hypermethylation of hMLH1, p16INK4A, and E-cadherin in gastric carcinomas with microsatellite instability. J. Pathol. 2003, 200, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Kusano, M.; Toyota, M.; Suzuki, H.; Akino, K.; Aoki, F.; Fujita, M.; Hosokawa, M.; Shinomura, Y.; Imai, K.; Tokino, T. Genetic, epigenetic, and clinicopathologic features of gastric carcinomas with the CpG island methylator phenotype and an association with Epstein-Barr virus. Cancer 2006, 106, 1467–1479. [Google Scholar] [CrossRef] [PubMed]
- Oue, N.; Oshimo, Y.; Nakayama, H.; Ito, R.; Yoshida, K.; Matsusaki, K.; Yasui, W. DNA methylation of multiple genes in gastric carcinoma: Association with histological type and CpG island methylator phenotype. Cancer Sci. 2003, 94, 901–905. [Google Scholar] [CrossRef] [PubMed]
- Toyota, M.; Ahuja, N.; Suzuki, H.; Itoh, F.; Ohe-Toyota, M.; Imai, K.; Baylin, S.B.; Issa, J.P. Aberrant methylation in gastric cancer associated with the CpG island methylator phenotype. Cancer Res. 1999, 59, 5438–5442. [Google Scholar] [PubMed]
- Cheng, Y.; Zhang, C.; Zhao, J.; Wang, C.; Xu, Y.; Han, Z.; Jiang, G.; Guo, X.; Li, R.; Bu, X.; et al. Correlation of CpG island methylator phenotype with poor prognosis in hepatocellular carcinoma. Exp. Mol. Pathol. 2010, 88, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.B.; Zhang, Y.X.; Zhou, S.H.; Shi, M.X.; Cai, J.; Liu, Y.; Chen, K.P.; Qiang, F.L. CpG island methylator phenotype in plasma is associated with hepatocellular carcinoma prognosis. World J. Gastroenterol. 2011, 17, 4718–4724. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Ahuja, N.; Shen, Y.; Habib, N.A.; Toyota, M.; Rashid, A.; Issa, J.P. DNA methylation and environmental exposures in human hepatocellular carcinoma. J. Natl. Cancer Inst. 2002, 94, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Li, Z.; Cheng, Y.; Jia, F.; Li, R.; Wu, M.; Li, K.; Wei, L. CpG island methylator phenotype association with elevated serum alpha-fetoprotein level in hepatocellular carcinoma. Clin. Cancer Res. 2007, 13, 944–952. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhao, J.; Chen, X.F.; Li, W.; Liu, R.; Lei, Z.; Liu, X.; Peng, X.; Xu, K.; Chen, J.; et al. CpG island methylator phenotype involving tumor suppressor genes located on chromosome 3p in non-small cell lung cancer. Lung Cancer 2008, 62, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Shigematsu, H.; Iizasa, T.; Hiroshima, K.; Nakatani, Y.; Minna, J.D.; Gazdar, A.F.; Fujisawa, T. Exclusive mutation in epidermal growth factor receptor gene, HER-2, and KRAS, and synchronous methylation of nonsmall cell lung cancer. Cancer 2006, 106, 2200–2207. [Google Scholar] [CrossRef] [PubMed]
- Strathdee, G.; Appleton, K.; Illand, M.; Millan, D.W.; Sargent, J.; Paul, J.; Brown, R. Primary ovarian carcinomas display multiple methylator phenotypes involving known tumor suppressor genes. Am. J. Pathol. 2001, 158, 1121–1127. [Google Scholar] [CrossRef]
- Kolbe, D.L.; DeLoia, J.A.; Porter-Gill, P.; Strange, M.; Petrykowska, H.M.; Guirguis, A.; Krivak, T.C.; Brody, L.C.; Elnitski, L. Differential analysis of ovarian and endometrial cancers identifies a methylator phenotype. PLoS ONE 2012, 7, e32941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueki, T.; Toyota, M.; Sohn, T.; Yeo, C.J.; Issa, J.P.; Hruban, R.H.; Goggins, M. Hypermethylation of multiple genes in pancreatic adenocarcinoma. Cancer Res. 2000, 60, 1835–1839. [Google Scholar] [PubMed]
- Maruyama, R.; Toyooka, S.; Toyooka, K.O.; Virmani, A.K.; Zochbauer-Muller, S.; Farinas, A.J.; Minna, J.D.; McConnell, J.; Frenkel, E.P.; Gazdar, A.F. Aberrant promoter methylation profile of prostate cancers and its relationship to clinicopathological features. Clin. Cancer Res. 2002, 8, 514–519. [Google Scholar] [PubMed]
- Arai, E.; Chiku, S.; Mori, T.; Gotoh, M.; Nakagawa, T.; Fujimoto, H.; Kanai, Y. Single-CpG-resolution methylome analysis identifies clinicopathologically aggressive CpG island methylator phenotype clear cell renal cell carcinomas. Carcinogenesis 2012, 33, 1487–1493. [Google Scholar] [CrossRef] [PubMed]
- Noushmehr, H.; Weisenberger, D.J.; Diefes, K.; Phillips, H.S.; Pujara, K.; Berman, B.P.; Pan, F.; Pelloski, C.E.; Sulman, E.P.; Bhat, K.P.; et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 2010, 17, 510–522. [Google Scholar] [CrossRef] [PubMed]
- Turcan, S.; Rohle, D.; Goenka, A.; Walsh, L.A.; Fang, F.; Yilmaz, E.; Campos, C.; Fabius, A.W.; Lu, C.; Ward, P.S.; et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012, 483, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Van den Bent, M.J.; Gravendeel, L.A.; Gorlia, T.; Kros, J.M.; Lapre, L.; Wesseling, P.; Teepen, J.L.; Idbaih, A.; Sanson, M.; Smitt, P.A.; et al. A hypermethylated phenotype is a better predictor of survival than MGMT methylation in anaplastic oligodendroglial brain tumors: A report from EORTC study 26951. Clin. Cancer Res. 2011, 17, 7148–7155. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Manero, G.; Daniel, J.; Smith, T.L.; Kornblau, S.M.; Lee, M.S.; Kantarjian, H.M.; Issa, J.P. DNA methylation of multiple promoter-associated CpG islands in adult acute lymphocytic leukemia. Clin. Cancer Res. 2002, 8, 2217–2224. [Google Scholar] [PubMed]
- Roman-Gomez, J.; Jimenez-Velasco, A.; Agirre, X.; Castillejo, J.A.; Navarro, G.; Calasanz, M.J.; Garate, L.; San Jose-Eneriz, E.; Cordeu, L.; Prosper, F.; et al. CpG island methylator phenotype redefines the prognostic effect of t(12;21) in childhood acute lymphoblastic leukemia. Clin. Cancer Res. 2006, 12, 4845–4850. [Google Scholar] [CrossRef] [PubMed]
- Roman-Gomez, J.; Jimenez-Velasco, A.; Agirre, X.; Prosper, F.; Heiniger, A.; Torres, A. Lack of CpG island methylator phenotype defines a clinical subtype of T-cell acute lymphoblastic leukemia associated with good prognosis. J. Clin. Oncol. 2005, 23, 7043–7049. [Google Scholar] [CrossRef] [PubMed]
- Toyota, M.; Kopecky, K.J.; Toyota, M.O.; Jair, K.W.; Willman, C.L.; Issa, J.P. Methylation profiling in acute myeloid leukemia. Blood 2001, 97, 2823–2829. [Google Scholar] [CrossRef] [PubMed]
- Tanemura, A.; Terando, A.M.; Sim, M.S.; van Hoesel, A.Q.; de Maat, M.F.; Morton, D.L.; Hoon, D.S. CpG island methylator phenotype predicts progression of malignant melanoma. Clin. Cancer Res. 2009, 15, 1801–1807. [Google Scholar] [CrossRef] [PubMed]
- Fu, T.; Pappou, E.P.; Guzzetta, A.A.; Jeschke, J.; Kwak, R.; Dave, P.; Hooker, C.M.; Morgan, R.; Baylin, S.B.; Iacobuzio-Donahue, C.A.; et al. CpG island methylator phenotype-positive tumors in the absence of MLH1 methylation constitute a distinct subset of duodenal adenocarcinomas and are associated with poor prognosis. Clin. Cancer Res. 2012, 18, 4743–4752. [Google Scholar] [CrossRef] [PubMed]
- Barreau, O.; Assie, G.; Wilmot-Roussel, H.; Ragazzon, B.; Baudry, C.; Perlemoine, K.; Rene-Corail, F.; Bertagna, X.; Dousset, B.; Hamzaoui, N.; et al. Identification of a CpG island methylator phenotype in adrenocortical carcinomas. J. Clin. Endocrinol. Metab. 2013, 98, E174–E184. [Google Scholar] [CrossRef] [PubMed]
- Abe, M.; Ohira, M.; Kaneda, A.; Yagi, Y.; Yamamoto, S.; Kitano, Y.; Takato, T.; Nakagawara, A.; Ushijima, T. CpG island methylator phenotype is a strong determinant of poor prognosis in neuroblastomas. Cancer Res. 2005, 65, 828–834. [Google Scholar] [PubMed]
- Abe, M.; Westermann, F.; Nakagawara, A.; Takato, T.; Schwab, M.; Ushijima, T. Marked and independent prognostic significance of the CpG island methylator phenotype in neuroblastomas. Cancer Lett. 2007, 247, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Tomczak, K.; Czerwinska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. (Pozn.) 2015, 19, A68–A77. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Vega, F.; Gotea, V.; Margolin, G.; Elnitski, L. Pan-cancer stratification of solid human epithelial tumors and cancer cell lines reveals commonalities and tissue-specific features of the CpG island methylator phenotype. Epigenetics Chromatin 2015, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- Moarii, M.; Reyal, F.; Vert, J.P. Integrative DNA methylation and gene expression analysis to assess the universality of the CpG island methylator phenotype. Hum. Genom. 2015, 9, 26. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.K.; Brenner, C. Suppression of TET1-dependent DNA demethylation is essential for KRAS-mediated transformation. Cell Rep. 2014, 9, 1827–1840. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Ye, D.; Guan, K.L.; Xiong, Y. IDH1 and IDH2 mutations in tumorigenesis: Mechanistic insights and clinical perspectives. Clin. Cancer Res. 2012, 18, 5562–5571. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar]
- The Cancer Genome Atlas Research Network; Kandoth, C.; Schultz, N.; Cherniack, A.D.; Akbani, R.; Liu, Y.; Shen, H.; Robertson, A.G.; Pashtan, I.; Shen, R.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [PubMed]
- Ciriello, G.; Miller, M.L.; Aksoy, B.A.; Senbabaoglu, Y.; Schultz, N.; Sander, C. Emerging landscape of oncogenic signatures across human cancers. Nat. Genet. 2013, 45, 1127–1133. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Esteller, M.; Rountree, M.R.; Bachman, K.E.; Schuebel, K.; Herman, J.G. Aberrant patterns of DNA methylation, chromatin formation and gene expression in cancer. Hum. Mol. Genet. 2001, 10, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Duncan, C.G.; Barwick, B.G.; Jin, G.; Rago, C.; Kapoor-Vazirani, P.; Powell, D.R.; Chi, J.T.; Bigner, D.D.; Vertino, P.M.; Yan, H. A heterozygous IDH1R132H/WT mutation induces genome-wide alterations in DNA methylation. Genome Res. 2012, 22, 2339–2355. [Google Scholar] [CrossRef] [PubMed]
- Weisenberger, D.J. Characterizing DNA methylation alterations from the cancer genome atlas. J. Clin. Investig. 2014, 124, 17–23. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even Warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Scourzic, L.; Mouly, E.; Bernard, O.A. TET proteins and the control of cytosine demethylation in cancer. Genome Med. 2015, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Zhang, Y. Reversing DNA methylation: Mechanisms, genomics, and biological functions. Cell 2014, 156, 45–68. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B. DNA methylation and gene silencing in cancer. Nat. Clin. Pract. Oncol. 2005, 2 (Suppl. 1), S4–S11. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Wang, K.Y.; Shen, C.K. DNA 5-methylcytosine demethylation activities of the mammalian DNA methyltransferases. J. Biol. Chem. 2013, 288, 9084–9091. [Google Scholar] [CrossRef] [PubMed]
- Snover, D.C. Update on the serrated pathway to colorectal carcinoma. Hum. Pathol. 2011, 42, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Vega, F.; Gotea, V.; Chen, Y.-C.; Elnitski, L. CpG island methylator phenotype in adenocarcinomas from the digestive tract: Methods, conclusions, and controversies. World J. Gastrointest. Oncol. 2016, in press. [Google Scholar]
- Sproul, D.; Meehan, R.R. Genomic insights into cancer-associated aberrant CpG island hypermethylation. Brief. Funct. Genom. 2013, 12, 174–190. [Google Scholar] [CrossRef] [PubMed]
- Sproul, D.; Kitchen, R.R.; Nestor, C.E.; Dixon, J.M.; Sims, A.H.; Harrison, D.J.; Ramsahoye, B.H.; Meehan, R.R. Tissue of origin determines cancer-associated CpG island promoter hypermethylation patterns. Genome Biol. 2012, 13, R84. [Google Scholar] [CrossRef] [PubMed]
- Nejman, D.; Straussman, R.; Steinfeld, I.; Ruvolo, M.; Roberts, D.; Yakhini, Z.; Cedar, H. Molecular rules governing de novo methylation in cancer. Cancer Res. 2014, 74, 1475–1483. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Breeze, C.E.; Zhen, S.; Beck, S.; Teschendorff, A.E. Tissue-independent and tissue-specific patterns of DNA methylation alteration in cancer. Epigenetics Chromatin 2016, 9, 10. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Vega, F.; Gotea, V.; Petrykowska, H.M.; Margolin, G.; Krivak, T.C.; DeLoia, J.A.; Bell, D.W.; Elnitski, L. Recurrent patterns of DNA methylation in the ZNF154, CASP8, and VHL promoters across a wide spectrum of human solid epithelial tumors and cancer cell lines. Epigenetics 2013, 8, 1355–1372. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Wan, J.; Su, Y.; Song, Q.; Zeng, Y.; Nguyen, H.N.; Shin, J.; Cox, E.; Rho, H.S.; Woodard, C.; et al. DNA methylation presents distinct binding sites for human transcription factors. eLife 2013, 2, e00726. [Google Scholar] [CrossRef] [PubMed]
- Flavahan, W.A.; Drier, Y.; Liau, B.B.; Gillespie, S.M.; Venteicher, A.S.; Stemmer-Rachamimov, A.O.; Suva, M.L.; Bernstein, B.E. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 2016, 529, 110–114. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, W.A.; Sachani, S.S.; White, C.R.; Mann, M.R. A role for chromatin topology in imprinted domain regulation. Biochem. Cell Biol. 2016, 94, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Berman, B.P.; Weisenberger, D.J.; Aman, J.F.; Hinoue, T.; Ramjan, Z.; Liu, Y.; Noushmehr, H.; Lange, C.P.; van Dijk, C.M.; Tollenaar, R.A.; et al. Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina-associated domains. Nat. Genet. 2012, 44, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Wen, B.; Wu, H.; Shinkai, Y.; Irizarry, R.A.; Feinberg, A.P. Large histone H3 lysine 9 dimethylated chromatin blocks distinguish differentiated from embryonic stem cells. Nat. Genet. 2009, 41, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Choi, J.Y.; Lee, K.M.; Sung, H.; Park, S.K.; Oze, I.; Pan, K.F.; You, W.C.; Chen, Y.X.; Fang, J.Y.; et al. DNA methylation in peripheral blood: A potential biomarker for cancer molecular epidemiology. J. Epidemiol. 2012, 22, 384–394. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miller, B.F.; Sánchez-Vega, F.; Elnitski, L. The Emergence of Pan-Cancer CIMP and Its Elusive Interpretation. Biomolecules 2016, 6, 45. https://doi.org/10.3390/biom6040045
Miller BF, Sánchez-Vega F, Elnitski L. The Emergence of Pan-Cancer CIMP and Its Elusive Interpretation. Biomolecules. 2016; 6(4):45. https://doi.org/10.3390/biom6040045
Chicago/Turabian StyleMiller, Brendan F., Francisco Sánchez-Vega, and Laura Elnitski. 2016. "The Emergence of Pan-Cancer CIMP and Its Elusive Interpretation" Biomolecules 6, no. 4: 45. https://doi.org/10.3390/biom6040045