
Emerging Roles for Ciz1 in Cell Cycle Regulation and as a Driver of Tumorigenesis


Abstract
:1. Introduction
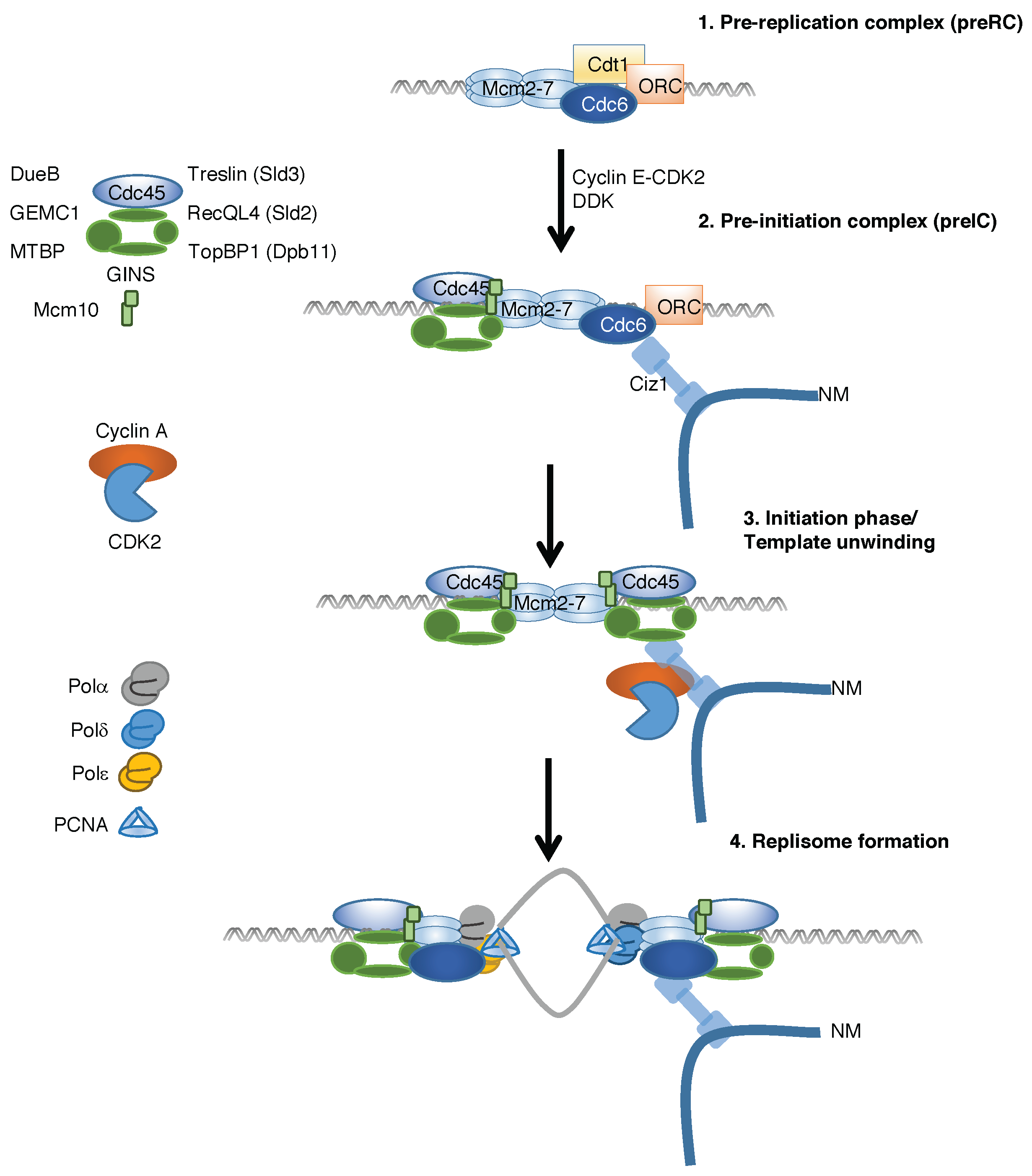
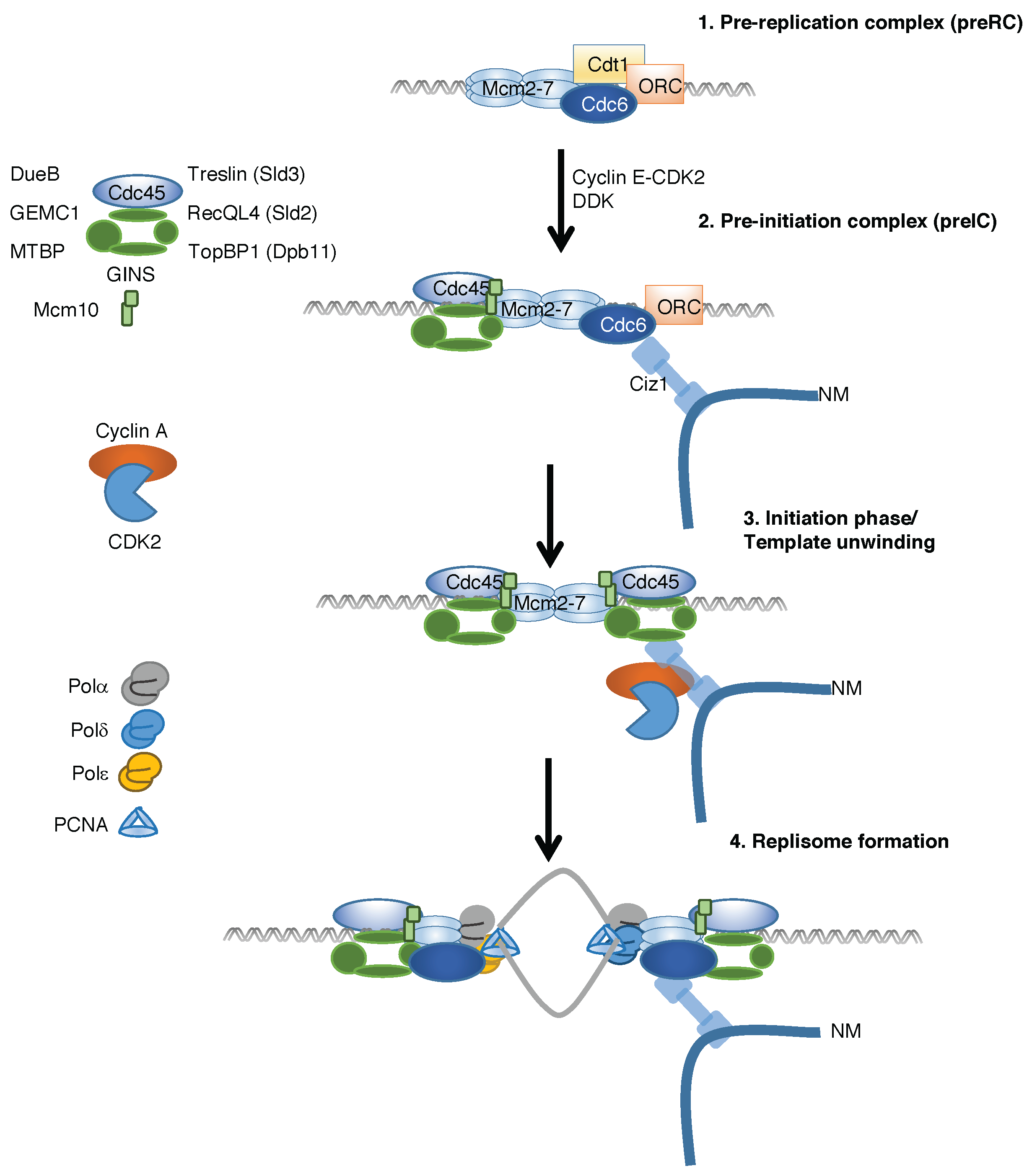
2. Temporal Regulation of Replication Complex Assembly
3. Discovery of Ciz1 and Its Role in Cell Cycle Regulation
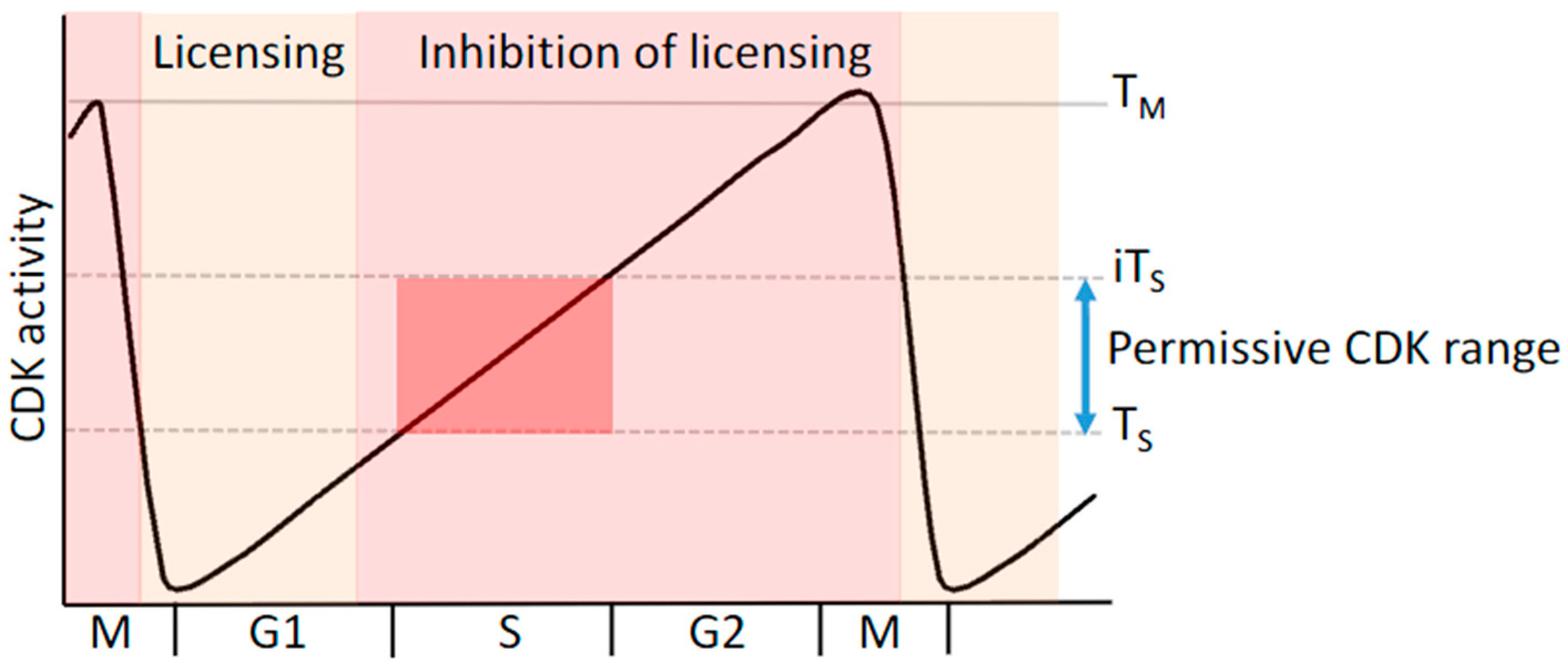
4. Ciz1 Is a CDK Sensor That Promotes Initiation of DNA Replication and Prevention of Re-Replication
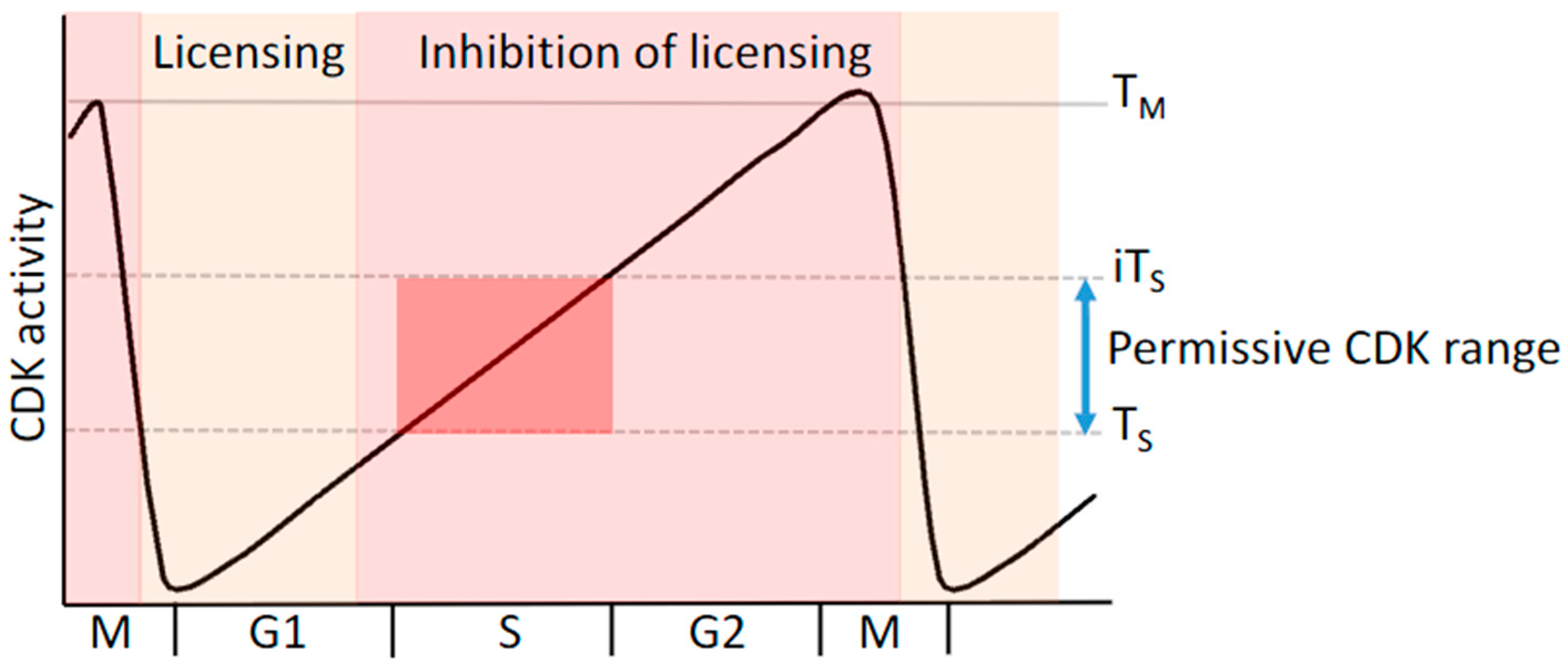
5. Replication Licensing and Prevention of Re-Replication Is Regulated by Cyclin-CDK Complexes
6. Ciz1 Is a Driver of Tumor Growth
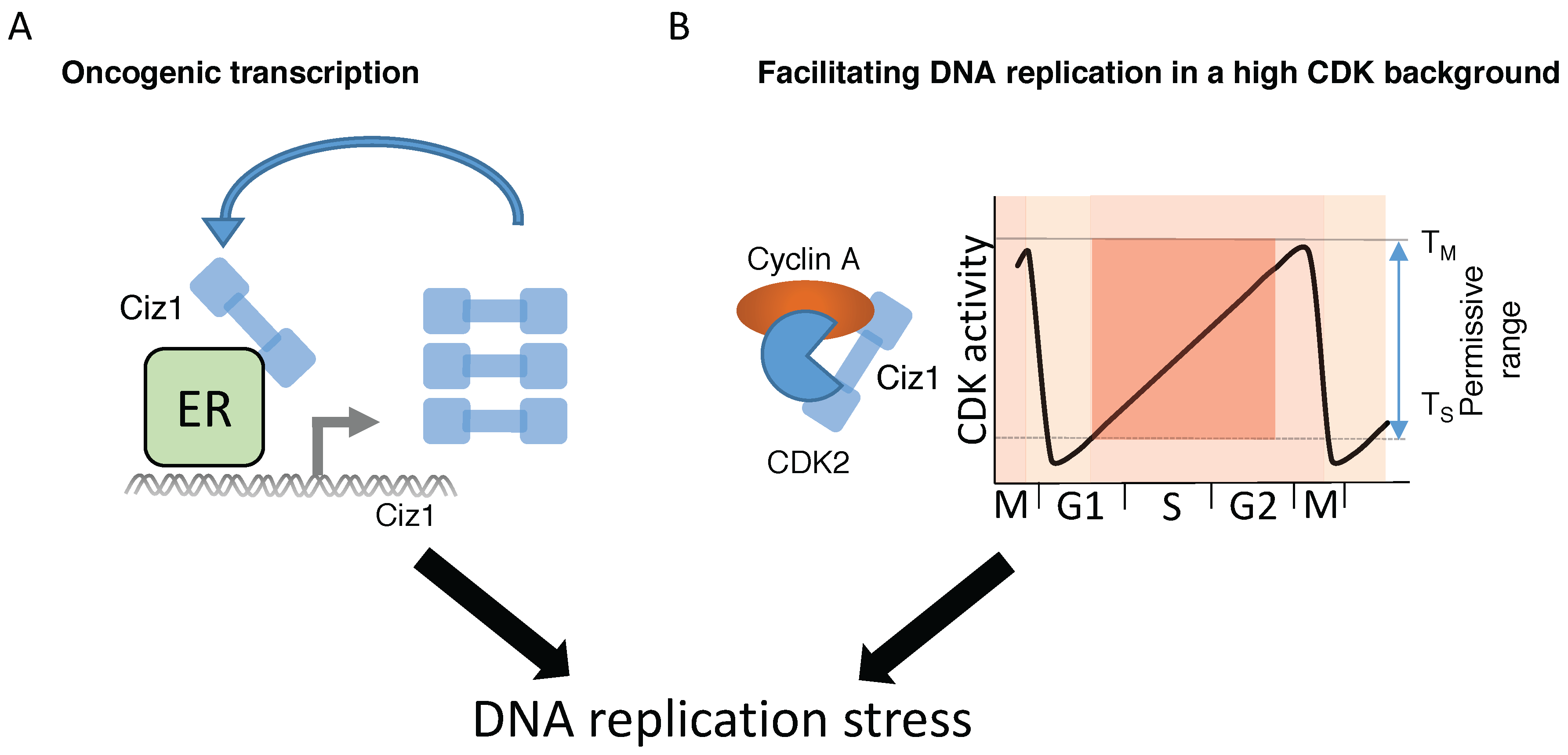
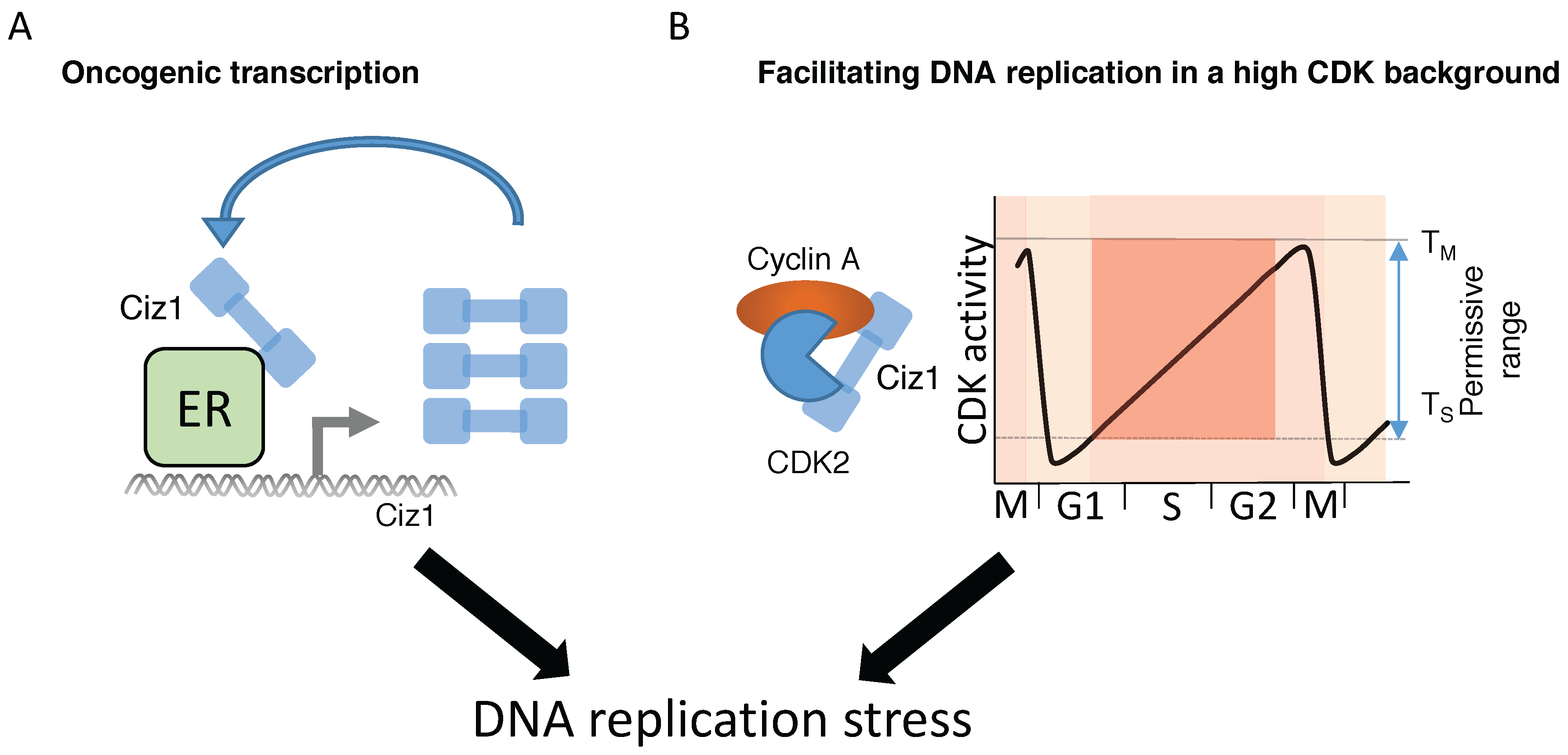
7. Ciz1-Mediated Transcriptional Regulation of Tumorigenesis
8. Future Perspectives
8.1. Deregulation of CDK Activity Promotes DNA Replication Stress
8.2. Does Ciz1 Contribute to Tumorigenesis by Inducing DNA Replication Stress?
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
| Ciz1 Binding Partner | Reference |
|---|---|
| p21 | [47] |
| Cell division cycle 6 (Cdc6) | [23] |
| Cyclin E | [42] |
| Cyclin A | [23,42] |
| Cyclin-dependent kinase 2 (CDK2) | [47] |
| Dynein light chain | [76,102,103,104] |
| Estrogen receptor | [77] |
| Histone cluster 1 | [104] |
| B cell Chronic lymphoid leukemia 7C | [105] |
| Kelch-like member 22 | [105] |
| Scaffold attachment regulator | [105] |
| Mitogen activated protein kinase 14 (MAPK14) | [105] |
| Acid phosphatase 5, tartrate resistant (ACP5) | [105] |
| B lymphoma Mo-MLV insertion region 1 homolog (Bmi1) | [106] |
| Obscurin-like 1 | [107] |
| Cullin 7 (Cul7) | [107] |
| Upf2 | [108] |
| Enhancer of rudimentary homologue (ERH) | [109] |
| SH3 homology domain kinase binding protein 1 | [110] |
| Polypyrimidine tract binding protein 1 | [102] |
| Ectodysplasin A | [105] |
| SH3-domain binding protein 4 | [111] |
References
- Bebenek, K.; Kunkel, T.A. Functions of DNA polymerases. Adv. Protein Chem. 2004, 69, 137–165. [Google Scholar] [PubMed]
- Mehanna, A.; Diffley, J.F. Pre-replicative complex assembly with purified proteins. Methods 2012, 57, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Duzdevich, D.; Warner, M.D.; Ticau, S.; Ivica, N.A.; Bell, S.P.; Greene, E.C. The dynamics of eukaryotic replication initiation: Origin specificity, licensing, and firing at the single-molecule level. Mol. Cell 2015, 58, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Fragkos, M.; Ganier, O.; Coulombe, P.; Mechali, M. DNA replication origin activation in space and time. Nat. Rev. Mol. Cell Biol. 2015, 16, 360–374. [Google Scholar] [CrossRef] [PubMed]
- Heller, R.C.; Kang, S.; Lam, W.M.; Chen, S.; Chan, C.S.; Bell, S.P. Eukaryotic origin-dependent DNA replication in vitro reveals sequential action of DDK and S-CDK kinases. Cell 2011, 146, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Yeeles, J.T.; Deegan, T.D.; Janska, A.; Early, A.; Diffley, J.F. Regulated eukaryotic DNA replication origin firing with purified proteins. Nature 2015, 519, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Perez-Arnaiz, P.; Bruck, I.; Kaplan, D.L. Mcm10 coordinates the timely assembly and activation of the replication fork helicase. Nucleic Acids Res. 2016, 44, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Onesti, S.; Macneill, S.A. Structure and evolutionary origins of the CMG complex. Chromosoma 2013, 122, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Shi, Y.; Georgescu, R.E.; Yuan, Z.; Chait, B.T.; Li, H.; O’Donnell, M.E. The architecture of a eukaryotic replisome. Nat. Struct. Mol. Biol. 2015, 22, 976–982. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; van Deursen, F.; de Piccoli, G.; Labib, K. Dpb2 Integrates the Leading-Strand DNA Polymerase into the Eukaryotic Replisome. Curr. Biol. 2013, 23, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Cvetic, C.; Walter, J.C. Eukaryotic origins of DNA replication: Could you please be more specific? Semin. Cell Dev. Biol. 2005, 16, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, D.M. In search of the holy replicator. Nat. Rev. Mol. Cell Biol. 2004, 5, 848–855. [Google Scholar] [CrossRef] [PubMed]
- Cayrou, C.; Ballester, B.; Peiffer, I.; Fenouil, R.; Coulombe, P.; Andrau, J.C.; van Helden, J.; Mechali, M. The chromatin environment shapes DNA replication origin organization and defines origin classes. Genome Res. 2015, 25, 1873–1885. [Google Scholar] [CrossRef] [PubMed]
- Tatsumi, Y.; Ohta, S.; Kimura, H.; Tsurimoto, T.; Obuse, C. The ORC1 cycle in human cells: I. cell cycle-regulated oscillation of human ORC1. J. Biol. Chem. 2003, 278, 41528–41534. [Google Scholar] [CrossRef] [PubMed]
- Thomae, A.W.; Pich, D.; Brocher, J.; Spindler, M.P.; Berens, C.; Hock, R.; Hammerschmidt, W.; Schepers, A. Interaction between HMGA1a and the Origin Recognition Complex Creates Site-Specific Replication Origins. Proc. Natl. Acad. Sci. USA 2008, 105, 1692–1697. [Google Scholar] [CrossRef] [PubMed]
- Edwards, M.C.; Tutter, A.V.; Cvetic, C.; Gilbert, C.H.; Prokhorova, T.A.; Walter, J.C. Mcm2-7 Complexes Bind Chromatin in a Distributed Pattern Surrounding the Origin Recognition Complex in Xenopus Egg Extracts. J. Biol. Chem. 2002, 277, 33049–33057. [Google Scholar] [CrossRef] [PubMed]
- Yardimci, H.; Walter, J.C. Prereplication-complex formation: A molecular double take? Nat. Struct. Mol. Biol. 2014, 21, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Ticau, S.; Friedman, L.J.; Ivica, N.A.; Gelles, J.; Bell, S.P. Single-molecule studies of origin licensing reveal mechanisms ensuring bidirectional helicase loading. Cell 2015, 161, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Das, S.P.; Borrman, T.; Liu, V.W.; Yang, S.C.; Bechhoefer, J.; Rhind, N. Replication timing is regulated by the number of Mcms loaded at origins. Genome Res. 2015, 25, 1886–1892. [Google Scholar] [CrossRef] [PubMed]
- Evrin, C.; Fernandez-Cid, A.; Riera, A.; Zech, J.; Clarke, P.; Herrera, M.C.; Tognetti, S.; Lurz, R.; Speck, C. The ORC/Cdc6/Mcm2-7 complex facilitates Mcm2-7 dimerization during prereplicative complex formation. Nucleic Acids Res. 2014, 42, 2257–2269. [Google Scholar] [CrossRef] [PubMed]
- Chang, F.; Riera, A.; Evrin, C.; Sun, J.; Li, H.; Speck, C.; Weinreich, M. Cdc6 ATPase activity disengages Cdc6 from the pre-replicative complex to promote DNA replication. eLife 2015, 4, e05795. [Google Scholar] [CrossRef] [PubMed]
- Deegan, T.D.; Yeeles, J.T.; Diffley, J.F. Phosphopeptide binding by Sld3 links Dbf4-dependent kinase to Mcm replicative helicase activation. EMBO J. 2016, 35, 961–973. [Google Scholar] [CrossRef] [PubMed]
- Copeland, N.A.; Sercombe, H.E.; Wilson, R.H.; Coverley, D. Cyclin-A-CDK2-mediated phosphorylation of CIZ1 blocks replisome formation and initiation of mammalian DNA replication. J. Cell Sci. 2015, 128, 1518–1527. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Fernandez-Cid, A.; Riera, A.; Tognetti, S.; Yuan, Z.; Stillman, B.; Speck, C.; Li, H. Structural and mechanistic insights into Mcm2-7 double-hexamer assembly and function. Genes Dev. 2014, 28, 2291–2303. [Google Scholar] [CrossRef] [PubMed]
- Boos, D.; Sanchez-Pulido, L.; Rappas, M.; Pearl, L.H.; Oliver, A.W.; Ponting, C.P.; Diffley, J.F. Regulation of DNA replication through Sld3-Dpb11 interaction is conserved from yeast to humans. Curr. Biol. 2011, 21, 1152–1157. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, A.; Shevchenko, A.; Dunphy, W.G. Treslin collaborates with TopBP1 in triggering the initiation of DNA replication. Cell 2010, 140, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, A.; Shevchenko, A.; Dunphy, W.G. Direct regulation of treslin by cyclin-dependent kinase is essential for the onset of DNA replication. J. Cell Biol. 2011, 193, 995–1007. [Google Scholar] [CrossRef] [PubMed]
- Boos, D.; Yekezare, M.; Diffley, J.F. Identification of a heteromeric complex that promotes DNA replication origin firing in human cells. Science 2013, 340, 981–984. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, A.; Liu, G.; Kemp, M.; Chen, X.; Katrangi, N.; Myers, S.; Ghosh, M.; Yao, J.; Gao, Y.; Bubulya, P.; et al. The DNA unwinding element binding protein DUE-B interacts with Cdc45 in preinitiation complex formation. Mol. Cell. Biol. 2010, 30, 1495–1507. [Google Scholar] [CrossRef] [PubMed]
- Balestrini, A.; Cosentino, C.; Errico, A.; Garner, E.; Costanzo, V. GEMC1 is a TopBP1-interacting protein required for chromosomal DNA replication. Nat. Cell Biol. 2010, 12, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Greaves, E.A.; Copeland, N.A.; Coverley, D.; Ainscough, J.F. Cancer-associated variant expression and interaction of CIZ1 with cyclin A1 in differentiating male germ cells. J. Cell Sci. 2012, 125, 2466–2477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villa, F.; Simon, A.C.; Ortiz Bazan, M.A.; Kilkenny, M.L.; Wirthensohn, D.; Wightman, M.; Matak-Vinkovic, D.; Pellegrini, L.; Labib, K. Ctf4 is a Hub in the Eukaryotic Replisome that Links Multiple CIP-Box Proteins to the CMG Helicase. Mol. Cell 2016, 63, 385–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacek, M.; Tutter, A.V.; Kubota, Y.; Takisawa, H.; Walter, J.C. Localization of Mcm2–7, Cdc45, and gins to the site of DNA unwinding during eukaryotic DNA replication. Mol. Cell 2006, 21, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Takeda, D.Y.; Dutta, A. DNA replication and progression through s phase. Oncogene 2005, 24, 2827–2843. [Google Scholar] [CrossRef] [PubMed]
- Chuang, L.C.; Teixeira, L.K.; Wohlschlegel, J.A.; Henze, M.; Yates, J.R.; Mendez, J.; Reed, S.I. Phosphorylation of Mcm2 by Cdc7 promotes pre-replication complex assembly during cell-cycle re-entry. Mol. Cell 2009, 35, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Umemori, T.; Hirai, K.; Muramatsu, S.; Kamimura, Y.; Araki, H. CDK-dependent phosphorylation of Sld2 and Sld3 initiates DNA replication in budding yeast. Nature 2007, 445, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Zegerman, P.; Diffley, J.F. Phosphorylation of Sld2 and Sld3 by cyclin-dependent kinases promotes DNA replication in budding yeast. Nature 2007, 445, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Bruck, I.; Kaplan, D.L. Conserved mechanism for coordinating replication fork helicase assembly with phosphorylation of the helicase. Proc. Natl. Acad. Sci. USA 2015, 112, 11223–11228. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.; Moore, K.; Murray, J.; Aves, S.J.; Price, C. Mcm10 interacts with Rad4/Cut5(TopBP1) and its association with origins of DNA replication is dependent on Rad4/Cut5(TopBP1). DNA Repair 2011, 10, 1154–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araki, H. Cyclin-dependent kinase-dependent initiation of chromosomal DNA replication. Curr. Opin. Cell Biol. 2010, 22, 766–771. [Google Scholar] [CrossRef] [PubMed]
- Douglas, M.E.; Diffley, J.F. Recruitment of Mcm10 to sites of replication initiation requires direct binding to the minichromosome maintenance (Mcm) complex. J. Biol. Chem. 2016, 291, 5879–5888. [Google Scholar] [CrossRef] [PubMed]
- Copeland, N.A.; Sercombe, H.E.; Ainscough, J.F.; Coverley, D. Ciz1 cooperates with cyclin-A-CDK2 to activate mammalian DNA replication in vitro. J. Cell Sci. 2010, 123, 1108–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellison, V.; Stillman, B. Opening of the clamp: An intimate view of an ATP-driven biological machine. Cell 2001, 106, 655–660. [Google Scholar] [CrossRef]
- Simon, A.C.; Zhou, J.C.; Perera, R.L.; van Deursen, F.; Evrin, C.; Ivanova, M.E.; Kilkenny, M.L.; Renault, L.; Kjaer, S.; Matak-Vinkovic, D.; et al. A Ctf4 trimer couples the CMG helicase to DNA polymerase alpha in the eukaryotic replisome. Nature 2014, 510, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Samora, C.P.; Saksouk, J.; Goswami, P.; Wade, B.O.; Singleton, M.R.; Bates, P.A.; Lengronne, A.; Costa, A.; Uhlmann, F. Ctf4 links DNA replication with Sister Chromatid Cohesion Establishment by Recruiting the Chl1 Helicase to the Replisome. Mol. Cell 2016, 63, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Stillman, B. Reconsidering DNA polymerases at the replication fork in eukaryotes. Mol. Cell 2015, 59, 139–141. [Google Scholar] [CrossRef] [PubMed]
- Mitsui, K.; Matsumoto, A.; Ohtsuka, S.; Ohtsubo, M.; Yoshimura, A. Cloning and Characterization of a Novel p21Cip1/Waf1-Interacting Zinc Finger Protein, Ciz1. Biochem. Biophys. Res. Commun. 1999, 264, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Nishibe, R.; Watanabe, W.; Ueda, T.; Yamasaki, N.; Koller, R.; Wolff, L.; Honda, Z.; Ohtsubo, M.; Honda, H. Ciz1, a p21Cip1/Waf1-interacting protein, functions as a tumor suppressor in vivo. FEBS Lett. 2013, 587, 1529–1535. [Google Scholar] [CrossRef] [PubMed]
- Ainscough, J.F.; Rahman, F.A.; Sercombe, H.; Sedo, A.; Gerlach, B.; Coverley, D. C-terminal domains deliver the DNA replication factor Ciz1 to the nuclear matrix. J. Cell Sci. 2007, 120, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Berezney, R.; Coffey, D.S. Nuclear protein matrix: Association with newly synthesized DNA. Science 1975, 189, 291–293. [Google Scholar] [CrossRef] [PubMed]
- Radichev, I.; Parashkevova, A.; Anachkova, B. Initiation of DNA replication at a nuclear matrix-attached chromatin fraction. J. Cell. Physiol. 2005, 203, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Munkley, J.; Copeland, N.A.; Moignard, V.; Knight, J.R.; Greaves, E.; Ramsbottom, S.A.; Pownall, M.E.; Southgate, J.; Ainscough, J.F.; Coverley, D. Cyclin E is recruited to the nuclear matrix during differentiation, but is not recruited in cancer cells. Nucleic Acids Res. 2011, 39, 2671–2677. [Google Scholar] [CrossRef] [PubMed]
- Coverley, D.; Marr, J.; Ainscough, J. Ciz1 promotes mammalian DNA replication. J. Cell Sci. 2005, 118, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Takeda, D.Y.; Wohlschlegel, J.A.; Dutta, A. A bipartite substrate recognition motif for cyclin-dependent kinases. J. Biol. Chem. 2001, 276, 1993–1997. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, K.; On, K.F.; Diffley, J.F. Regulating DNA replication in eukarya. Cold Spring Harb. Perspect. Biol. 2013, 5, a012930. [Google Scholar] [CrossRef] [PubMed]
- Reusswig, K.U.; Zimmermann, F.; Galanti, L.; Pfander, B. Robust replication control is generated by temporal gaps between licensing and firing phases and depends on degradation of firing factor Sld2. Cell Rep. 2016, 17, 556–569. [Google Scholar] [CrossRef] [PubMed]
- Diffley, J.F.; Cocker, J.H.; Dowell, S.J.; Rowley, A. Two steps in the assembly of complexes at yeast replication origins in vivo. Cell 1994, 78, 303–316. [Google Scholar] [CrossRef]
- Mailand, N.; Diffley, J.F. Cdks promote DNA replication origin licensing in human cells by protecting Cdc6 from APC/C-dependent proteolysis. Cell 2005, 122, 915–926. [Google Scholar] [CrossRef] [PubMed]
- Coverley, D.; Laman, H.; Laskey, R.A. Distinct roles for cyclins e and a during DNA replication complex assembly and activation. Nat. Cell Biol. 2002, 4, 523–528. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.; Lee, Y.M.; Welcker, M.; Swanger, J.; Zagozdzon, A.; Winer, J.D.; Roberts, J.M.; Kaldis, P.; Clurman, B.E.; Sicinski, P. Kinase-Independent Function of Cyclin E. Mol. Cell 2007, 25, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Coudreuse, D.; Nurse, P. Driving the cell cycle with a minimal CDK control network. Nature 2010, 468, 1074–1079. [Google Scholar] [CrossRef] [PubMed]
- Uhlmann, F.; Bouchoux, C.; Lopez-Aviles, S. A quantitative model for cyclin-dependent kinase control of the cell cycle: revisited. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2011, 366, 3572–3583. [Google Scholar] [CrossRef] [PubMed]
- Stern, B.; Nurse, P. A quantitative model for the Cdc2 control of S phase and mitosis in fission yeast. Trends Genet. 1996, 12, 345–350. [Google Scholar] [CrossRef]
- Wilmes, G.M.; Archambault, V.; Austin, R.J.; Jacobson, M.D.; Bell, S.P.; Cross, F.R. Interaction of the S-phase cyclin Clb5 with an “RXL” docking sequence in the initiator protein Orc6 provides an origin-localized replication control switch. Genes Dev. 2004, 18, 981–991. [Google Scholar] [CrossRef] [PubMed]
- Mimura, S.; Seki, T.; Tanaka, S.; Diffley, J.F. Phosphorylation-dependent binding of mitotic cyclins to Cdc6 contributes to DNA replication control. Nature 2004, 431, 1118–1123. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Bell, S.P. CDK prevents Mcm2-7 helicase loading by inhibiting Cdt1 interaction with Orc6. Genes Dev. 2011, 25, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Diril, M.K.; Ratnacaram, C.K.; Padmakumar, V.C.; Du, T.; Wasser, M.; Coppola, V.; Tessarollo, L.; Kaldis, P. Cyclin-dependent kinase 1 (Cdk1) is essential for cell division and suppression of DNA re-replication but not for liver regeneration. Proc. Natl. Acad. Sci. USA 2012, 109, 3826–3831. [Google Scholar] [CrossRef] [PubMed]
- Walter, D.; Hoffmann, S.; Komseli, E.S.; Rappsilber, J.; Gorgoulis, V.; Sorensen, C.S. SCF(Cyclin F)-dependent degradation of CDC6 suppresses DNA re-replication. Nat. Commun. 2016, 7, 10530. [Google Scholar] [CrossRef] [PubMed]
- Johansson, P.; Jeffery, J.; Al-Ejeh, F.; Schulz, R.B.; Callen, D.F.; Kumar, R.; Khanna, K.K. SCF-FBXO31 E3 Ligase Targets DNA Replication Factor Cdt1 for Proteolysis in the G2 Phase of Cell Cycle to Prevent Re-replication. J. Biol. Chem. 2014, 289, 18514–18525. [Google Scholar] [CrossRef] [PubMed]
- Drury, L.S.; Perkins, G.; Diffley, J.F. The cyclin-dependent kinase Cdc28p regulates distinct modes of Cdc6P proteolysis during the budding yeast cell cycle. Curr. Biol. 2000, 10, 231–240. [Google Scholar] [CrossRef]
- Voitenleitner, C.; Fanning, E.; Nasheuer, H.P. Phosphorylation of DNA polymerase alpha-primase by cyclin A-dependent kinases regulates initiation of DNA replication in vitro. Oncogene 1997, 14, 1611–1615. [Google Scholar] [CrossRef] [PubMed]
- Voitenleitner, C.; Rehfuess, C.; Hilmes, M.; O’Rear, L.; Liao, P.C.; Gage, D.A.; Ott, R.; Nasheuer, H.P.; Fanning, E. Cell cycle-dependent regulation of human DNA polymerase alpha-primase activity by phosphorylation. Mol. Cell. Biol. 1999, 19, 646–656. [Google Scholar] [CrossRef] [PubMed]
- Higgins, G.; Roper, K.M.; Watson, I.J.; Blackhall, F.H.; Rom, W.N.; Pass, H.I.; Ainscough, J.F.; Coverley, D. Variant Ciz1 is a circulating biomarker for early-stage lung cancer. Proc. Natl. Acad. Sci. USA 2012, 109, E3128–E3135. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Wang, C.; Tang, X.; Sun, H.; Shao, Q.; Yang, X.; Qu, X. CIZ1 regulates the proliferation, cycle distribution and colony formation of RKO human colorectal cancer cells. Mol. Med. Rep. 2013, 8, 1630–1634. [Google Scholar] [PubMed]
- Wang, D.Q.; Wang, K.; Yan, D.W.; Liu, J.; Wang, B.; Li, M.X.; Wang, X.W.; Liu, J.; Peng, Z.H.; Li, G.X.; et al. Ciz1 is a novel predictor of survival in human colon cancer. Exp. Biol. Med. 2014, 239, 862–870. [Google Scholar] [CrossRef] [PubMed]
- Den Hollander, P.; Kumar, R. Dynein light chain 1 contributes to cell cycle progression by increasing cyclin-dependent kinase 2 activity in estrogen-stimulated cells. Cancer Res. 2006, 66, 5941–5949. [Google Scholar] [CrossRef] [PubMed]
- Den Hollander, P.; Rayala, S.K.; Coverley, D.; Kumar, R. Ciz1, a novel DNA-binding coactivator of the estrogen receptor alpha, confers hypersensitivity to estrogen action. Cancer Res. 2006, 66, 11021–11029. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Ren, X.; Li, L.; Yin, L.; Liang, K.; Yu, H.; Ren, H.; Zhou, W.; Jing, H.; Kong, C. Ciz1 promotes tumorigenicity of prostate carcinoma cells. Front. Biosci. 2015, 20, 705–715. [Google Scholar] [CrossRef]
- Lei, L.; Wu, J.; Gu, D.; Liu, H.; Wang, S. Ciz1 interacts with yap and activates its transcriptional activity in hepatocellular carcinoma cells. Tumour Biol. 2016, 37, 11073–11079. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wang, Y.; Dai, Y.; Wang, J.; Suo, T.; Pan, H.; Liu, H.; Shen, S.; Liu, H. Ciz1 promoted the growth and migration of gallbladder cancer cells. Tumour Biol. 2015, 36, 2583–2591. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Lei, L.; Gu, D.; Liu, H.; Wang, S. Ciz1 is upregulated in hepatocellular carcinoma and promotes the growth and migration of the cancer cells. Tumour Biol. 2015, 3, 4735–4742. [Google Scholar] [CrossRef] [PubMed]
- Pobbati, A.V.; Hong, W. Emerging roles of TEAD transcription factors and its coactivators in cancers. Cancer Biol. Ther. 2013, 14, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Powers, S.; Zhu, W.; Hannun, Y.A. Substantial contribution of extrinsic risk factors to cancer development. Nature 2016, 529, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Burrell, R.A.; McClelland, S.E.; Endesfelder, D.; Groth, P.; Weller, M.C.; Shaikh, N.; Domingo, E.; Kanu, N.; Dewhurst, S.M.; Gronroos, E.; et al. Replication stress links structural and numerical cancer chromosomal instability. Nature 2013, 494, 492–496. [Google Scholar] [CrossRef] [PubMed]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Macheret, M.; Halazonetis, T.D. DNA replication stress as a hallmark of cancer. Annu. Rev. Pathol. 2015, 10, 425–448. [Google Scholar] [CrossRef] [PubMed]
- Shimura, T.; Ochiai, Y.; Noma, N.; Oikawa, T.; Sano, Y.; Fukumoto, M. Cyclin D1 overexpression perturbs DNA replication and induces replication-associated DNA double-strand breaks in acquired radioresistant cells. Cell Cycle 2013, 12, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Bester, A.C.; Roniger, M.; Oren, Y.S.; Im, M.M.; Sarni, D.; Chaoat, M.; Bensimon, A.; Zamir, G.; Shewach, D.S.; Kerem, B. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 2011, 145, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, L.K.; Wang, X.; Li, Y.; Ekholm-Reed, S.; Wu, X.; Wang, P.; Reed, S.I. Cyclin E deregulation promotes loss of specific genomic regions. Curr. Biol. 2015, 25, 1327–1333. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.M.; Mortusewicz, O.; Afzal, I.; Lorvellec, M.; Garcia, P.; Helleday, T.; Petermann, E. Increased replication initiation and conflicts with transcription underlie cyclin E-induced replication stress. Oncogene 2013, 32, 3744–3753. [Google Scholar] [CrossRef] [PubMed]
- Quereda, V.; Porlan, E.; Canamero, M.; Dubus, P.; Malumbres, M. An essential role for Ink4 and Cip/Kip cell-cycle inhibitors in preventing replicative stress. Cell Death Differ. 2016, 23, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Hughes, B.T.; Sidorova, J.; Swanger, J.; Monnat, R.J., Jr.; Clurman, B.E. Essential role for Cdk2 inhibitory phosphorylation during replication stress revealed by a human Cdk2 knockin mutation. Proc. Natl. Acad. Sci. USA 2013, 110, 8954–8959. [Google Scholar] [CrossRef] [PubMed]
- Beck, H.; Nahse-Kumpf, V.; Larsen, M.S.; O’Hanlon, K.A.; Patzke, S.; Holmberg, C.; Mejlvang, J.; Groth, A.; Nielsen, O.; Syljuasen, R.G.; et al. Cyclin-dependent kinase suppression by WEE1 kinase protects the genome through control of replication initiation and nucleotide consumption. Mol. Cell. Biol. 2012, 32, 4226–4236. [Google Scholar] [CrossRef] [PubMed]
- Greil, C.; Krohs, J.; Schnerch, D.; Follo, M.; Felthaus, J.; Engelhardt, M.; Wasch, R. The role of APC/C(Cdh1) in replication stress and origin of genomic instability. Oncogene 2016, 35, 3062–3070. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Lin, J.R.; Vannier, J.B.; Slaats, G.G.; Kile, A.C.; Paulsen, R.D.; Manning, D.K.; Beier, D.R.; Giles, R.H.; Boulton, S.J.; et al. NEK8 links the ATR-regulated replication stress response and S phase CDK activity to renal ciliopathies. Mol. Cell 2013, 51, 423–439. [Google Scholar] [CrossRef] [PubMed]
- Spencer, S.L.; Cappell, S.D.; Tsai, F.C.; Overton, K.W.; Wang, C.L.; Meyer, T. The proliferation-quiescence decision is controlled by a bifurcation in CDK2 activity at mitotic exit. Cell 2013, 155, 369–383. [Google Scholar] [CrossRef] [PubMed]
- Barr, A.R.; Heldt, F.S.; Zhang, T.; Bakal, C.; Novak, B. A dynamical framework for the all-or-none G1/S transition. Cell Syst. 2016, 2, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Kotsantis, P.; Silva, L.M.; Irmscher, S.; Jones, R.M.; Folkes, L.; Gromak, N.; Petermann, E. Increased global transcription activity as a mechanism of replication stress in cancer. Nat. Commun. 2016, 7, 13087. [Google Scholar] [CrossRef] [PubMed]
- Bertoli, C.; Herlihy, A.E.; Pennycook, B.R.; Kriston-Vizi, J.; de Bruin, R.A. Sustained E2F-dependent transcription is a key mechanism to prevent replication-stress-induced DNA damage. Cell Rep. 2016, 15, 1412–1422. [Google Scholar] [CrossRef] [PubMed]
- Chatr-Aryamontri, A.; Oughtred, R.; Boucher, L.; Rust, J.; Chang, C.; Kolas, N.K.; O’Donnell, L.; Oster, S.; Theesfeld, C.; Sellam, A.; et al. The BioGRID interaction database: 2017 Update. Nucleic Acids Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Chatr-Aryamontri, A.; Breitkreutz, B.J.; Oughtred, R.; Boucher, L.; Heinicke, S.; Chen, D.; Stark, C.; Breitkreutz, A.; Kolas, N.; O’Donnell, L.; et al. The BioGRID interaction database: 2015 Update. Nucleic Acids Res. 2015, 43, D470–D478. [Google Scholar] [CrossRef] [PubMed]
- Hein, M.Y.; Hubner, N.C.; Poser, I.; Cox, J.; Nagaraj, N.; Toyoda, Y.; Gak, I.A.; Weisswange, I.; Mansfeld, J.; Buchholz, F.; et al. A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell 2015, 163, 712–723. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.D.; Coyaud, E.; Goncalves, J.; Mojarad, B.A.; Liu, Y.; Wu, Q.; Gheiratmand, L.; Comartin, D.; Tkach, J.M.; Cheung, S.W.; et al. A dynamic protein interaction landscape of the human centrosome-cilium interface. Cell 2015, 163, 1484–1499. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.P.; Tucholska, M.; Go, C.; Knight, J.D.; Gingras, A.C. Proximity biotinylation and affinity purification are complementary approaches for the interactome mapping of chromatin-associated protein complexes. J. Proteom. 2015, 118, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Huttlin, E.L.; Ting, L.; Bruckner, R.J.; Gebreab, F.; Gygi, M.P.; Szpyt, J.; Tam, S.; Zarraga, G.; Colby, G.; Baltier, K.; et al. The bioplex network: A systematic exploration of the human interactome. Cell 2015, 162, 425–440. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Wang, X.; Zhao, M.; Yang, R.; Malik, R.; Qiao, Y.; Poliakov, A.; Yocum, A.K.; Li, Y.; Chen, W.; et al. The central role of EED in the orchestration of polycomb group complexes. Nat. Commun. 2014, 5, 3127. [Google Scholar] [CrossRef] [PubMed]
- Hanson, D.; Stevens, A.; Murray, P.G.; Black, G.C.; Clayton, P.E. Identifying biological pathways that underlie primordial short stature using network analysis. J. Mol. Endocrinol. 2014, 52, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Lehner, B.; Sanderson, C.M. A protein interaction framework for human mRNA degradation. Genome Res. 2004, 14, 1315–1323. [Google Scholar] [CrossRef] [PubMed]
- Krzyzanowski, M.K.; Kozlowska, E.; Kozlowski, P. Identification and Functional Analysis of the erh1+ Gene Encoding Enhancer of Rudimentary Homolog from the Fission Yeast Schizosaccharomyces pombe. PLoS ONE 2012, 7, e49059. [Google Scholar] [CrossRef] [PubMed]
- Havrylov, S.; Rzhepetskyy, Y.; Malinowska, A.; Drobot, L.; Redowicz, M.J. Proteins recruited by SH3 domains of Ruk/CIN85 adaptor identified by LC-MS/MS. Proteome Sci. 2009, 7, 21. [Google Scholar] [CrossRef] [PubMed]
- Thalappilly, S.; Suliman, M.; Gayet, O.; Soubeyran, P.; Hermant, A.; Lecine, P.; Iovanna, J.L.; Dusetti, N.J. Identification of multi-SH3 domain-containing protein interactome in pancreatic cancer: A yeast two-hybrid approach. Proteomics 2008, 8, 3071–3081. [Google Scholar] [CrossRef] [PubMed]
| Cancer | Cancer-Specific Ciz1 Alteration | Mode of Intervention | Result of Intervention | Ref. |
|---|---|---|---|---|
| Lung cancer | Alternative splicing | shRNA | Reduced tumor growth in xenograft models | [73] |
| Colorectal carcinoma (CRC) | Overexpression | siRNA | Reduced proliferation, and colony formation in vitro | [74] |
| Gall bladder carcinoma (GBC) | Overexpression | siRNA | Reduced xenograft tumor growth. Reduced tumor migration in vivo | [80] |
| Prostate cancer | Overexpression | siRNA | Reduced tumorigenesis in xenograft models, reduced proliferation, G1 checkpoint activation | [78] |
| Breast cancer | Overexpression | siRNA | Reduced tumorigenesis, proliferation and anchorage dependence | [76,77] |
| Breast cancer | Overexpression of Ciz1 increases estrogen sensitivity | Ciz1 overexpression | Increased estrogen sensitivity and increased tumor size in xenograft models. | [77] |
| Hepatocellular carcinoma | Overexpression | Ciz1 overexpression | Increased proliferation, migration | [79,81] |
| siRNA | Reduced growth, tumorigenesis, metastasis |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pauzaite, T.; Thacker, U.; Tollitt, J.; Copeland, N.A. Emerging Roles for Ciz1 in Cell Cycle Regulation and as a Driver of Tumorigenesis. Biomolecules 2017, 7, 1. https://doi.org/10.3390/biom7010001
Pauzaite T, Thacker U, Tollitt J, Copeland NA. Emerging Roles for Ciz1 in Cell Cycle Regulation and as a Driver of Tumorigenesis. Biomolecules. 2017; 7(1):1. https://doi.org/10.3390/biom7010001
Chicago/Turabian StylePauzaite, Tekle, Urvi Thacker, James Tollitt, and Nikki A. Copeland. 2017. "Emerging Roles for Ciz1 in Cell Cycle Regulation and as a Driver of Tumorigenesis" Biomolecules 7, no. 1: 1. https://doi.org/10.3390/biom7010001