
The Genomic Impact of DNA CpG Methylation on Gene Expression; Relationships in Prostate Cancer

{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Methylation of DNA Cytosine
2. The Control of CpG Methylation and Its Impact on Transcription
3. The Transition from Epigenetic to Epigenomic Analyses of Cancer Genomes
3.1. Development of Technologies and Computational Approaches for Epigenomic Analyses
3.2. Analyses of CpG Methylation in Large Cohorts of Publically Available Data
4. Prostate Cancer as a Model of the Interplay between Genomic and Epigenomic Cancer Drivers
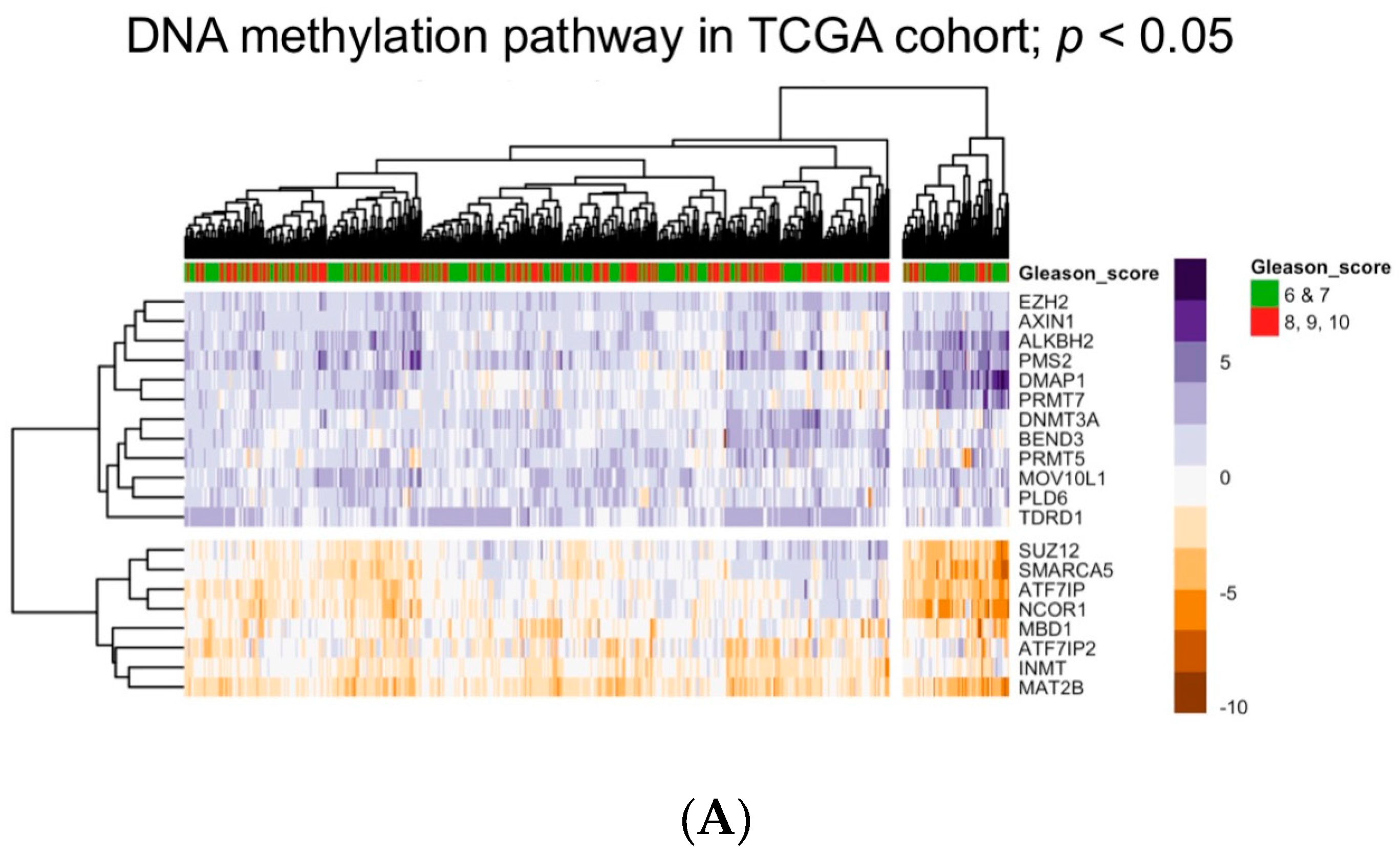
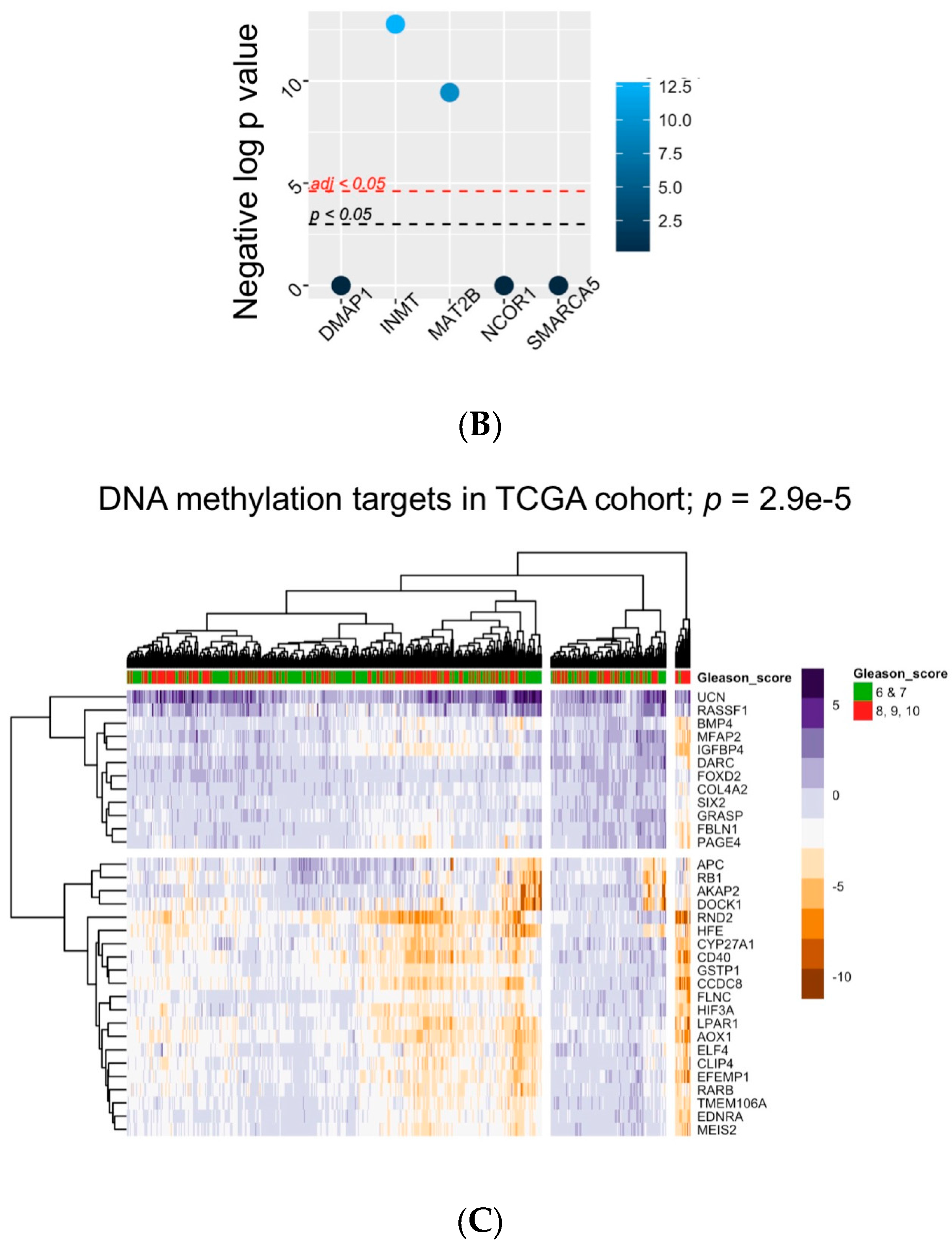
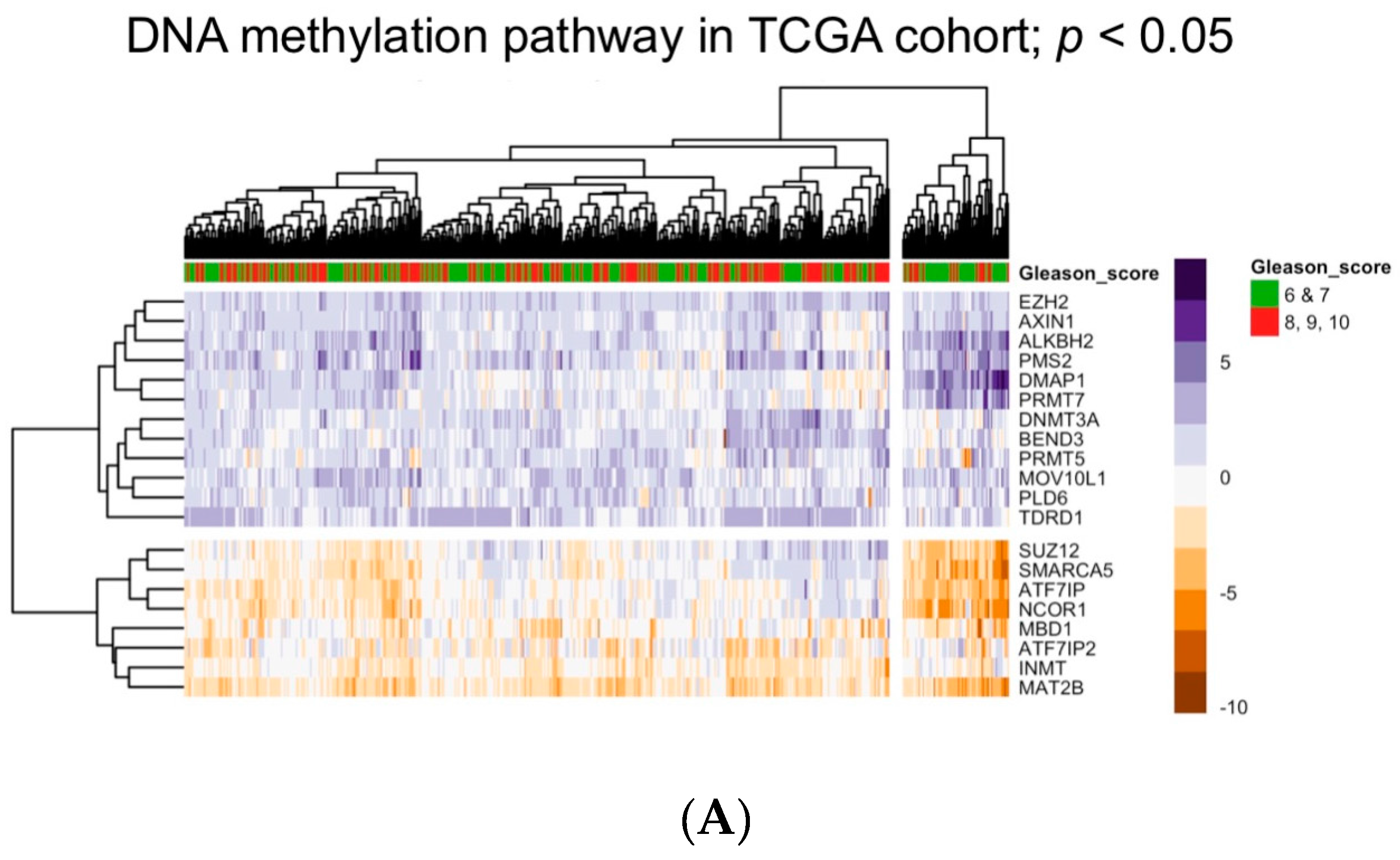
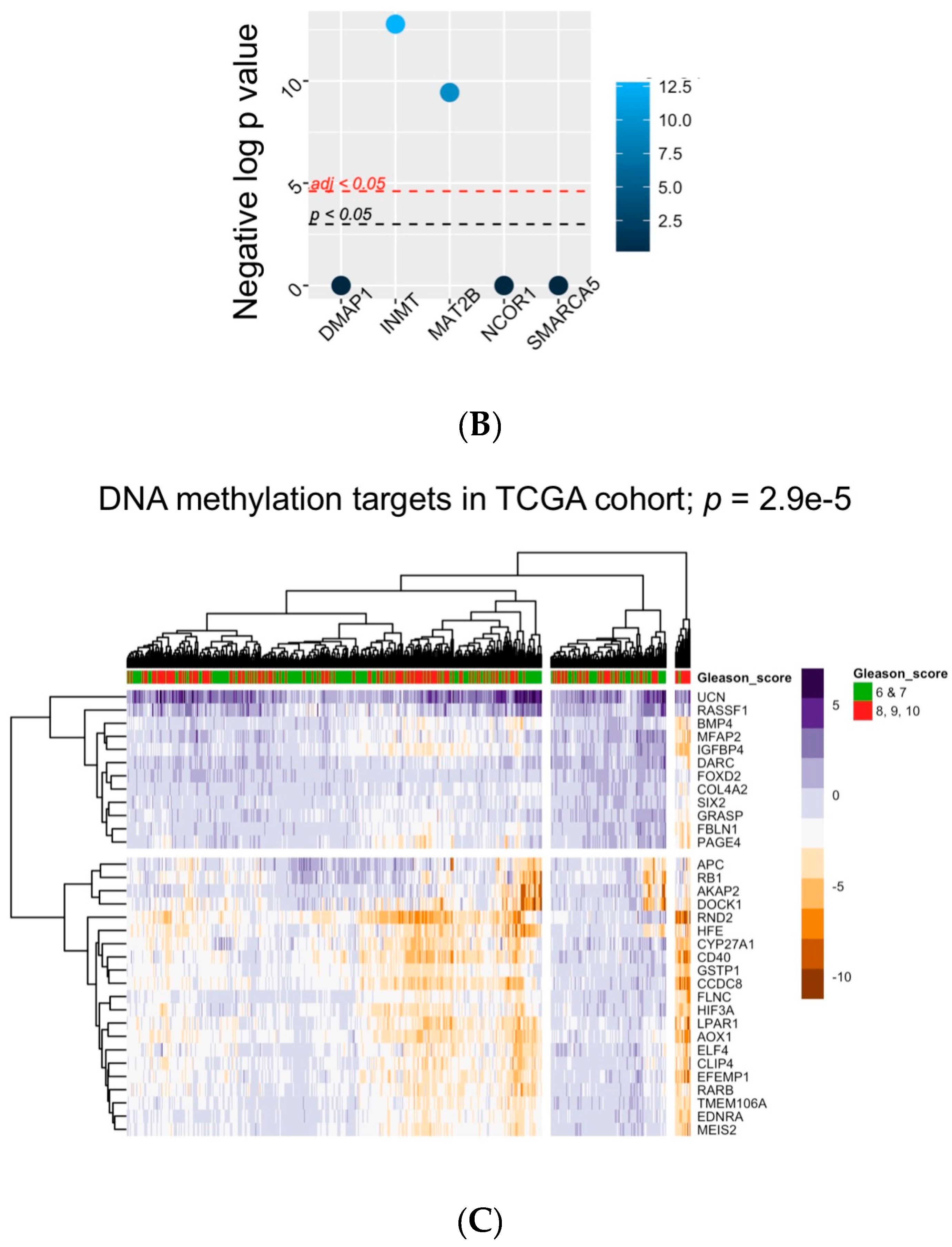
5. Bioinformatic Approaches to Reveal Associations between DNA Methylation and Prostate Tumor Status
6. Summary
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Mukamel, E.A. Turning over DNA methylation in the mind. Front. Neurosci. 2015, 9, 252. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.J.; Goodman, S.J.; Kobor, M.S. DNA methylation and healthy human aging. Aging Cell 2015, 14, 924–932. [Google Scholar] [CrossRef] [PubMed]
- Niculescu, M.D.; Lupu, D.S. Nutritional influence on epigenetics and effects on longevity. Curr. Opin. Clin. Nutr. Metab. Care 2011, 14, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat. Rev. Genet. 2007, 8, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Cheng, X.; Klimasauskas, S.; Mi, S.; Posfai, J.; Roberts, R.J.; Wilson, G.G. The DNA (cytosine-5) methyltransferases. Nucleic Acids Res. 1994, 22, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Matzke, M.A.; Mosher, R.A. RNA-directed DNA methylation: An epigenetic pathway of increasing complexity. Nat. Rev. Genet. 2014, 15, 394–408. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Histone methylation in transcriptional control. Curr. Opin. Genet. Dev. 2002, 12, 198–209. [Google Scholar] [CrossRef]
- Guenther, M.G.; Jenner, R.G.; Chevalier, B.; Nakamura, T.; Croce, C.M.; Canaani, E.; Young, R.A. Global and Hox-specific roles for the MLL1 methyltransferase. Proc. Natl. Acad. Sci. USA 2005, 102, 8603–8608. [Google Scholar] [CrossRef] [PubMed]
- Mentch, S.J.; Locasale, J.W. One-carbon metabolism and epigenetics: Understanding the specificity. Ann. N. Y. Acad. Sci. 2016, 1363, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Mattocks, D.A.; Mentch, S.J.; Shneyder, J.; Ables, G.P.; Sun, D.; Richie, J.P., Jr.; Locasale, J.W.; Nichenametla, S.N. Short term methionine restriction increases hepatic global DNA methylation in adult but not young male C57BL/6J mice. Exp. Gerontol. 2016, 88, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Nazki, F.H.; Sameer, A.S.; Ganaie, B.A. Folate: Metabolism, genes, polymorphisms and the associated diseases. Gene 2014, 533, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Scarano, E.; Geraci, G.; Rossi, M. Deoxycytidylate aminohydrolase. II. Kinetic properties. The activatory effect of deoxycytidine triphosphate and the inhibitory effect of deoxythymidine triphosphate. Biochemistry 1967, 6, 192–201. [Google Scholar] [PubMed]
- Geraci, G.; Rossi, M.; Scarano, E. Deoxycytidylate aminohydrolase. I. Preparation and properties of the homogeneous enzyme. Biochemistry 1967, 6, 183–191. [Google Scholar] [PubMed]
- He, X.; Tillo, D.; Vierstra, J.; Syed, K.S.; Deng, C.; Ray, G.J.; Stamatoyannopoulos, J.; FitzGerald, P.C.; Vinson, C. Methylated Cytosines Mutate to Transcription Factor Binding Sites that Drive Tetrapod Evolution. Genome Biol. Evol. 2015, 7, 3155–3169. [Google Scholar] [CrossRef] [PubMed]
- Stier, I.; Kiss, A. Cytosine-to-Uracil Deamination by SssI DNA Methyltransferase. PLoS ONE 2013, 8, e79003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannistraro, V.J.; Taylor, J.S. Acceleration of 5-methylcytosine deamination in cyclobutane dimers by G and its implications for UV-induced C-to-T mutation hotspots. J. Mol. Biol. 2009, 392, 1145–1157. [Google Scholar] [CrossRef] [PubMed]
- Hermann, A.; Gowher, H.; Jeltsch, A. Biochemistry and biology of mammalian DNA methyltransferases. Cell. Mol. Life Sci. 2004, 61, 2571–2587. [Google Scholar] [CrossRef] [PubMed]
- Sandelin, A.; Carninci, P.; Lenhard, B.; Ponjavic, J.; Hayashizaki, Y.; Hume, D.A. Mammalian RNA polymerase II core promoters: Insights from genome-wide studies. Nat. Rev. Genet. 2007, 8, 424–436. [Google Scholar] [CrossRef] [PubMed]
- Zemach, A.; McDaniel, I.E.; Silva, P.; Zilberman, D. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 2010, 328, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Doi, A.; Park, I.H.; Wen, B.; Murakami, P.; Aryee, M.J.; Irizarry, R.; Herb, B.; Ladd-Acosta, C.; Rho, J.; Loewer, S.; et al. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat. Genet. 2009, 41, 1350–1353. [Google Scholar] [CrossRef] [PubMed]
- Guenther, M.G.; Levine, S.S.; Boyer, L.A.; Jaenisch, R.; Young, R.A. A chromatin landmark and transcription initiation at most promoters in human cells. Cell 2007, 130, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, T.S.; Ku, M.; Jaffe, D.B.; Issac, B.; Lieberman, E.; Giannoukos, G.; Alvarez, P.; Brockman, W.; Kim, T.K.; Koche, R.P.; et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 2007, 448, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009, 458, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Medvedeva, Y.A.; Fridman, M.V.; Oparina, N.J.; Malko, D.B.; Ermakova, E.O.; Kulakovskiy, I.V.; Heinzel, A.; Makeev, V.J. Intergenic, gene terminal, and intragenic CpG islands in the human genome. BMC Genom. 2010, 11, 48. [Google Scholar] [CrossRef] [PubMed]
- Jin, B.; Robertson, K.D. DNA methyltransferases, DNA damage repair, and cancer. Adv. Exp. Med. Biol. 2013, 754, 3–29. [Google Scholar] [PubMed]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Dai, H.; Martos, S.N.; Xu, B.; Gao, Y.; Li, T.; Zhu, G.; Schones, D.E.; Wang, Z. Distinct roles of DNMT1-dependent and DNMT1-independent methylation patterns in the genome of mouse embryonic stem cells. Genome Biol. 2015, 16, 115. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Karnik, R.; Gu, H.; Ziller, M.J.; Clement, K.; Tsankov, A.M.; Akopian, V.; Gifford, C.A.; Donaghey, J.; Galonska, C.; et al. Targeted disruption of DNMT1, DNMT3A and DNMT3B in human embryonic stem cells. Nat. Genet. 2015, 47, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Egger, G.; Jeong, S.; Escobar, S.G.; Cortez, C.C.; Li, T.W.; Saito, Y.; Yoo, C.B.; Jones, P.A.; Liang, G. Identification of DNMT1 (DNA methyltransferase 1) hypomorphs in somatic knockouts suggests an essential role for DNMT1 in cell survival. Proc. Natl. Acad. Sci. USA 2006, 103, 14080–14085. [Google Scholar] [CrossRef] [PubMed]
- Oka, M.; Meacham, A.M.; Hamazaki, T.; Rodic, N.; Chang, L.J.; Terada, N. De novo DNA methyltransferases Dnmt3a and Dnmt3b primarily mediate the cytotoxic effect of 5-aza-2′-deoxycytidine. Oncogene 2005, 24, 3091–3099. [Google Scholar] [CrossRef] [PubMed]
- Hattori, N.; Abe, T.; Hattori, N.; Suzuki, M.; Matsuyama, T.; Yoshida, S.; Li, E.; Shiota, K. Preference of DNA methyltransferases for CpG islands in mouse embryonic stem cells. Genome Res. 2004, 14, 1733–1740. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Gonzalez-Avalos, E.; Chawla, A.; Jeong, M.; Lopez-Moyado, I.F.; Li, W.; Goodell, M.A.; Chavez, L.; Ko, M.; Rao, A. Acute loss of TET function results in aggressive myeloid cancer in mice. Nat. Commun. 2015, 6, 10071. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Liu, Y.; Jiang, L.; Yamaguchi, S.; Zhang, Y. Role of Tet proteins in enhancer activity and telomere elongation. Genes Dev. 2014, 28, 2103–2119. [Google Scholar] [CrossRef] [PubMed]
- Dawlaty, M.M.; Breiling, A.; Le, T.; Barrasa, M.I.; Raddatz, G.; Gao, Q.; Powell, B.E.; Cheng, A.W.; Faull, K.F.; Lyko, F.; et al. Loss of Tet enzymes compromises proper differentiation of embryonic stem cells. Dev. Cell 2014, 29, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Shi, Y.G. Tet family proteins and 5-hydroxymethylcytosine in development and disease. Development 2012, 139, 1895–1902. [Google Scholar] [CrossRef] [PubMed]
- Ono, R.; Kumagai, H.; Nakajima, H.; Hishiya, A.; Taki, T.; Horikawa, K.; Takatsu, K.; Satoh, T.; Hayashi, Y.; Kitamura, T.; et al. Mixed-lineage-leukemia (MLL) fusion protein collaborates with Ras to induce acute leukemia through aberrant Hox expression and Raf activation. Leukemia 2009, 23, 2197–2209. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; D’Alessio, A.C.; Taranova, O.V.; Hong, K.; Sowers, L.C.; Zhang, Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 2010, 466, 1129–1133. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, S.; Hong, K.; Liu, R.; Shen, L.; Inoue, A.; Diep, D.; Zhang, K.; Zhang, Y. Tet1 controls meiosis by regulating meiotic gene expression. Nature 2012, 492, 443–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, C.; Lu, Y.; Jelinek, J.; Liang, S.; Estecio, M.R.; Barton, M.C.; Issa, J.P. TET1 is a maintenance DNA demethylase that prevents methylation spreading in differentiated cells. Nucleic Acids Res. 2014, 42, 6956–6971. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Zhang, Y. Mechanisms and functions of Tet protein-mediated 5-methylcytosine oxidation. Genes Dev. 2011, 25, 2436–2452. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; D’Alessio, A.C.; Ito, S.; Xia, K.; Wang, Z.; Cui, K.; Zhao, K.; Sun, Y.E.; Zhang, Y. Dual functions of Tet1 in transcriptional regulation in mouse embryonic stem cells. Nature 2011, 473, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Pastor, W.A.; Pape, U.J.; Huang, Y.; Henderson, H.R.; Lister, R.; Ko, M.; McLoughlin, E.M.; Brudno, Y.; Mahapatra, S.; Kapranov, P.; et al. Genome-wide mapping of 5-hydroxymethylcytosine in embryonic stem cells. Nature 2011, 473, 394–397. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Wu, H.; Diep, D.; Yamaguchi, S.; D’Alessio, A.C.; Fung, H.L.; Zhang, K.; Zhang, Y. Genome-wide analysis reveals TET- and TDG-dependent 5-methylcytosine oxidation dynamics. Cell 2013, 153, 692–706. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, S.; Shen, L.; Liu, Y.; Sendler, D.; Zhang, Y. Role of Tet1 in erasure of genomic imprinting. Nature 2013, 504, 460–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Zhao, K.; Shen, Q.; Han, Y.; Gu, Y.; Li, X.; Zhao, D.; Liu, Y.; Wang, C.; Zhang, X.; et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 2015, 525, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Abdel-Wahab, O.; Cervantes, F.; Crispino, J.D.; Finazzi, G.; Girodon, F.; Gisslinger, H.; Gotlib, J.; Kiladjian, J.J.; Levine, R.L.; et al. Mutations with epigenetic effects in myeloproliferative neoplasms and recent progress in treatment: Proceedings from the 5th International Post-ASH Symposium. Blood Cancer J. 2011, 1, e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watt, F.; Molloy, P.L. Cytosine methylation prevents binding to DNA of a HeLa cell transcription factor required for optimal expression of the adenovirus major late promoter. Genes Dev. 1988, 2, 1136–1143. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.D.; Meehan, R.R.; Henzel, W.J.; Maurer-Fogy, I.; Jeppesen, P.; Klein, F.; Bird, A. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell 1992, 69, 905–914. [Google Scholar] [CrossRef]
- Jones, P.A.; Laird, P.W. Cancer epigenetics comes of age. Nat. Genet. 1999, 21, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Makhlouf, M.; Ouimette, J.F.; Oldfield, A.; Navarro, P.; Neuillet, D.; Rougeulle, C. A prominent and conserved role for YY1 in Xist transcriptional activation. Nat. Commun. 2014, 5, 4878. [Google Scholar] [CrossRef] [PubMed]
- Nora, E.P.; Lajoie, B.R.; Schulz, E.G.; Giorgetti, L.; Okamoto, I.; Servant, N.; Piolot, T.; van Berkum, N.L.; Meisig, J.; Sedat, J.; et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 2012, 485, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Umlauf, D.; Goto, Y.; Cao, R.; Cerqueira, F.; Wagschal, A.; Zhang, Y.; Feil, R. Imprinting along the Kcnq1 domain on mouse chromosome 7 involves repressive histone methylation and recruitment of Polycomb group complexes. Nat. Genet. 2004, 36, 1296–1300. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Barger, C.J.; Eng, K.H.; Klinkebiel, D.; Link, P.A.; Omilian, A.; Bshara, W.; Odunsi, K.; Karpf, A.R. PRAME expression and promoter hypomethylation in epithelial ovarian cancer. Oncotarget 2016, 7, 45352–45369. [Google Scholar] [CrossRef] [PubMed]
- Liang, P.; Song, F.; Ghosh, S.; Morien, E.; Qin, M.; Mahmood, S.; Fujiwara, K.; Igarashi, J.; Nagase, H.; Held, W.A. Genome-wide survey reveals dynamic widespread tissue-specific changes in DNA methylation during development. BMC Genom. 2011, 12, 231. [Google Scholar] [CrossRef] [PubMed]
- Meklat, F.; Li, Z.; Wang, Z.; Zhang, Y.; Zhang, J.; Jewell, A.; Lim, S.H. Cancer-testis antigens in haematological malignancies. Br. J. Haematol. 2007, 136, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.A.; Kang, Y.; Abdullaev, Z.; Flanagan, P.T.; Pack, S.D.; Fischette, M.R.; Adnani, M.T.; Loukinov, D.I.; Vatolin, S.; Risinger, J.I.; et al. Reciprocal binding of CTCF and BORIS to the NY-ESO-1 promoter coincides with derepression of this cancer-testis gene in lung cancer cells. Cancer Res. 2005, 65, 7763–7774. [Google Scholar] [PubMed]
- Honda, T.; Tamura, G.; Waki, T.; Kawata, S.; Terashima, M.; Nishizuka, S.; Motoyama, T. Demethylation of MAGE promoters during gastric cancer progression. Br. J. Cancer 2004, 90, 838–843. [Google Scholar] [CrossRef] [PubMed]
- Sigalotti, L.; Coral, S.; Nardi, G.; Spessotto, A.; Cortini, E.; Cattarossi, I.; Colizzi, F.; Altomonte, M.; Maio, M. Promoter methylation controls the expression of MAGE2, 3 and 4 genes in human cutaneous melanoma. J. Immunother. 2002, 25, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Sieber, O.M.; Tomlinson, S.R.; Tomlinson, I.P. Tissue, cell and stage specificity of (epi)mutations in cancers. Nat. Rev. Cancer 2005, 5, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P.; Vogelstein, B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983, 301, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Hoppener, J.W.; de Bustros, A.; Steenbergh, P.H.; Lips, C.J.; Nelkin, B.D. DNA methylation patterns of the calcitonin gene in human lung cancers and lymphomas. Cancer Res. 1986, 46, 2917–2922. [Google Scholar] [PubMed]
- Kushwaha, G.; Dozmorov, M.; Wren, J.D.; Qiu, J.; Shi, H.; Xu, D. Hypomethylation coordinates antagonistically with hypermethylation in cancer development: A case study of leukemia. Hum. Genom. 2016, 10, 18. [Google Scholar] [CrossRef] [PubMed]
- Abdelfatah, E.; Kerner, Z.; Nanda, N.; Ahuja, N. Epigenetic therapy in gastrointestinal cancer: The right combination. Therap. Adv. Gastroenterol. 2016, 9, 560–579. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, E.A.; Gore, S.D. DNA methyltransferase and histone deacetylase inhibitors in the treatment of myelodysplastic syndromes. Semin. Hematol. 2008, 45, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Costello, J.F.; Fruhwald, M.C.; Smiraglia, D.J.; Rush, L.J.; Robertson, G.P.; Gao, X.; Wright, F.A.; Feramisco, J.D.; Peltomaki, P.; Lang, J.C.; et al. Aberrant CpG-island methylation has non-random and tumour-type-specific patterns. Nat. Genet. 2000, 24, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.M.; Bird, A. DNA methylation landscapes: Provocative insights from epigenomics. Nat. Rev. Genet. 2008, 9, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Wang, Y.W.; Zhang, M.Q.; Gazdar, A.F. DNA methylation data analysis and its application to cancer research. Epigenomics 2013, 5, 301–316. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, V.N.; Lingjaerde, O.C.; Russnes, H.G.; Vollan, H.K.; Frigessi, A.; Borresen-Dale, A.L. Principles and methods of integrative genomic analyses in cancer. Nat. Rev. Cancer 2014, 14, 299–313. [Google Scholar] [CrossRef] [PubMed]
- Dedeurwaerder, S.; Defrance, M.; Bizet, M.; Calonne, E.; Bontempi, G.; Fuks, F. A comprehensive overview of Infinium HumanMethylation450 data processing. Brief. Bioinform. 2014, 15, 929–941. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Fang, F.; Miller, D.F.; Pilrose, J.M.; Matei, D.; Huang, T.H.; Nephew, K.P. Global DNA methylation profiling technologies and the ovarian cancer methylome. Methods Mol. Biol. 2015, 1238, 653–675. [Google Scholar] [PubMed]
- Sarda, S.; Hannenhalli, S. Next-generation sequencing and epigenomics research: A hammer in search of nails. Genom. Inf. 2014, 12, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Le Meur, N.; Gentleman, R. Analyzing biological data using R: Methods for graphs and networks. Methods Mol. Biol. 2012, 804, 343–373. [Google Scholar] [PubMed]
- Dudoit, S.; Gentleman, R.C.; Quackenbush, J. Open source software for the analysis of microarray data. Biotechniques 2003, 3, 45–51. [Google Scholar]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, W.; Carey, V.J.; Gentleman, R.; Anders, S.; Carlson, M.; Carvalho, B.S.; Bravo, H.C.; Davis, S.; Gatto, L.; Girke, T.; et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 2015, 12, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Maksimovic, J.; Phipson, B.; Oshlack, A. A cross-package Bioconductor workflow for analysing methylation array data. F1000Reserch 2016, 5, 1281. [Google Scholar] [CrossRef]
- Birney, E. The making of ENCODE: Lessons for big-data projects. Nature 2012, 489, 49–51. [Google Scholar] [CrossRef] [PubMed]
- Consortium, E.P.; Birney, E.; Stamatoyannopoulos, J.A.; Dutta, A.; Guigo, R.; Gingeras, T.R.; Margulies, E.H.; Weng, Z.; Snyder, M.; Dermitzakis, E.T.; et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 2007, 447, 799–816. [Google Scholar]
- Roadmap Epigenomics, C.; Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar]
- Sanli, K.; Karlsson, F.H.; Nookaew, I.; Nielsen, J. FANTOM: Functional and taxonomic analysis of metagenomes. BMC Bioinform. 2013, 14, 38. [Google Scholar] [CrossRef] [PubMed]
- Weinhold, N.; Jacobsen, A.; Schultz, N.; Sander, C.; Lee, W. Genome-wide analysis of noncoding regulatory mutations in cancer. Nat. Genet. 2014, 46, 1160–1165. [Google Scholar] [CrossRef] [PubMed]
- Ongen, H.; Andersen, C.L.; Bramsen, J.B.; Oster, B.; Rasmussen, M.H.; Ferreira, P.G.; Sandoval, J.; Vidal, E.; Whiffin, N.; Planchon, A.; et al. Putative cis-regulatory drivers in colorectal cancer. Nature 2014, 512, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.D.; Desai, K.; Kron, K.J.; Mazrooei, P.; Sinnott-Armstrong, N.A.; Treloar, A.E.; Dowar, M.; Thu, K.L.; Cescon, D.W.; Silvester, J.; et al. Noncoding somatic and inherited single-nucleotide variants converge to promote ESR1 expression in breast cancer. Nat. Genet. 2016, 48, 1260–1266. [Google Scholar] [CrossRef] [PubMed]
- Oldridge, D.A.; Wood, A.C.; Weichert-Leahey, N.; Crimmins, I.; Sussman, R.; Winter, C.; McDaniel, L.D.; Diamond, M.; Hart, L.S.; Zhu, S.; et al. Genetic predisposition to neuroblastoma mediated by a LMO1 super-enhancer polymorphism. Nature 2015, 528, 418–421. [Google Scholar] [CrossRef] [PubMed]
- Chng, K.R.; Chang, C.W.; Tan, S.K.; Yang, C.; Hong, S.Z.; Sng, N.Y.; Cheung, E. A transcriptional repressor co-regulatory network governing androgen response in prostate cancers. EMBO J. 2012, 31, 2810–2823. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.; Qi, J.; Filipp, F.V. Refinement of the androgen response element based on ChIP-Seq in androgen-insensitive and androgen-responsive prostate cancer cell lines. Sci. Rep. 2016, 6, 32611. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, W.; Liu, X.S.; Carroll, J.S.; Janne, O.A.; Keeton, E.K.; Chinnaiyan, A.M.; Pienta, K.J.; Brown, M. A hierarchical network of transcription factors governs androgen receptor-dependent prostate cancer growth. Mol. Cell 2007, 27, 380–392. [Google Scholar] [CrossRef] [PubMed]
- Witte, T.; Plass, C.; Gerhauser, C. Pan-cancer patterns of DNA methylation. Genome Med. 2014, 6, 66. [Google Scholar] [CrossRef] [PubMed]
- Bock, C.; Beerman, I.; Lien, W.H.; Smith, Z.D.; Gu, H.; Boyle, P.; Gnirke, A.; Fuchs, E.; Rossi, D.J.; Meissner, A. DNA Methylation Dynamics during In Vivo Differentiation of Blood and Skin Stem Cells. Mol. Cell 2012, 47, 633–647. [Google Scholar] [CrossRef] [PubMed]
- Krum, S.A.; Miranda-Carboni, G.A.; Lupien, M.; Eeckhoute, J.; Carroll, J.S.; Brown, M. Unique ERα Cistromes Control Cell Type-Specific Gene Regulation. Mol. Endocrinol. 2008, 22, 2393–2406. [Google Scholar] [CrossRef] [PubMed]
- Ogoshi, K.; Hashimoto, S.; Nakatani, Y.; Qu, W.; Oshima, K.; Tokunaga, K.; Sugano, S.; Hattori, M.; Morishita, S.; Matsushima, K. Genome-wide profiling of DNA methylation in human cancer cells. Genomics 2011, 98, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Irizarry, R.A.; Ladd-Acosta, C.; Wen, B.; Wu, Z.; Montano, C.; Onyango, P.; Cui, H.; Gabo, K.; Rongione, M.; Webster, M.; et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet. 2009, 41, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.C.; Houseman, E.A.; King, J.E.; von Herrmann, K.M.; Fadul, C.E.; Christensen, B.C. 5-Hydroxymethylcytosine localizes to enhancer elements and is associated with survival in glioblastoma patients. Nat. Commun. 2016, 7, 13177. [Google Scholar] [CrossRef] [PubMed]
- Bogdanovic, O.; Smits, A.H.; Mustienes, E.D.; Tena, J.J.; Ford, E.; Williams, R.; Senanayake, U.; Schultz, M.D.; Hontelez, S.; van Kruijsbergen, I.; et al. Active DNA demethylation at enhancers during the vertebrate phylotypic period. Nat. Genet. 2016, 48, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Charlet, J.; Duymich, C.E.; Lay, F.D.; Mundbjerg, K.; Sorensen, K.D.; Liang, G.; Jones, P.A. Bivalent Regions of Cytosine Methylation and H3K27 Acetylation Suggest an Active Role for DNA Methylation at Enhancers. Mol. Cell 2016, 62, 422–431. [Google Scholar] [CrossRef] [PubMed]
- Engreitz, J.M.; Haines, J.E.; Perez, E.M.; Munson, G.; Chen, J.; Kane, M.; McDonel, P.E.; Guttman, M.; Lander, E.S. Local regulation of gene expression by lncRNA promoters, transcription and splicing. Nature 2016, 539, 452–455. [Google Scholar] [CrossRef] [PubMed]
- Ebisuya, M.; Yamamoto, T.; Nakajima, M.; Nishida, E. Ripples from neighbouring transcription. Nat. Cell Biol. 2008, 10, 1106–1113. [Google Scholar] [CrossRef] [PubMed]
- Kosak, S.T.; Scalzo, D.; Alworth, S.V.; Li, F.; Palmer, S.; Enver, T.; Lee, J.S.; Groudine, M. Coordinate Gene Regulation During Hematopoiesis Is Related to Genomic Organization. PLoS Biol. 2007, 5, e309. [Google Scholar] [CrossRef] [PubMed]
- Medvedeva, Y.A.; Khamis, A.M.; Kulakovskiy, I.V.; Ba-Alawi, W.; Bhuyan, M.S.; Kawaji, H.; Lassmann, T.; Harbers, M.; Forrest, A.R.; Bajic, V.B.; et al. Effects of cytosine methylation on transcription factor binding sites. BMC Genom. 2014, 15, 119. [Google Scholar] [CrossRef] [PubMed]
- Aran, D.; Hellman, A. DNA methylation of transcriptional enhancers and cancer predisposition. Cell 2013, 154, 11–13. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.C.; Kim, N.Y.; Seo, Y.R.; Kim, Y. An Integrated Analysis of the Genome-Wide Profiles of DNA Methylation and mRNA Expression Defining the Side Population of a Human Malignant Mesothelioma Cell Line. J. Cancer 2016, 7, 1668–1679. [Google Scholar] [CrossRef] [PubMed]
- He, D.X.; Gu, F.; Gao, F.; Hao, J.J.; Gong, D.; Gu, X.T.; Mao, A.Q.; Jin, J.; Fu, L.; Ma, X. Genome-wide profiles of methylation, microRNAs, and gene expression in chemoresistant breast cancer. Sci. Rep. 2016, 6, 24706. [Google Scholar] [CrossRef] [PubMed]
- Bjaanaes, M.M.; Fleischer, T.; Halvorsen, A.R.; Daunay, A.; Busato, F.; Solberg, S.; Jorgensen, L.; Kure, E.; Edvardsen, H.; Borresen-Dale, A.L.; et al. Genome-wide DNA methylation analyses in lung adenocarcinomas: Association with EGFR, KRAS and TP53 mutation status, gene expression and prognosis. Mol. Oncol. 2016, 10, 330–343. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Peng, H.; Huang, X.; Zhao, M.; Li, Z.; Yin, N.; Wang, X.; Yu, F.; Yin, B.; Yuan, Y.; et al. Genome-wide profiling of DNA methylation and gene expression in esophageal squamous cell carcinoma. Oncotarget 2016, 7, 4507–4521. [Google Scholar] [PubMed]
- Singhal, S.K.; Usmani, N.; Michiels, S.; Metzger-Filho, O.; Saini, K.S.; Kovalchuk, O.; Parliament, M. Towards understanding the breast cancer epigenome: A comparison of genome-wide DNA methylation and gene expression data. Oncotarget 2016, 7, 3002–3017. [Google Scholar] [PubMed]
- Schroder, F.H.; Hugosson, J.; Roobol, M.J.; Tammela, T.L.; Ciatto, S.; Nelen, V.; Kwiatkowski, M.; Lujan, M.; Lilja, H.; Zappa, M.; et al. Screening and prostate-cancer mortality in a randomized European study. N. Engl. J. Med. 2009, 360, 1320–1328. [Google Scholar] [CrossRef] [PubMed]
- Andriole, G.L.; Crawford, E.D.; Grubb, R.L., III; Buys, S.S.; Chia, D.; Church, T.R.; Fouad, M.N.; Gelmann, E.P.; Kvale, P.A.; Reding, D.J.; et al. Mortality results from a randomized prostate-cancer screening trial. N. Engl. J. Med. 2009, 360, 1310–1319. [Google Scholar] [CrossRef] [PubMed]
- Freedland, S.J. Screening, risk assessment, and the approach to therapy in patients with prostate cancer. Cancer 2011, 117, 1123–1135. [Google Scholar] [CrossRef] [PubMed]
- Cookson, M.S.; Aus, G.; Burnett, A.L.; Canby-Hagino, E.D.; D’Amico, A.V.; Dmochowski, R.R.; Eton, D.T.; Forman, J.D.; Goldenberg, S.L.; Hernandez, J.; et al. Variation in the definition of biochemical recurrence in patients treated for localized prostate cancer: The American Urological Association Prostate Guidelines for Localized Prostate Cancer Update Panel report and recommendations for a standard in the reporting of surgical outcomes. J. Urol. 2007, 177, 540–545. [Google Scholar] [PubMed]
- Yamoah, K.; Deville, C.; Vapiwala, N.; Spangler, E.; Zeigler-Johnson, C.M.; Malkowicz, B.; Lee, D.I.; Kattan, M.; Dicker, A.P.; Rebbeck, T.R. African American men with low-grade prostate cancer have increased disease recurrence after prostatectomy compared with Caucasian men. Urol. Oncol. 2015, 33, 70.e15–70.e22. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, M.S.; Kuban, D.A.; Strom, S.S.; Du, X.L.; Lopez, D.S.; Yamal, J.M. Assessing the Optimum Use of Androgen-Deprivation Therapy in High-Risk Prostate Cancer Patients Undergoing External Beam Radiation Therapy. Am. J. Mens. Health 2015. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.; Grizzle, W.; Wang, H.; Yates, C. MicroRNAs that affect prostate cancer: Emphasis on prostate cancer in African Americans. Biotechnol. Histochem. 2013, 88, 410–424. [Google Scholar] [CrossRef] [PubMed]
- Powell, I.J.; Bollig-Fischer, A. Minireview: The molecular and genomic basis for prostate cancer health disparities. Mol. Endocrinol. 2013, 27, 879–891. [Google Scholar] [CrossRef] [PubMed]
- Drake, B.F.; Keane, T.E.; Mosley, C.M.; Adams, S.A.; Elder, K.T.; Modayil, M.V.; Ureda, J.R.; Hebert, J.R. Prostate cancer disparities in South Carolina: Early detection, special programs, and descriptive epidemiology. J.S.C. Med. Assoc. 2006, 102, 241–249. [Google Scholar] [PubMed]
- Cooney, K.A. Hereditary prostate cancer in African-American families. Semin. Urol. Oncol. 1998, 16, 202–206. [Google Scholar] [PubMed]
- Fraser, M.; Sabelnykova, V.Y.; Yamaguchi, T.N.; Heisler, L.E.; Livingstone, J.; Huang, V.; Shiah, Y.J.; Yousif, F.; Lin, X.; Masella, A.P.; et al. Genomic hallmarks of localized, non-indolent prostate cancer. Nature 2017, 541, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Li, P.E.; Nelson, P.S. Prostate cancer genomics. Curr. Urol. Rep. 2001, 2, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, W.B. Molecular genetics of prostate cancer. Cancer Surv. 1995, 25, 357–379. [Google Scholar] [PubMed]
- Baca, S.C.; Prandi, D.; Lawrence, M.S.; Mosquera, J.M.; Romanel, A.; Drier, Y.; Park, K.; Kitabayashi, N.; MacDonald, T.Y.; Ghandi, M.; et al. Punctuated evolution of prostate cancer genomes. Cell 2013, 153, 666–677. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, C.E.; Baca, S.C.; Lawrence, M.S.; Demichelis, F.; Blattner, M.; Theurillat, J.P.; White, T.A.; Stojanov, P.; van Allen, E.; Stransky, N.; et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat. Genet. 2012, 44, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, J.; Esteller, M. Cancer epigenomics: Beyond genomics. Curr. Opin. Genet. Dev. 2012, 22, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Varambally, S.; Dhanasekaran, S.M.; Zhou, M.; Barrette, T.R.; Kumar-Sinha, C.; Sanda, M.G.; Ghosh, D.; Pienta, K.J.; Sewalt, R.G.; Otte, A.P.; et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002, 419, 624–629. [Google Scholar] [CrossRef] [PubMed]
- Saramaki, O.R.; Tammela, T.L.; Martikainen, P.M.; Vessella, R.L.; Visakorpi, T. The gene for polycomb group protein enhancer of zeste homolog 2 (EZH2) is amplified in late-stage prostate cancer. Genes Chromosom. Cancer 2006, 45, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Ho, V.T.; Vanneman, M.; Kim, H.; Sasada, T.; Kang, Y.J.; Pasek, M.; Cutler, C.; Koreth, J.; Alyea, E.; Sarantopoulos, S.; et al. Biologic activity of irradiated, autologous, GM-CSF-secreting leukemia cell vaccines early after allogeneic stem cell transplantation. Proc. Natl. Acad. Sci. USA 2009, 106, 15825–15830. [Google Scholar] [CrossRef] [PubMed]
- Borno, S.T.; Fischer, A.; Kerick, M.; Falth, M.; Laible, M.; Brase, J.C.; Kuner, R.; Dahl, A.; Grimm, C.; Sayanjali, B.; et al. Genome-wide DNA methylation events in TMPRSS2-ERG fusion-negative prostate cancers implicate an EZH2-dependent mechanism with miR-26a hypermethylation. Cancer Discov. 2012, 2, 1024–1035. [Google Scholar] [CrossRef] [PubMed]
- Kasinski, A.L.; Slack, F.J. Epigenetics and genetics. MicroRNAs en route to the clinic: Progress in validating and targeting microRNAs for cancer therapy. Nat. Rev. Cancer 2011, 11, 849–864. [Google Scholar] [PubMed]
- Singh, P.K.; Preus, L.; Hu, Q.; Yan, L.; Long, M.D.; Morrison, C.D.; Nesline, M.; Johnson, C.S.; Koochekpour, S.; Kohli, M.; et al. Serum microRNA expression patterns that predict early treatment failure in prostate cancer patients. Oncotarget 2014, 5, 824–840. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.K.; Campbell, M.J. The Interactions of microRNA and Epigenetic Modifications in Prostate Cancer. Cancers 2013, 5, 998–1019. [Google Scholar] [CrossRef] [PubMed]
- Massie, C.E.; Mills, I.G.; Lynch, A.G. The importance of DNA methylation in prostate cancer development. J. Steroid Biochem. Mol. Biol. 2017, 166, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.J.; Park, S.; Uskokovic, M.R.; Dawson, M.I.; Koeffler, H.P. Expression of retinoic acid receptor-beta sensitizes prostate cancer cells to growth inhibition mediated by combinations of retinoids and a 19-nor hexafluoride vitamin D3 analog. Endocrinology 1998, 139, 1972–1980. [Google Scholar] [PubMed]
- Litovkin, K.; van Eynde, A.; Joniau, S.; Lerut, E.; Laenen, A.; Gevaert, T.; Gevaert, O.; Spahn, M.; Kneitz, B.; Gramme, P.; et al. DNA Methylation-Guided Prediction of Clinical Failure in High-Risk Prostate Cancer. PLoS ONE 2015, 10, e0130651. [Google Scholar] [CrossRef] [PubMed]
- Haldrup, C.; Mundbjerg, K.; Vestergaard, E.M.; Lamy, P.; Wild, P.; Schulz, W.A.; Arsov, C.; Visakorpi, T.; Borre, M.; Hoyer, S.; et al. DNA methylation signatures for prediction of biochemical recurrence after radical prostatectomy of clinically localized prostate cancer. J. Clin. Oncol. 2013, 31, 3250–3258. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jadhav, R.R.; Liu, J.; Wilson, D.; Chen, Y.; Thompson, I.M.; Troyer, D.A.; Hernandez, J.; Shi, H.; Leach, R.J.; et al. Roles of Distal and Genic Methylation in the Development of Prostate Tumorigenesis Revealed by Genome-wide DNA Methylation Analysis. Sci. Rep. 2016, 6, 22051. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, C.; Mao, H.; Du, Z.; Chan, W.Y.; Murray, P.; Luo, B.; Chan, A.T.; Mok, T.S.; Chan, F.K.; et al. Epigenetic inactivation of the CpG demethylase TET1 as a DNA methylation feedback loop in human cancers. Sci. Rep. 2016, 6, 26591. [Google Scholar] [CrossRef] [PubMed]
- Kinney, S.R.M.; Smiraglia, D.J.; James, S.R.; Moser, M.T.; Foster, B.A.; Karpf, A.R. Stage-specific alterations of DNA methyltransferase expression, DNA hypermethylation, and DNA hypomethylation during prostate cancer progression in the transgenic adenocarcinoma of mouse prostate model. Mol. Cancer Res. 2008, 6, 1365–1374. [Google Scholar] [CrossRef] [PubMed]
- Camoriano, M.; Kinney, S.R.; Moser, M.T.; Foster, B.A.; Mohler, J.L.; Trump, D.L.; Karpf, A.R.; Smiraglia, D.J. Phenotype-specific CpG island methylation events in a murine model of prostate cancer. Cancer Res. 2008, 68, 4173–4182. [Google Scholar] [CrossRef] [PubMed]
- Morey, S.R.; Smiraglia, D.J.; James, S.R.; Yu, J.; Moser, M.T.; Foster, B.A.; Karpf, A.R. DNA methylation pathway alterations in an autochthonous murine model of prostate cancer. Cancer Res. 2006, 66, 11659–11667. [Google Scholar] [CrossRef] [PubMed]
- Spans, L.; van den Broeck, T.; Smeets, E.; Prekovic, S.; Thienpont, B.; Lambrechts, D.; Karnes, R.J.; Erho, N.; Alshalalfa, M.; Davicioni, E.; et al. Genomic and epigenomic analysis of high-risk prostate cancer reveals changes in hydroxymethylation and TET1. Oncotarget 2016, 7, 24326–24338. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.A.; Zhao, J.C.; Fong, K.W.; Kim, J.; Li, S.; Song, C.; Song, B.; Zheng, B.; He, C.; Yu, J. FOXA1 potentiates lineage-specific enhancer activation through modulating TET1 expression and function. Nucleic Acids Res. 2016, 44, 8153–8164. [Google Scholar] [CrossRef] [PubMed]
- Labbe, D.P.; Zadra, G.; Ebot, E.M.; Mucci, L.A.; Kantoff, P.W.; Loda, M.; Brown, M. Role of diet in prostate cancer: The epigenetic link. Oncogene 2015, 34, 4683–4691. [Google Scholar] [CrossRef] [PubMed]
- Bistulfi, G.; Affronti, H.C.; Foster, B.A.; Karasik, E.; Gillard, B.; Morrison, C.; Mohler, J.; Phillips, J.G.; Smiraglia, D.J. The essential role of methylthioadenosine phosphorylase in prostate cancer. Oncotarget 2016, 7, 14380–14393. [Google Scholar] [PubMed]
- Bistulfi, G.; Vandette, E.; Matsui, S.; Smiraglia, D.J. Mild folate deficiency induces genetic and epigenetic instability and phenotype changes in prostate cancer cells. BMC Biol. 2010, 8, 6. [Google Scholar] [CrossRef] [PubMed]
- Bistulfi, G.; Diegelman, P.; Foster, B.A.; Kramer, D.L.; Porter, C.W.; Smiraglia, D.J. Polyamine biosynthesis impacts cellular folate requirements necessary to maintain S-adenosylmethionine and nucleotide pools. FASEB J. 2009, 23, 2888–2897. [Google Scholar] [CrossRef] [PubMed]
- Shabbeer, S.; Williams, S.A.; Simons, B.W.; Herman, J.G.; Carducci, M.A. Progression of prostate carcinogenesis and dietary methyl donors: Temporal dependence. Cancer Prev. Res. (Phila) 2012, 5, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Kutmon, M.; Riutta, A.; Nunes, N.; Hanspers, K.; Willighagen, E.L.; Bohler, A.; Melius, J.; Waagmeester, A.; Sinha, S.R.; Miller, R.; et al. WikiPathways: Capturing the full diversity of pathway knowledge. Nucleic Acids Res. 2016, 44, D488–D494. [Google Scholar] [CrossRef] [PubMed]
- Pundir, S.; Magrane, M.; Martin, M.J.; O’Donovan, C.; UniProt, C. Searching and Navigating UniProt Databases. Curr. Protoc. Bioinform. 2015, 50, 1.27.1–10. [Google Scholar]
- Long, M.D.; Campbell, M.J. Pan-cancer analyses of the nuclear receptor superfamily. Nucl. Recept. Res. 2015, 2, 101182. [Google Scholar] [CrossRef] [PubMed]
- Doig, C.L.; Singh, P.K.; Dhiman, V.K.; Thorne, J.L.; Battaglia, S.; Sobolewski, M.; Maguire, O.; O’Neill, L.P.; Turner, B.M.; McCabe, C.J.; et al. Recruitment of NCOR1 to VDR target genes is enhanced in prostate cancer cells and associates with altered DNA methylation patterns. Carcinogenesis 2013, 34, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Larkin, S.E.; Holmes, S.; Cree, I.A.; Walker, T.; Basketter, V.; Bickers, B.; Harris, S.; Garbis, S.D.; Townsend, P.A.; Aukim-Hastie, C. Identification of markers of prostate cancer progression using candidate gene expression. Br. J. Cancer 2012, 106, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.; Leha, A.; Salinas-Riester, G. Treatment of prostate cancer cells with S-adenosylmethionine leads to genome-wide alterations in transcription profiles. Gene 2016, 595, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Shukeir, N.; Stefanska, B.; Parashar, S.; Chik, F.; Arakelian, A.; Szyf, M.; Rabbani, S.A. Pharmacological methyl group donors block skeletal metastasis in vitro and in vivo. Br. J. Pharmacol. 2015, 172, 2769–2781. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Huang, Y.L.; Huang, Y.C.; Shui, I.M.; Giovannucci, E.; Chen, Y.C.; Chen, Y.M. Genetic Polymorphisms of the Glycine N-Methyltransferase and Prostate Cancer risk in the Health Professionals Follow-Up Study. PLoS ONE 2014, 9, e94683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Liu, X.; Wang, W.; Sun, H.; Hu, Y.; Lei, H.; Liu, G.; Gao, Y. Effects of antisense RNA targeting of ODC and AdoMetDC on the synthesis of polyamine synthesis and cell growth in prostate cancer cells using a prostatic androgen-dependent promoter in adenovirus. Prostate 2008, 68, 1354–1361. [Google Scholar] [CrossRef] [PubMed]
- Kee, K.; Vujcic, S.; Merali, S.; Diegelman, P.; Kisiel, N.; Powell, C.T.; Kramer, D.L.; Porter, C.W. Metabolic and antiproliferative consequences of activated polyamine catabolism in LNCaP prostate carcinoma cells. J. Biol. Chem. 2004, 279, 27050–27058. [Google Scholar] [CrossRef] [PubMed]
- Mamont, P.S.; Danzin, C.; Wagner, J.; Siat, M.; Joder-Ohlenbusch, A.M.; Claverie, N. Accumulation of Decarboxylated S-adenosyl-l-Methionine in Mammalian Cells as a Consequence of the Inhibition of Putrescine Biosynthesis. Eur. J. Biochem. 1982, 123, 499–504. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Long, M.D.; Smiraglia, D.J.; Campbell, M.J. The Genomic Impact of DNA CpG Methylation on Gene Expression; Relationships in Prostate Cancer. Biomolecules 2017, 7, 15. https://doi.org/10.3390/biom7010015
Long MD, Smiraglia DJ, Campbell MJ. The Genomic Impact of DNA CpG Methylation on Gene Expression; Relationships in Prostate Cancer. Biomolecules. 2017; 7(1):15. https://doi.org/10.3390/biom7010015
Chicago/Turabian StyleLong, Mark D., Dominic J. Smiraglia, and Moray J. Campbell. 2017. "The Genomic Impact of DNA CpG Methylation on Gene Expression; Relationships in Prostate Cancer" Biomolecules 7, no. 1: 15. https://doi.org/10.3390/biom7010015