
Dealing with an Unconventional Genetic Code in Mitochondria: The Biogenesis and Pathogenic Defects of the 5‐Formylcytosine Modification in Mitochondrial tRNAMet

{kind=link}
{kind=link}
Abstract
:1. Introduction
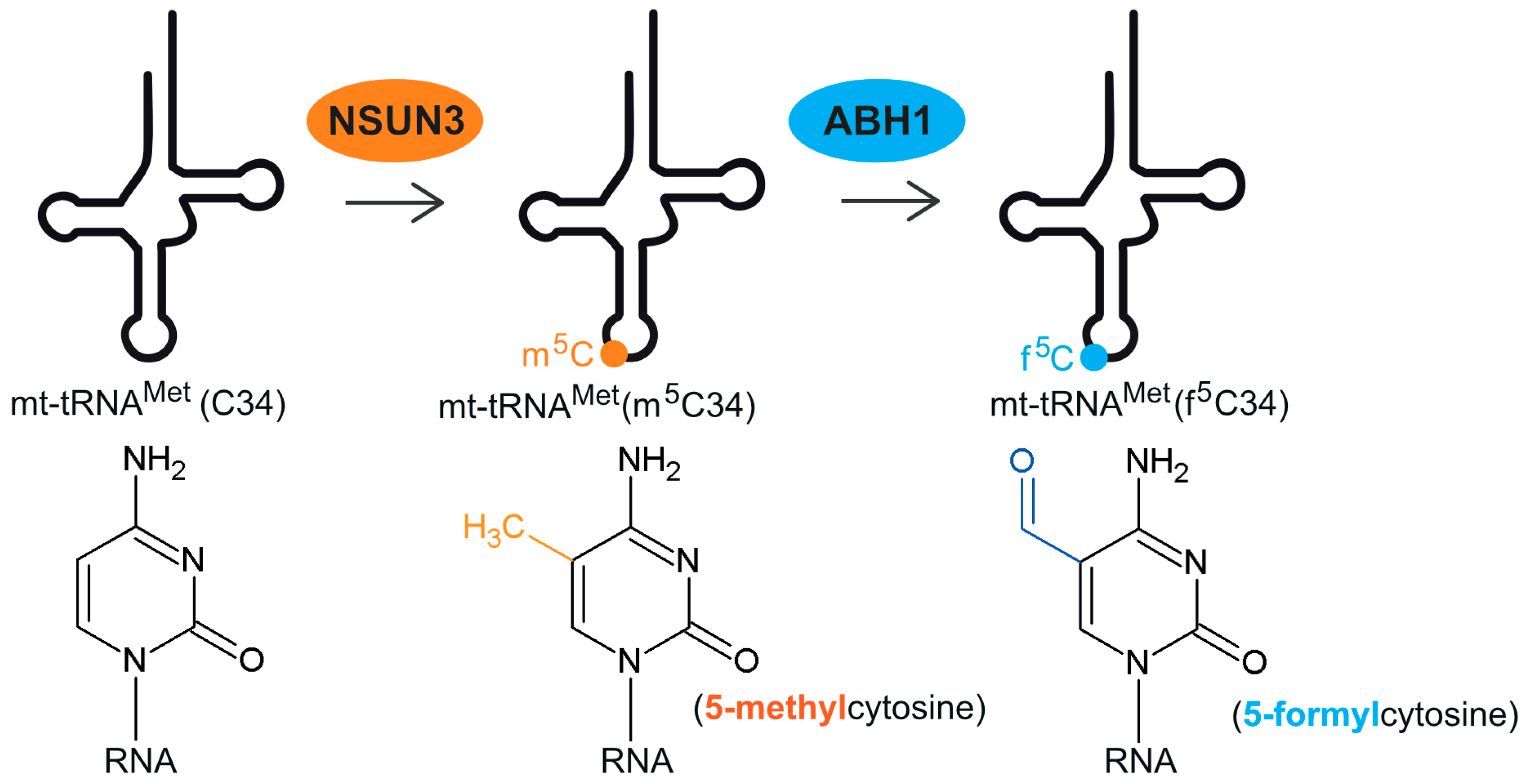
2. Discovery and Formation of 5-Formylcytosine at Position C34 of mt-tRNAMet
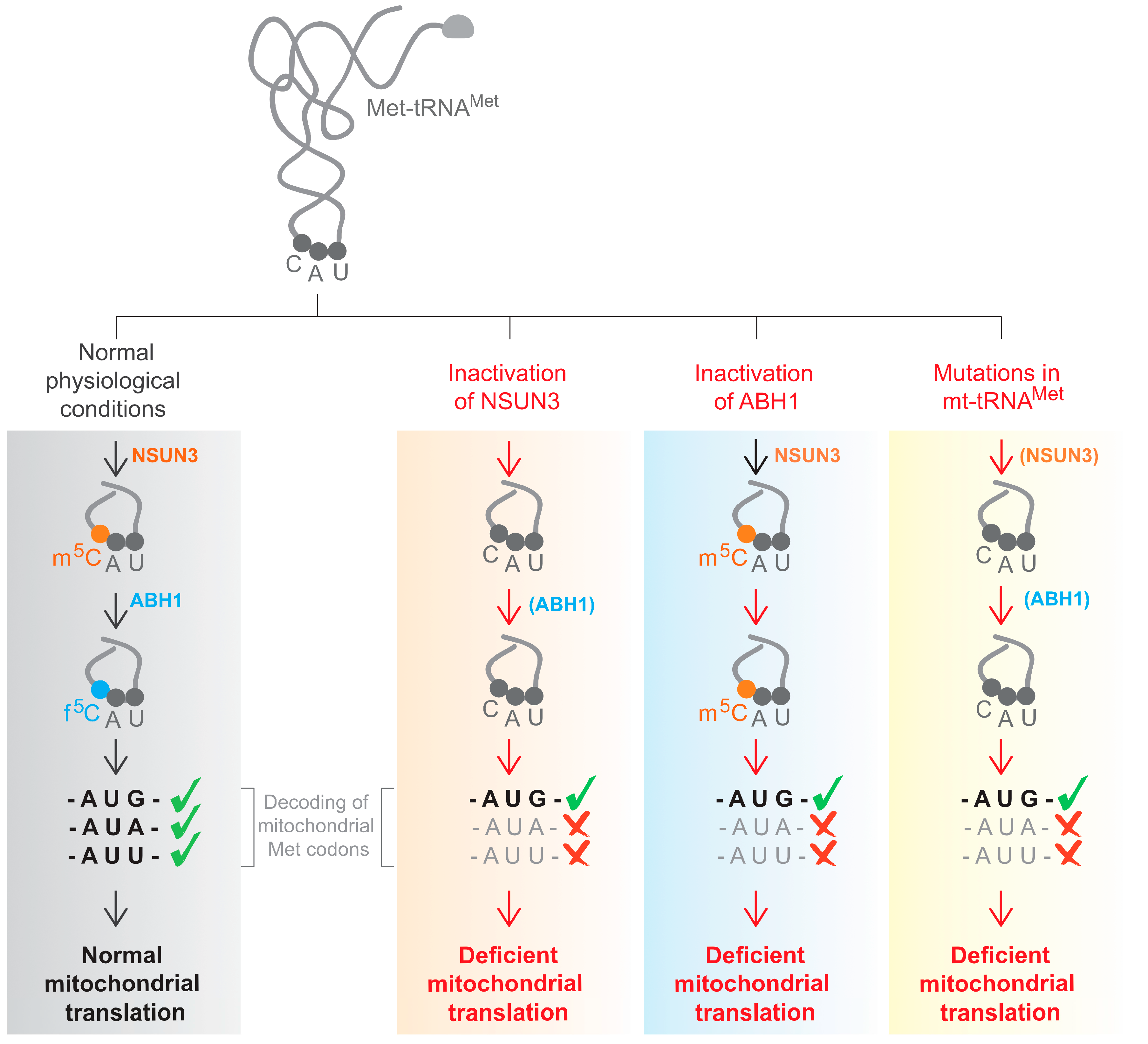
3. The Role f5C34 in mt-tRNAMet
4. The Role of f5C34 in mt-tRNAMet in Human Disease
5. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Van Haute, L.; Pearce, S.F.; Powell, C.A.; D’Souza, A.R.; Nicholls, T.J.; Minczuk, M. Mitochondrial transcript maturation and its disorders. J. Inherit. Metab. Dis. 2015, 38, 655–680. [Google Scholar] [CrossRef] [PubMed]
- Rorbach, J.; Minczuk, M. The post-transcriptional life of mammalian mitochondrial RNA. Biochem. J. 2012, 444, 357–373. [Google Scholar] [CrossRef] [PubMed]
- Hallberg, B.M.; Larsson, N.G. Making proteins in the powerhouse. Cell Metab. 2014, 20, 226–240. [Google Scholar] [CrossRef] [PubMed]
- Boczonadi, V.; Horvath, R. Mitochondria: Impaired mitochondrial translation in human disease. Int. J. Biochem. Cell Biol. 2014, 48, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, T.J.; Rorbach, J.; Minczuk, M. Mitochondria: Mitochondrial RNA metabolism and human disease. Int. J. Biochem. Cell Biol. 2013, 45, 845–849. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, C.M.; Falkenberg, M.; Larsson, N.G. Maintenance and expression of mammalian mitochondrial DNA. Annu. Rev. Biochem. 2016, 85, 133–160. [Google Scholar] [CrossRef] [PubMed]
- Ott, M.; Amunts, A.; Brown, A. Organization and regulation of mitochondrial protein synthesis. Annu. Rev. Biochem. 2016, 85, 77–101. [Google Scholar] [CrossRef] [PubMed]
- Rogalski, M.; Karcher, D.; Bock, R. Superwobbling facilitates translation with reduced tRNA sets. Nat. Struct. Mol. Biol. 2008, 15, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Young, I.G.; Anderson, S. The genetic code in bovine mitochondria: Sequence of genes for the cytochrome oxidase subunit II and two tRNAs. Gene 1980, 12, 257–265. [Google Scholar] [CrossRef]
- Temperley, R.; Richter, R.; Dennerlein, S.; Lightowlers, R.N.; Chrzanowska-Lightowlers, Z.M. Hungry codons promote frameshifting in human mitochondrial ribosomes. Science 2010, 327, 301. [Google Scholar] [CrossRef] [PubMed]
- Akabane, S.; Ueda, T.; Nierhaus, K.H.; Takeuchi, N. Ribosome Rescue and Translation Termination at Non-standard Stop Codons by ICT1 in Mammalian Mitochondria. PLoS Genet. 2014, 10, e1004616. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, N.; Nierhaus, K.H. Response to the Formal Letter of Z. Chrzanowska-Lightowlers and R. N. Lightowlers Regarding Our Article “Ribosome Rescue and Translation Termination at Non-Standard Stop Codons by ICT1 in Mammalian Mitochondria”. PLoS Genet. 2015, 11, e1005218. [Google Scholar] [CrossRef] [PubMed]
- Chrzanowska-Lightowlers, Z.M.; Lightowlers, R.N. Response to “Ribosome Rescue and Translation Termination at Non-sSandard Stop Codons byICT1 in Mammalian Mitochondria”. PLoS Genet. 2015, 11, e1005227. [Google Scholar] [CrossRef] [PubMed]
- Takemoto, C.; Koike, T.; Yokogawa, T.; Benkowski, L.; Spremulli, L.L.; Ueda, T.A.; Nishikawa, K.; Watanabe, K. The ability of bovine mitochondrial transfer RNAMet to decode AUG and AUA codons. Biochimie 1995, 77, 104–108. [Google Scholar] [CrossRef]
- Suzuki, T.; Nagao, A.; Suzuki, T. Human mitochondrial tRNAs: Biogenesis, function, structural aspects, and diseases. Annu. Rev. Genet. 2011, 45, 299–329. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Suzuki, T. A complete landscape of post-transcriptional modifications in mammalian mitochondrial tRNAs. Nucleic Acids Res. 2014, 42, 7346–7357. [Google Scholar] [CrossRef] [PubMed]
- Powell, C.A.; Nicholls, T.J.; Minczuk, M. Nuclear-encoded factors involved in post-transcriptional processing and modification of mitochondrial tRNAs in human disease. Front. Genet. 2015, 6, 79. [Google Scholar] [CrossRef] [PubMed]
- Yarham, J.W.; Lamichhane, T.N.; Pyle, A.; Mattijssen, S.; Baruffini, E.; Bruni, F.; Donnini, C.; Vassilev, A.; He, L.; Blakely, E.L.; et al. Defective i6A37 Modification of Mitochondrial and Cytosolic tRNAs Results from Pathogenic Mutations in TRIT1 and Its Substrate tRNA. PLoS Genet. 2014, 10, e1004424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghezzi, D.; Baruffini, E.; Haack, T.B.; Invernizzi, F.; Melchionda, L.; Dallabona, C.; Strom, T.M.; Parini, R.; Burlina, A.B.; Meitinger, T.; et al. Mutations of the Mitochondrial-tRNA Modifier MTO1 Cause Hypertrophic Cardiomyopathy and Lactic Acidosis. Am. J. Hum. Genet. 2012, 90, 1079–1087. [Google Scholar] [CrossRef] [PubMed]
- Powell, C.A.; Kopajtich, R.; D’Souza, A.R.; Rorbach, J.; Kremer, L.S.; Husain, R.A.; Dallabona, C.; Donnini, C.; Alston, C.L.; Griffin, H.; et al. TRMT5 Mutations Cause a Defect in Post-transcriptional Modification of Mitochondrial tRNA Associated with Multiple Respiratory-Chain Deficiencies. Am. J. Hum. Genet. 2015, 97, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Kopajtich, R.; Nicholls, T.J.; Rorbach, J.; Metodiev, M.D.; Freisinger, P.; Mandel, H.; Vanlander, A.; Ghezzi, D.; Carrozzo, R.; Taylor, R.W.; et al. Mutations in GTPBP3 cause a mitochondrial translation defect associated with hypertrophic cardiomyopathy, lactic acidosis, and encephalopathy. Am. J. Hum. Genet. 2014, 95, 708–720. [Google Scholar] [CrossRef] [PubMed]
- Umeda, N.; Suzuki, T.; Yukawa, M.; Ohya, Y.; Shindo, H.; Watanabe, K. Mitochondria-specific RNA-modifying enzymes responsible for the biosynthesis of the wobble base in mitochondrial tRNAs. Implications for the molecular pathogenesis of human mitochondrial diseases. J. Biol. Chem. 2005, 280, 1613–1624. [Google Scholar] [CrossRef] [PubMed]
- Bykhovskaya, Y.; Casas, K.; Mengesha, E.; Inbal, A.; Fischel-Ghodsian, N. Missense mutation in pseudouridine synthase 1 (PUS1) causes mitochondrial myopathy and sideroblastic anemia (MLASA). Am. J. Hum. Genet. 2004, 74, 1303–1308. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Vizarra, E.; Berardinelli, A.; Valente, L.; Tiranti, V.; Zeviani, M. Nonsense mutation in pseudouridylate synthase 1 (PUS1) in two brothers affected by myopathy, lactic acidosis and sideroblastic anaemia (MLASA). J. Med. Genet. 2007, 44, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Baruffini, E.; Dallabona, C.; Invernizzi, F.; Yarham, J.W.; Melchionda, L.; Blakely, E.L.; Lamantea, E.; Donnini, C.; Santra, S.; Vijayaraghavan, S.; et al. MTO1 mutations are associated with hypertrophic cardiomyopathy and lactic acidosis and cause respiratory chain deficiency in humans and yeast. Hum. Mutat. 2013, 34, 1501–1509. [Google Scholar] [CrossRef] [PubMed]
- Najmabadi, H.; Hu, H.; Garshasbi, M.; Zemojtel, T.; Abedini, S.S.; Chen, W.; Hosseini, M.; Behjati, F.; Haas, S.; Jamali, P.; et al. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature 2011, 478, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Zeharia, A.; Shaag, A.; Pappo, O.; Mager-Heckel, A.M.; Saada, A.; Beinat, M.; Karicheva, O.; Mandel, H.; Ofek, N.; Segel, R.; et al. Acute Infantile Liver Failure Due to Mutations in the TRMU Gene. Am. J. Hum. Genet. 2009, 85, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Moriya, J.; Yokogawa, T.; Wakita, K.; Ueda, T.; Nishikawa, K.; Crain, P.F.; Hashizume, T.; Pomerantz, S.C.; McCloskey, J.A.; Kawai, G.; et al. A novel modified nucleoside found at the first position of the anticodon of methionine tRNA from bovine liver mitochondria. Biochemistry 1994, 33, 2234–2239. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Tsurui, H.; Ueda, T.; Furushima, R.; Takamiya, S.; Kita, K.; Nishikawa, K.; Watanabe, K. Primary and higher order structures of nematode (Ascaris suum) mitochondrial tRNAs lacking either the T or D stem. J. Biol. Chem. 1994, 269, 22902–22906. [Google Scholar] [PubMed]
- Tomita, K.; Ueda, T.; Watanabe, K. 5-formylcytidine (f5C) found at the wobble position of the anticodon of squid mitochondrial tRNA(Met)CAU. Nucleic Acids Symp. Ser. 1997, 197–198. [Google Scholar]
- Tomita, K.; Ueda, T.; Ishiwa, S.; Crain, P.F.; McCloskey, J.A.; Watanabe, K. Codon reading patterns in Drosophila melanogaster mitochondria based on their tRNA sequences: A unique wobble rule in animal mitochondria. Nucleic Acids Res. 1999, 27, 4291–4297. [Google Scholar] [CrossRef] [PubMed]
- Takemoto, C.; Ueda, T.; Miura, K.; Watanabe, K. Nucleotide sequences of animal mitochondrial tRNAs(Met) possibly recognizing both AUG and AUA codons. Nucleic Acids Symp. Ser. 1999, 42, 77–78. [Google Scholar] [CrossRef]
- Nakano, S.; Suzuki, T.; Kawarada, L.; Iwata, H.; Asano, K.; Suzuki, T. NSUN3 methylase initiates 5-formylcytidine biogenesis in human mitochondrial tRNA(Met). Nat. Chem. Biol. 2016, 12, 546–551. [Google Scholar] [CrossRef] [PubMed]
- Van Haute, L.; Dietmann, S.; Kremer, L.; Hussain, S.; Pearce, S.F.; Powell, C.A.; Rorbach, J.; Lantaff, R.; Blanco, S.; Sauer, S.; et al. Deficient methylation and formylation of mt-tRNAMet wobble cytosine in a patient carrying mutations in NSUN3. Nat. Commun. 2016, 7, 12039. [Google Scholar] [CrossRef] [PubMed]
- Haag, S.; Sloan, K.E.; Ranjan, N.; Warda, A.S.; Kretschmer, J.; Blessing, C.; Hubner, B.; Seikowski, J.; Dennerlein, S.; Rehling, P.; et al. NSUN3 and ABH1 modify the wobble position of mt-tRNAMet to expand codon recognition in mitochondrial translation. EMBO J. 2016, 35, 2104–2119. [Google Scholar] [CrossRef] [PubMed]
- Bachman, M.; Uribe-Lewis, S.; Yang, X.; Burgess, H.E.; Iurlaro, M.; Reik, W.; Murrell, A.; Balasubramanian, S. 5-formylcytosine can be a stable DNA modification in mammals. Nat. Chem. Biol. 2015, 11, 555–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brzezicha, B.; Schmidt, M.; Makalowska, I.; Jarmolowski, A.; Pienkowska, J.; Szweykowska-Kulinska, Z. Identification of human tRNA:m5C methyltransferase catalysing intron-dependent m5C formation in the first position of the anticodon of the pre-tRNA Leu (CAA). Nucleic Acids Res. 2006, 34, 6034–6043. [Google Scholar] [CrossRef] [PubMed]
- Haag, S.; Warda, A.S.; Kretschmer, J.; Gunnigmann, M.A.; Hobartner, C.; Bohnsack, M.T. NSUN6 is a human RNA methyltransferase that catalyzes formation of m5C72 in specific tRNAs. RNA 2015, 21, 1532–1543. [Google Scholar] [CrossRef] [PubMed]
- Sloan, K.E.; Leisegang, M.S.; Doebele, C.; Ramirez, A.S.; Simm, S.; Safferthal, C.; Kretschmer, J.; Schorge, T.; Markoutsa, S.; Haag, S.; et al. The association of late-acting snoRNPs with human pre-ribosomal complexes requires the RNA helicaseDDX21. Nucleic Acids Res. 2015, 43, 553–564. [Google Scholar] [CrossRef] [PubMed]
- Schosserer, M.; Minois, N.; Angerer, T.B.; Amring, M.; Dellago, H.; Harreither, E.; Calle-Perez, A.; Pircher, A.; Gerstl, M.P.; Pfeifenberger, S.; et al. Methylation of ribosomal RNA by NSUN5 is a conserved mechanism modulating organismal lifespan. Nat. Commun. 2015, 6, 6158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camara, Y.; Asin-Cayuela, J.; Park, C.B.; Metodiev, M.D.; Shi, Y.; Ruzzenente, B.; Kukat, C.; Habermann, B.; Wibom, R.; Hultenby, K.; et al. MTERF4 regulates translation by targeting the methyltransferase NSUN4 to the mammalian mitochondrial ribosome. Cell Metab. 2011, 13, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Metodiev, M.D.; Spahr, H.; Loguercio Polosa, P.; Meharg, C.; Becker, C.; Altmueller, J.; Habermann, B.; Larsson, N.G.; Ruzzenente, B. NSUN4 is a Dual Function Mitochondrial Protein Required for Both Methylation of 12S rRNA and Coordination of Mitoribosomal Assembly. PLoS Genet. 2014, 10, e1004110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhee, H.W.; Zou, P.; Udeshi, N.D.; Martell, J.D.; Mootha, V.K.; Carr, S.A.; Ting, A.Y. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science 2013, 339, 1328–1331. [Google Scholar] [CrossRef] [PubMed]
- Fedeles, B.I.; Singh, V.; Delaney, J.C.; Li, D.; Essigmann, J.M. The AlkB family of Fe(II)/α-Ketoglutarate-dependent Dioxygenases: Repairing nucleic acid alkylation damage and beyond. J. Biol. Chem. 2015, 290, 20734–20742. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Clark, W.; Luo, G.; Wang, X.; Fu, Y.; Wei, J.; Wang, X.; Hao, Z.; Dai, Q.; Zheng, G.; et al. ALKBH1-Mediated tRNA Demethylation Regulates Translation. Cell 2016, 167, 816–828. [Google Scholar] [CrossRef] [PubMed]
- Nordstrand, L.M.; Svard, J.; Larsen, E.; Nilsen, A.; Ougland, R.; Furu, K.; Lien, G.F.; Rognes, T.; Namekawa, S.H.; Lee, J.T.; et al. Mice lacking ALKBH1 display sex-ratio distortion and unilateral eye defects. PLoS ONE 2010, 5, e13827. [Google Scholar] [CrossRef] [PubMed]
- Ougland, R.; Lando, D.; Jonson, I.; Dahl, J.A.; Moen, M.N.; Nordstrand, L.M.; Rognes, T.; Lee, J.T.; Klungland, A.; Kouzarides, T.; et al. ALKBH1 is a histone H2A dioxygenase involved in neural differentiation. Stem Cells 2012, 30, 2672–2682. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.P.; Wang, T.; Seetin, M.G.; Lai, Y.; Zhu, S.; Lin, K.; Liu, Y.; Byrum, S.D.; Mackintosh, S.G.; Zhong, M.; et al. DNA methylation on N(6)-adenine in mammalian embryonic stem cells. Nature 2016, 532, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Lusic, H.; Gustilo, E.M.; Vendeix, F.A.; Kaiser, R.; Delaney, M.O.; Graham, W.D.; Moye, V.A.; Cantara, W.A.; Agris, P.F.; Deiters, A. Synthesis and investigation of the 5-formylcytidine modified, anticodon stem and loop of the human mitochondrial tRNAMet. Nucleic Acids Res. 2008, 36, 6548–6557. [Google Scholar] [CrossRef] [PubMed]
- Bilbille, Y.; Gustilo, E.M.; Harris, K.A.; Jones, C.N.; Lusic, H.; Kaiser, R.J.; Delaney, M.O.; Spremulli, L.L.; Deiters, A.; Agris, P.F. The human mitochondrial tRNAMet: Structure/Function Relationship of a Unique Modification in the Decoding of Unconventional Codons. J. Mol. Biol. 2011, 406, 257–274. [Google Scholar] [CrossRef] [PubMed]
- Cantara, W.A.; Murphy, F.V.T.; Demirci, H.; Agris, P.F. Expanded use of sense codons is regulated by modified cytidines in tRNA. Proc. Natl. Acad. Sci. USA 2013, 110, 10964–10969. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Luo, Z.; He, K.; Delaney, M.O.; Chen, D.; Sheng, J. Base pairing and structural insights into the 5-formylcytosine in RNA duplex. Nucleic Acids Res. 2016, 44, 4968–4977. [Google Scholar] [CrossRef] [PubMed]
- Spencer, A.C.; Heck, A.; Takeuchi, N.; Watanabe, K.; Spremulli, L.L. Characterization of the human mitochondrial methionyl-tRNA synthetase. Biochemistry 2004, 43, 9743–9754. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Yarham, J.W.; Elson, J.L.; Blakely, E.L.; McFarland, R.; Taylor, R.W. Mitochondrial tRNA mutations and disease. Wiley Interdiscip. Rev. RNA 2010, 1, 304–324. [Google Scholar] [CrossRef] [PubMed]
- Brule, H.; Holmes, W.M.; Keith, G.; Giege, R.; Florentz, C. Effect of a mutation in the anticodon of human mitochondrial tRNAPro on its post-transcriptional modification pattern. Nucleic Acids Res. 1998, 26, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Yasukawa, T.; Kirino, Y.; Ishii, N.; Holt, I.J.; Jacobs, H.T.; Makifuchi, T.; Fukuhara, N.; Ohta, S.; Suzuki, T.; Watanabe, K. Wobble modification deficiency in mutant tRNAs in patients with mitochondrial diseases. FEBS Lett. 2005, 579, 2948–2952. [Google Scholar] [CrossRef] [PubMed]
- Kirino, Y.; Suzuki, T. Human mitochondrial diseases associated with tRNA wobble modification deficiency. RNA Biol. 2005, 2, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Levinger, L.; Morl, M.; Florentz, C. Mitochondrial tRNA 3’ end metabolism and human disease. Nucleic Acids Res. 2004, 32, 5430–5441. [Google Scholar] [CrossRef] [PubMed]
- Lott, M.T.; Leipzig, J.N.; Derbeneva, O.; Xie, H.M.; Chalkia, D.; Sarmady, M.; Procaccio, V.; Wallace, D.C. mtDNA Variation and Analysis Using MITOMAP and MITOMASTER. Curr. Protoc. Bioinform. 2013, 44, 1–23. [Google Scholar] [PubMed]
- Lu, Z.; Chen, H.; Meng, Y.; Wang, Y.; Xue, L.; Zhi, S.; Qiu, Q.; Yang, L.; Mo, J.Q.; Guan, M.X. The tRNAMet 4435A>G mutation in the mitochondrial haplogroup G2a1 is responsible for maternally inherited hypertension in a Chinese pedigree. Eur. J. Hum. Genet. 2011, 19, 1181–1186. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Li, R.; Zhou, X.; Tong, Y.; Lu, F.; Qian, Y.; Hu, Y.; Mo, J.Q.; West, C.E.; Guan, M.X. The Novel A4435G Mutation in the Mitochondrial tRNAMet May Modulate the Phenotypic Expression of the LHON-Associated ND4 G11778A Mutation. Investig. Ophthalmol. Vis. Sci. 2006, 47, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Vissing, J.; Salamon, M.B.; Arlien-Soborg, P.; Norby, S.; Manta, P.; DiMauro, S.; Schmalbruch, H. A new mitochondrial tRNAMet gene mutation in a patient with dystrophic muscle and exercise intolerance. Neurology 1998, 50, 1875–1878. [Google Scholar] [CrossRef] [PubMed]
- Sternberg, D.; Danan, C.; Lombes, A.; Laforet, P.; Girodon, E.; Goossens, M.; Amselem, S. Exhaustive scanning approach to screen all the mitochondrial tRNA genes for mutations and its application to the investigation of 35 independent patients with mitochondrial disorders. Hum. Mol. Genet. 1998, 7, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Wang, J.; Zhang, V.W.; Li, F.Y.; Landsverk, M.; Cui, H.; Truong, C.K.; Wang, G.; Chen, L.C.; Graham, B.; et al. Transition to next generation analysis of the whole mitochondrial genome: A summary of molecular defects. Hum. Mutat. 2013, 34, 882–893. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van Haute, L.; Powell, C.; Minczuk, M. Dealing with an Unconventional Genetic Code in Mitochondria: The Biogenesis and Pathogenic Defects of the 5‐Formylcytosine Modification in Mitochondrial tRNAMet. Biomolecules 2017, 7, 24. https://doi.org/10.3390/biom7010024
Van Haute L, Powell C, Minczuk M. Dealing with an Unconventional Genetic Code in Mitochondria: The Biogenesis and Pathogenic Defects of the 5‐Formylcytosine Modification in Mitochondrial tRNAMet. Biomolecules. 2017; 7(1):24. https://doi.org/10.3390/biom7010024
Chicago/Turabian StyleVan Haute, Lindsey, Christopher A. Powell, and Michal Minczuk. 2017. "Dealing with an Unconventional Genetic Code in Mitochondria: The Biogenesis and Pathogenic Defects of the 5‐Formylcytosine Modification in Mitochondrial tRNAMet" Biomolecules 7, no. 1: 24. https://doi.org/10.3390/biom7010024