
QueF-Like, a Non-Homologous Archaeosine Synthase from the Crenarchaeota
by
 and
and
Adriana Bon Ramos
1, 
Lide Bao
1,
Ben Turner
1,
Valérie De Crécy-Lagard
2 and
Dirk Iwata-Reuyl
1,* 1
Department of Chemistry, Portland State University, Portland, OR 97207, USA
2
The Department of Microbiology and Cell Science Department, University of Florida, Gainesville, FL 32611, USA
*
Author to whom correspondence should be addressed.
Biomolecules 2017, 7(2), 36; https://doi.org/10.3390/biom7020036
Submission received: 28 February 2017
/
Revised: 23 March 2017
/
Accepted: 24 March 2017
/
Published: 6 April 2017
(This article belongs to the Collection RNA Modifications)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Archaeosine (G+) is a structurally complex modified nucleoside ubiquitous to the Archaea, where it is found in the D-loop of virtually all archaeal transfer RNA (tRNA). Its unique structure, which includes a formamidine group that carries a formal positive charge, and location in the tRNA, led to the proposal that it serves a key role in stabilizing tRNA structure. Although G+ is limited to the Archaea, it is structurally related to the bacterial modified nucleoside queuosine, and the two share homologous enzymes for the early steps of their biosynthesis. In the Euryarchaeota, the last step of the archaeosine biosynthetic pathway involves the amidation of a nitrile group on an archaeosine precursor to give formamidine, a reaction catalyzed by the enzyme Archaeosine Synthase (ArcS). Most Crenarchaeota lack ArcS, but possess two proteins that inversely distribute with ArcS and each other, and are implicated in G+ biosynthesis. Here, we describe biochemical studies of one of these, the protein QueF-like (QueF-L) from Pyrobaculum calidifontis, that demonstrate the catalytic activity of QueF-L, establish where in the pathway QueF-L acts, and identify the source of ammonia in the reaction.
1. Introduction
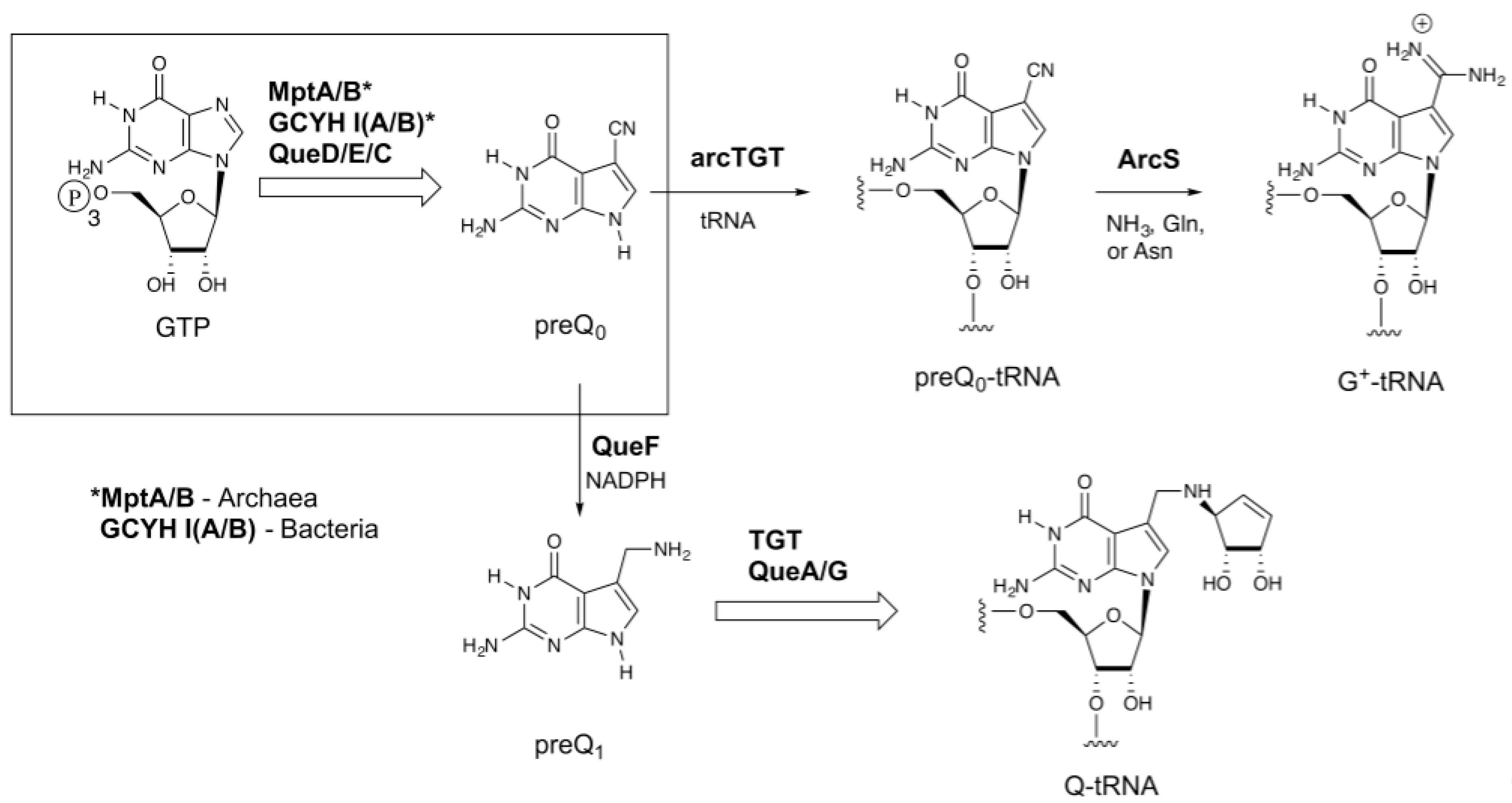
Transfer RNA (tRNA) is unique among nucleic acids in undergoing extensive nucleoside modification during maturation of the transcript. Over 100 modified nucleosides have been characterized [1], and these typically comprise roughly 10% of the nucleosides in a given tRNA, but can account for as much as 25% [2]. The 7-deazaguanosine nucleosides archaeosine (G+) and queuosine (Q) are two of the most structurally complex modified nucleosides found in tRNA [3]. Both share a 7-deazaguanosine core (Figure 1), but differ in the extent of further elaboration; archaeosine possesses an amidine functional group at the 7-position [4] of this core structure, while queuosine possesses a cyclopentenediol ring appended to an aminomethyl group at the 7-position [5,6], which in some mammalian tRNAs can be glycosylated with galactose or mannose at one of the hydroxyls of the cyclopentenediol ring [7,8], or in some bacteria aminoacylated with glutamate [9,10,11,12]. Queuosine is ubiquitous throughout eukaryotic and bacterial phyla and occurs exclusively at position 34 (the wobble position) in the anticodons of tRNAs coding for the amino acids asparagine, aspartic acid, histidine, and tyrosine [13]. Its location in the anticodon suggests a role in modulating translational fidelity and/or efficiency, and physiological studies are consistent with such a role [14,15,16,17]. In marked contrast, archaeosine is present only in archaeal tRNA, and is located at position 15 (and position 13 in Thermoplasma acidophilum tRNALeu [18]) in the dihydrouridine loop (D-loop) [19], where it is proposed to play a key role in tertiary structure stabilization [20].
Archaeosine is found quasi-universally along the archaeal tree, and while all Archaea that possess G+ possess genes encoding homologs of the biosynthetic enzymes up through arcTGT, the enzyme that catalyzes the exchange of the genetically encoded guanine for the precursor 7-cyano-7-deazaguanine (preQ0) in tRNA (Figure 1), Archaeosine Synthase (ArcS), which is the amidinotransferase responsible for converting preQ0-tRNA to G+-tRNA [25], is not universally distributed. arcS is ubiquitous in Euryarchaeota, but the majority of sequenced Crenarchaeota lack arcS homologs. Previously we identified two non-homologous enzymes that, when expressed in Escherichia coli, resulted in the production of G+-modified tRNA [26]. One of these, GAT-QueC, is a fusion of a glutamine amidotransferase domain (GAT) with QueC, the enzyme responsible for forming the precursor preQ0. Interestingly, the other enzyme, QueF-like (QueF-L), is a homolog of the bacterial enzyme QueF, which catalyzes the NADPH-dependent reduction of preQ0 to 7-aminomethyl-7-deazaguanine (preQ1) [27] in the queuosine pathway (Figure 1). QueF is a member of the tunneling-fold (T-fold) superfamily [27,28], a small structural superfamily that was already known to support an astonishing diversity of chemical reactions [29].
Recently, we reported the X-ray crystal structure of QueF-L [30], confirming that it is a member of the T-fold superfamily with significant structural homology to QueF. Here we describe biochemical studies of the recombinant QueF-L enzyme from Pyrobaculum calidifontis that clearly demonstrate the catalytic activity of QueF-L, establish where in the pathway QueF-L acts, and identify the source of ammonia in the reaction.
2. Results
2.1. Over-Expression of queF-L and Purification of Recombinant Pyrobaculum calidifontis QueF-L
The queF-L gene from P. calidifontis was synthesized (GenScript) with codon optimization for E. coli expression. We subcloned queF-L from this construct into a pET30-based vector for over-production of QueF-L as a His6-affinity tagged recombinant protein. The protein was expressed well, and was purified to >95% homogeneity by an initial heat treatment (80 °C) to precipitate heat labile proteins followed by affinity chromatography on Ni2+-nitrilotriacetic acid (NTA) resin.
2.2. Identification of the Substrates for P. calidifontis QueF-L
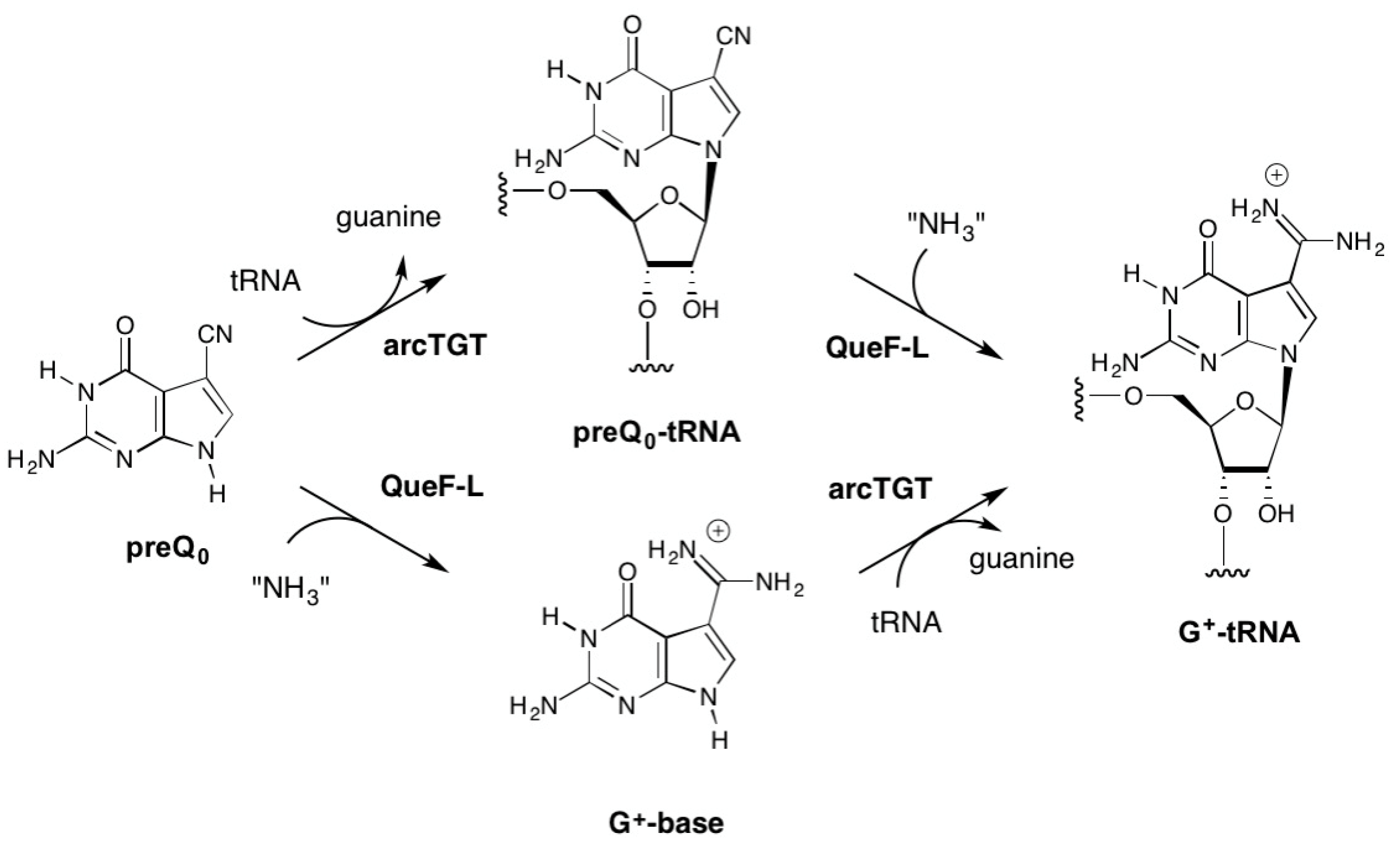
While our in vivo data clearly demonstrated that QueF-L functions as an amidinotransferase in the biosynthesis of G+-modified tRNA, even when expressed in E. coli [26], it was not known if the conversion of the nitrile to the formamidino group occurred before or after preQ0 is inserted into tRNA (Figure 2), or what the source of NH3 was.
Given that ArcS functions on preQ0-modifed tRNA in the final step of G+ biosynthesis (Figure 1), it was reasonable to propose that QueF-L functioned analogously (Figure 2, top). On the other hand, QueF acts on free preQ0 in bacterial Q biosynthesis, and given the high sequence and structural homologies with QueF-L [30] it was not unreasonable to consider that QueF-L might utilize preQ0 directly (Figure 2, bottom). The fact that G+ was formed in bacterial tRNA when QueF-L was expressed in a ΔqueF strain [26] is consistent with both proposals. Indeed, the bacterial TGT can utilize preQ0 as a substrate [31], and preQ0 nucleoside is detected in ΔqueF mutants [26,27]. While biochemical analysis of the canonical arcTGT has demonstrated that it is not able to utilize G+-base [32], our 3D homology models of the catalytic domains of arcTGT from Crenarchaeota that lack ArcS [26] revealed differences from the canonical arcTGT in the active sites [33,34] that might allow accommodation of the formamidino group of G+ base were it available. Thus, both free preQ0 and preQ0-tRNA were considered viable candidates as the natural substrate for QueF-L (Figure 2).
2.2.1. Thioimide Formation with preQ0 and preQ0-tRNA
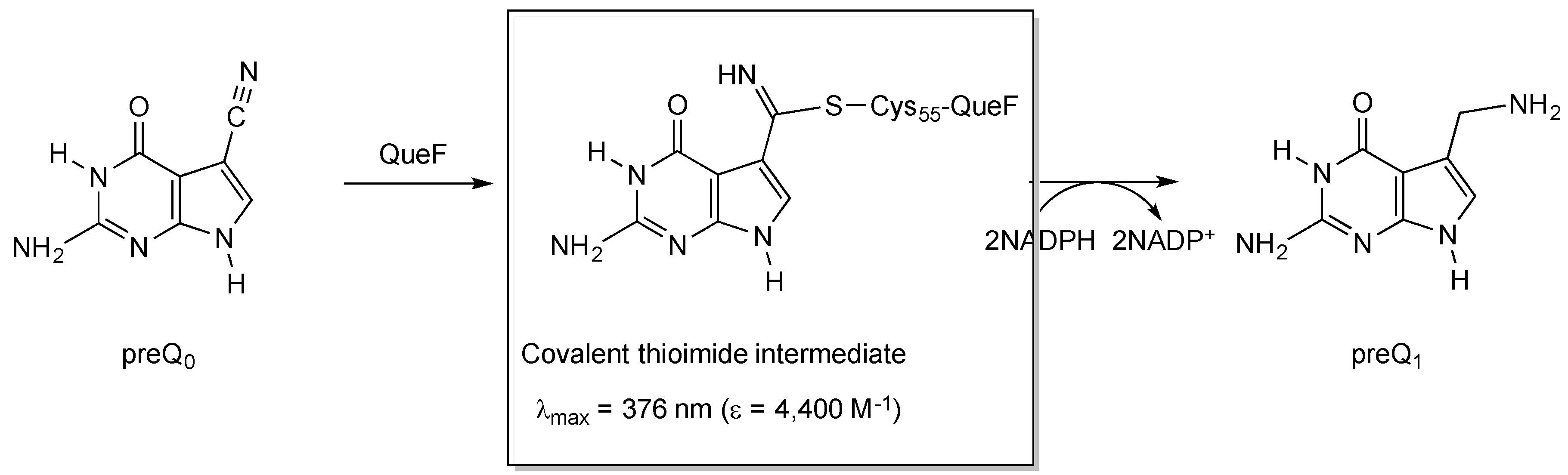
The conservation of Cys21 in QueF-L (P. calidifontis QueF-L numbering) and QueF (Cys55 in Bacillus subtilis QueF numbering) [26], which in QueF participates in the catalytic mechanism via nucleophilic attack of the thiol group on the nitrile of preQ0 to form a covalent thioimide intermediate (Figure 3) [28,35], suggested that QueF-L might utilize a similar intermediate in the mechanism to form the formamidine of G+. Given that the preQ0-QueF thioimide has a distinct absorption at 376 nm, we reasoned that probing for this absorption spectroscopically might allow us to determine whether preQ0 or preQ0-tRNA were capable of forming such an intermediate, and thus was the actual substrate.
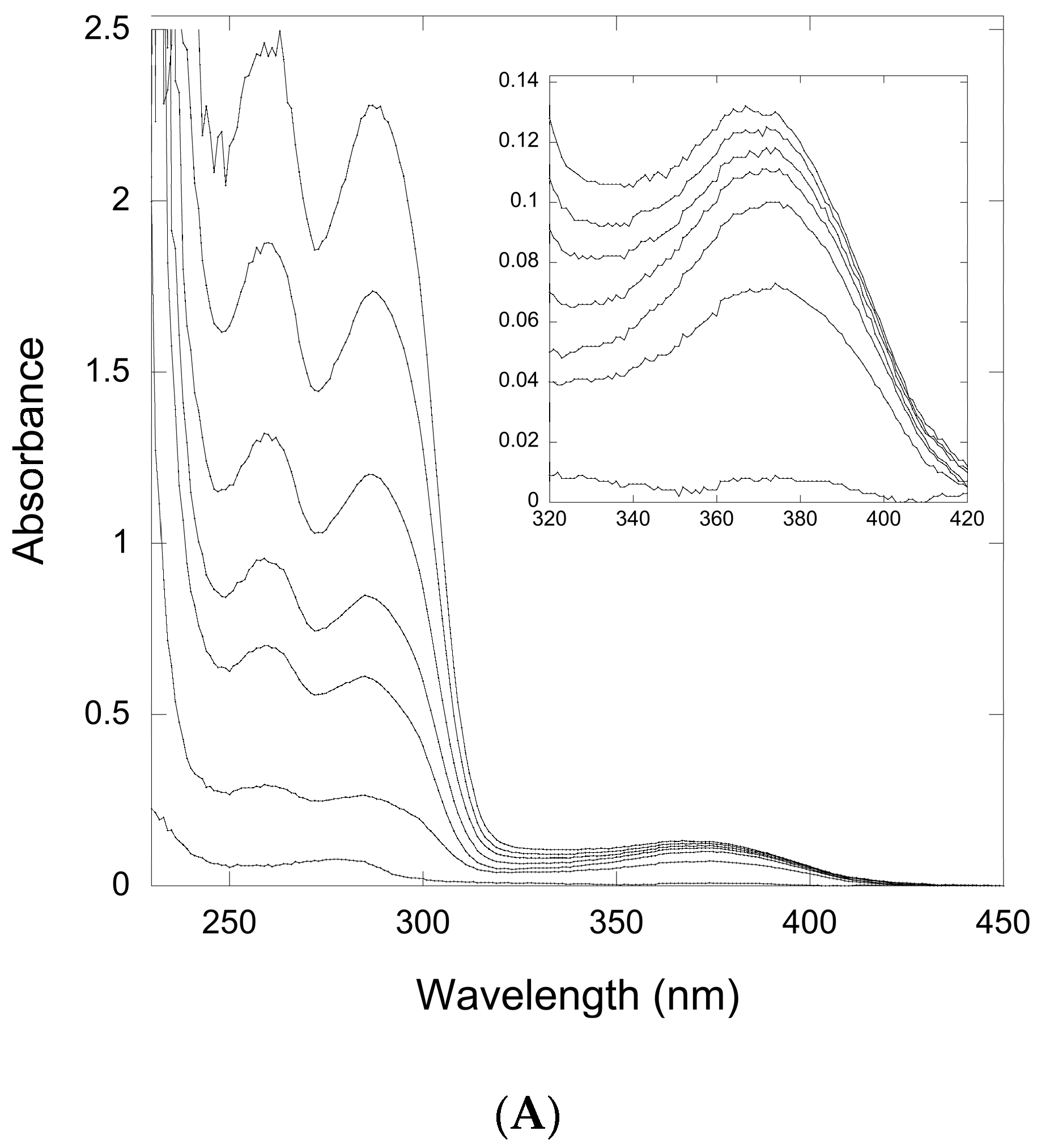
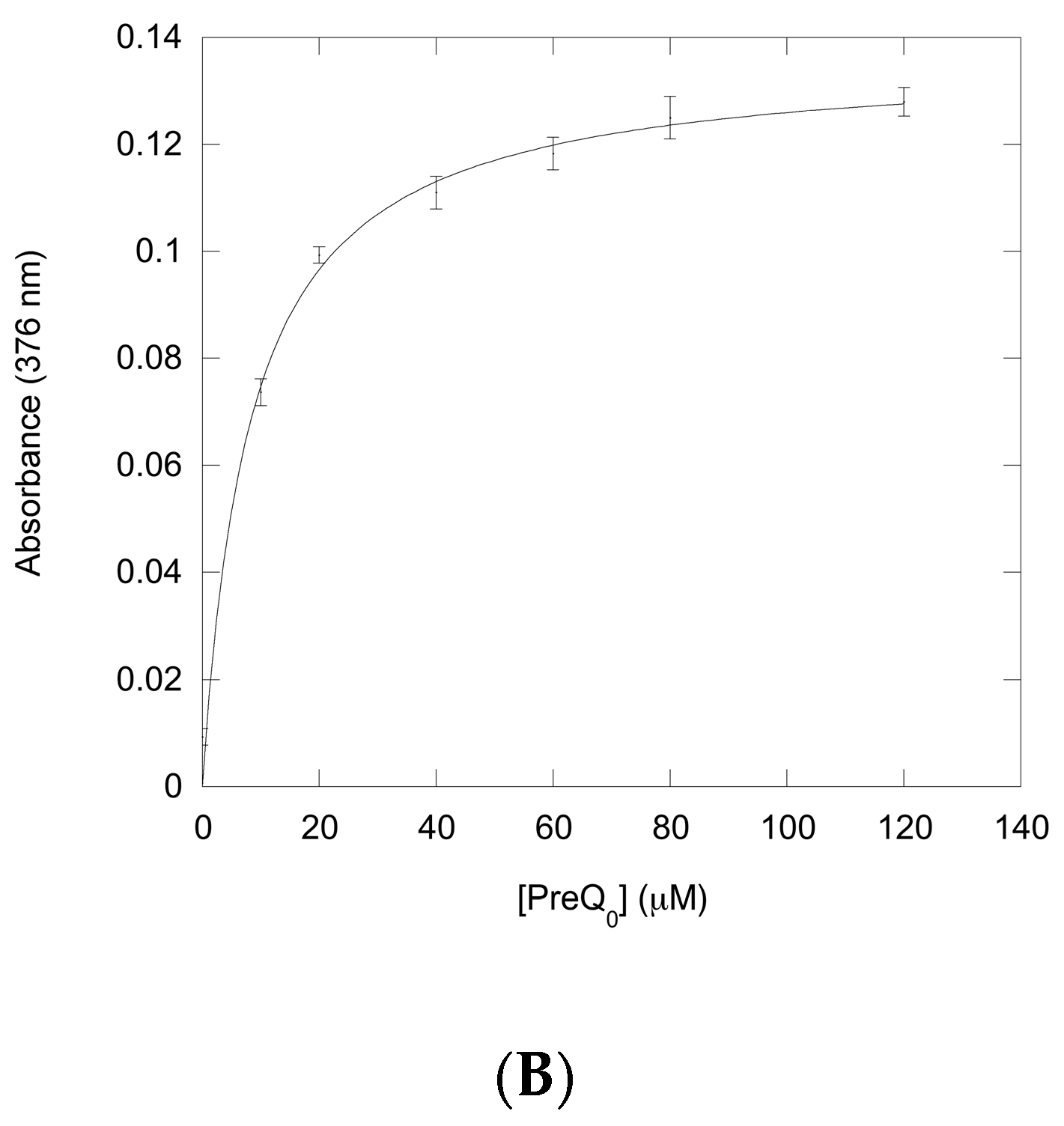
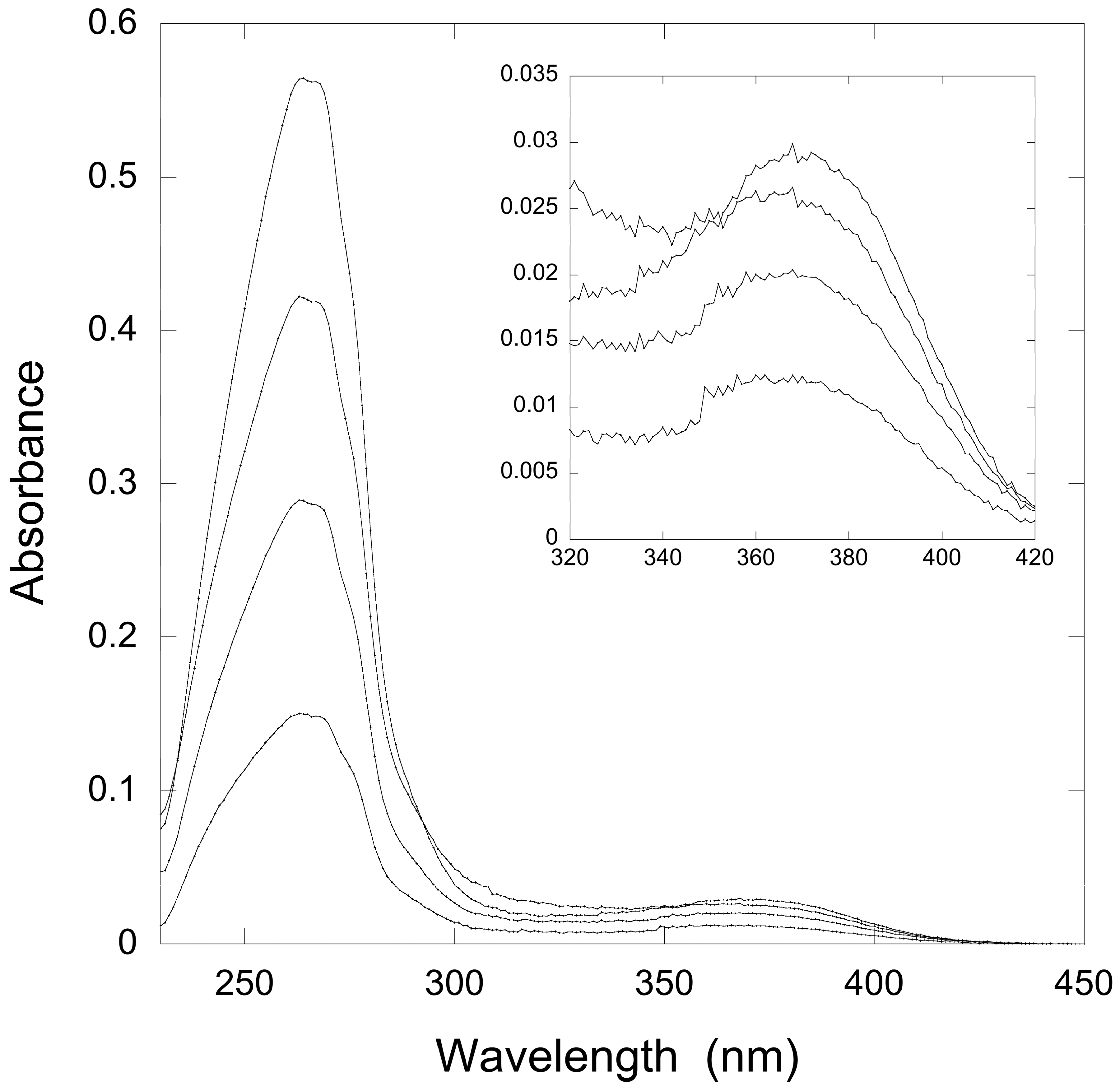
Therefore, we titrated solutions of QueF-L with preQ0 or preQ0-tRNA and measured the absorption from 200 to 450 nm. Interestingly, titration with either potential substrate resulted in the formation of a new absorption at 376 nm that grew in intensity with increasing preQ0 or preQ0-tRNA (Figure 4A and Figure 5), suggesting that thioimide adducts formed with both. In the case of the preQ0 titration, the absorption exhibited a saturation curve (Figure 4B), consistent with specific binding to QueF-L (apparent KD = 8 μM); due to limiting preQ0-tRNA we did not titrate this to saturation. Notably, these data are consistent with our subsequent observation of a thioimide intermediate in the X-ray structure of QueF-L crystalized in the presence of preQ0 [30].
2.2.2. Reactivity of preQ0 and preQ0-tRNA Thioimides with Various Nitrogen Sources
The archaeosine base is very unstable, readily undergoing deamination to form preQ0 [32], making the direct observation of archaeosine base problematic if it was in fact the product of the reaction. However, the thioimide intermediate formed in the QueF-catalyzed reaction is quite stable [28,35], and this adduct can be isolated and probed free of excess substrate. Therefore, if the QueF-L adducts with preQ0 and preQ0-tRNA were similarly stable, it would be possible to investigate their fate when incubated in the presence of different ammonia sources.
To test this, we preformed the thioimide adducts of QueF-L with preQ0 and preQ0-tRNA and investigated their behavior with potential NH3 sources. Glutamine, and occasionally asparagine, function as NH3 donors in virtually all amidotransferases [36,37] with the concomitant formation of glutamate and aspartate, respectively, and therefore were potential sources of NH3 for QueF-L. However, all amidotransferases that utilize these amino acids as a source of NH3 also possess a conserved cysteine residue that is essential in the chemical mechanism for the overall hydrolysis of the amide groups, and QueF-L possesses only one conserved cysteine, which, as discussed above, is implicated in thioimide formation. Therefore, we also investigated NH4Cl as a source of free ammonia.
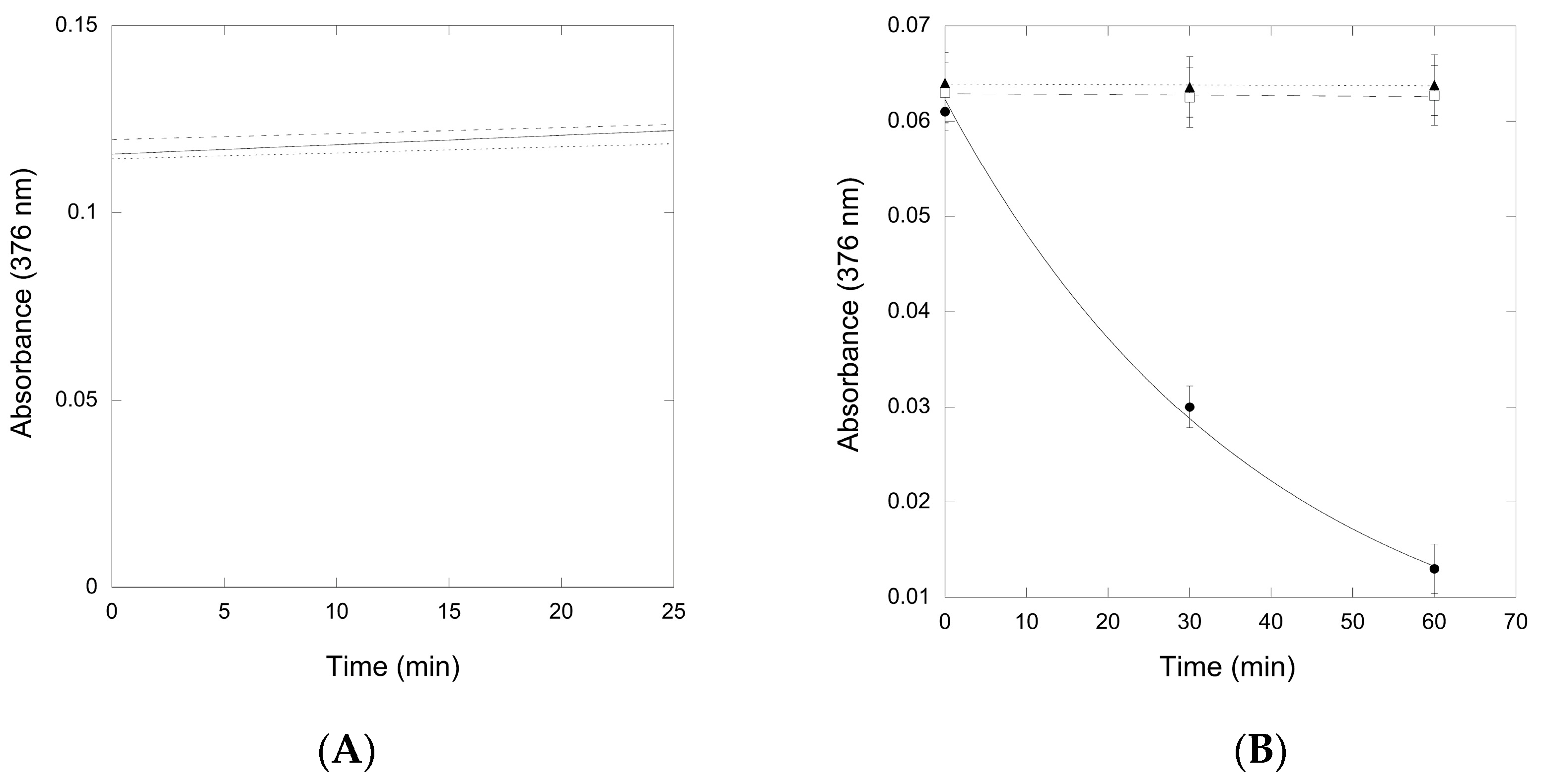
The thioimide absorbance due to the QueF-L/preQ0 adduct was stable in the presence of all three potential sources of ammonia (Figure 6A), indicating that the thioimide was unreactive even when incubated for prolonged periods of time. Similarly, the absorbance due to the QueF-L/preQ0-tRNA adduct was stable in the presence of glutamine and asparagine, but in contrast it decayed rapidly in the presence of NH4Cl, consistent with turnover to form G+-modified tRNA (Figure 6B).
2.2.3. Conversion of preQ0-Modified tRNA to G+-Modified tRNA by QueF-L
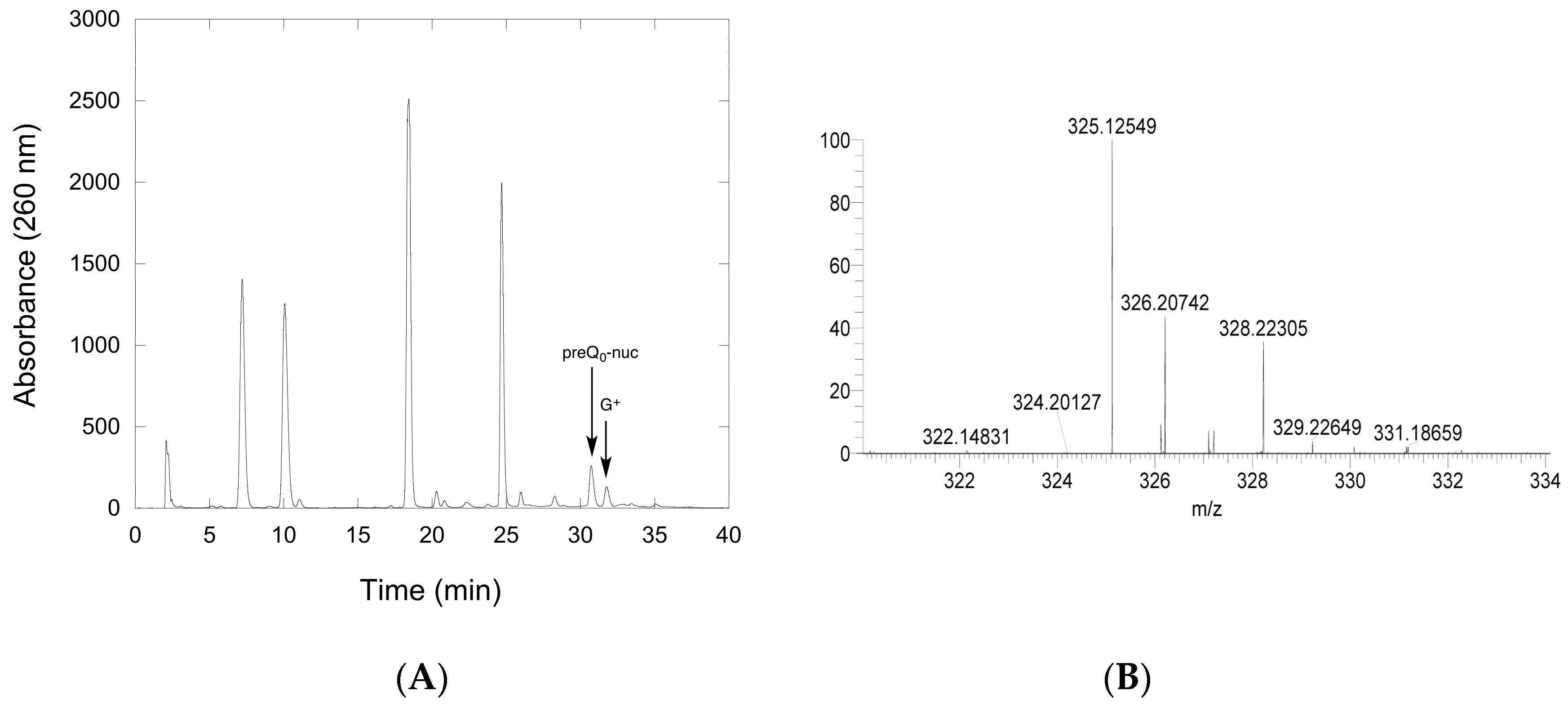
To provide unambiguous evidence that preQ0-modified tRNA was the substrate of QueF-L, and that it was turned over in the presence of NH4Cl to produce G+-modified tRNA, we carried out reactions of QueF-L with preQ0-tRNA and NH4Cl, followed by isolation of the tRNA, and, after hydrolysis and dephosphorylation, analysis of the constituent nucleosides by liquid chromatography mass spectrometry (LCMS) A peak with the expected retention time of G+ was observed in the chromatogram (Figure 7A), and mass spectrometry analysis of that component provided an m/z of 325.12549 consistent with G+ (Figure 7B).
3. Discussion
The biosynthetic pathway to the 7-deazaguanosine modified nucleosides of tRNA is one of the most complex of the known modifications, and the only one in which a significant portion occurs outside the context of tRNA. While homologs of virtually all of the enzymes that catalyze steps in the pathway can be readily identified in all organisms that possess these modifications, the first and last steps of the pathways exhibit considerable diversity. Three distinct enzymes have been identified that catalyze the first step, although all are members of the same protein superfamily [22,24] and are thus evolutionarily related. In contrast, the enzymes catalyzing the last step in both the archaeosine [25,26] and queuosine [38,39] branches are structurally unique and represent distinct evolutionary solutions to the reactions that they catalyze. QueF-L is especially interesting in this regard as it is closely related to the bacterial QueF enzyme, an NADPH-dependent oxido-reductase in the queuosine pathway, and further expands the already diverse chemistry catalyzed by enzymes of the T-fold superfamily.
Although in vivo data implicated P. calidifontis QueF-L as an amidinotransferase involved in the G+ pathway [26], there were two places in the pathway where it could conceivably function (Figure 2): in the last step analogous to ArcS [25], or earlier in the pathway prior to incorporation into the tRNA in analogy to QueF [27]. The results presented here clearly establish QueF-L as functionally analogous to ArcS, and preQ0-modified tRNA as the relevant substrate. Notably, in addition to the aforementioned new catalytic activity, this represents the first example of a T-fold enzyme utilizing a nucleic acid substrate.
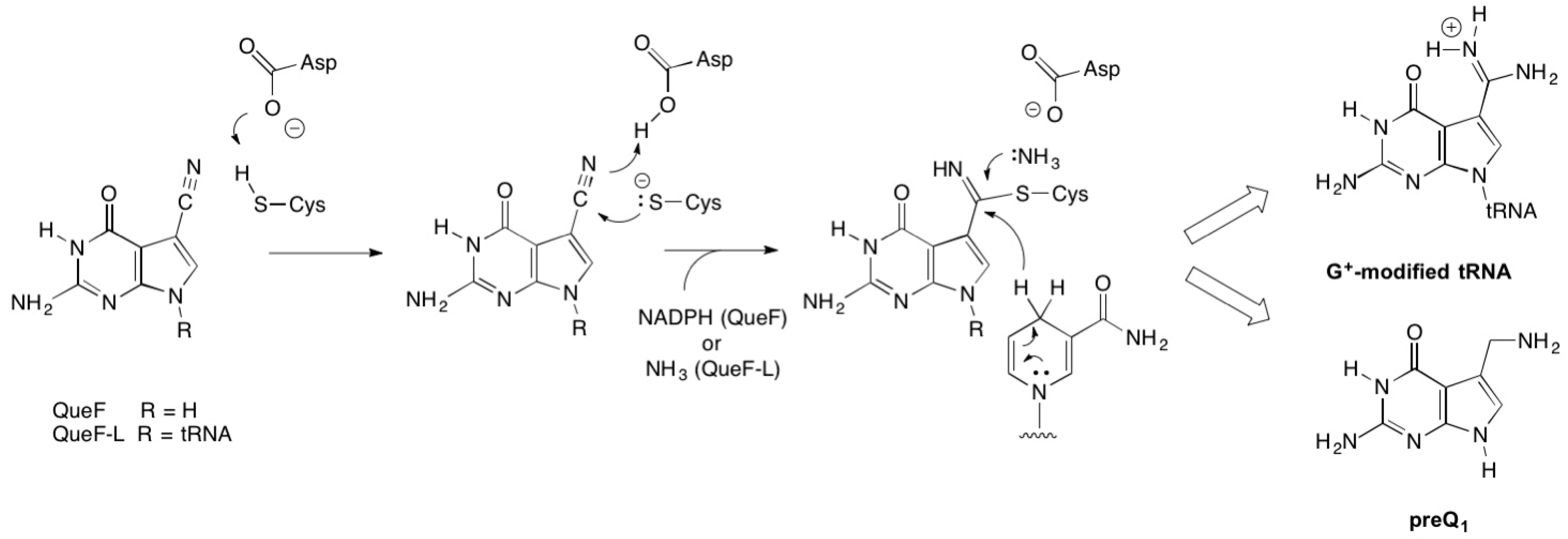
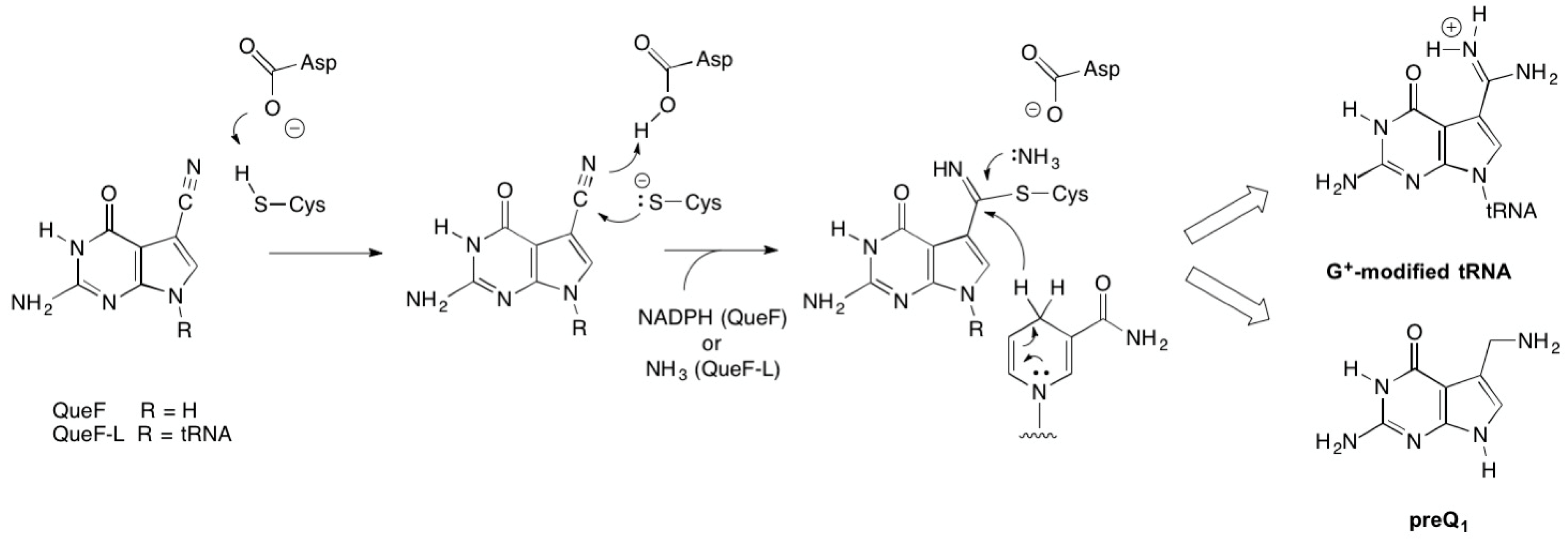
While the products of the QueF- and QueF-L-catalyzed reactions are markedly different, an aminomethyl and a formamidine, respectively, both enzymes share an identical mechanistic path to analogous covalent thioimide intermediates (Figure 8), a process mediated by conserved active-site residues that include the cysteine involved in the thioimide and an aspartic acid that serves as a general acid/base [30]. The paths diverge at the thioimide intermediate, and differ in the identity of the nucleophile that attacks the thioimide intermediate—a hydride from NADPH in the case of QueF, and ammonia in the QueF-L reaction—and in the stoichiometry of co-substrate binding: two equivalents of NADPH to carry out the four-electron reduction, and one equivalent of ammonia.
Finally, unlike ArcS, as well as transamidases as a whole, glutamine is not the ammonia donor in the reaction, and instead the enzyme is only able to utilize free NH4+. The structural basis for this is now evident, as the X-ray crystal structure [30] shows that NH4+ gains access to the putative ammonia binding site via the central tunnel of the QueF-L decamer, which displays a uniform surface that clearly lacks any architectural features of an active-site that might bind a substrate that could serve as an ammonia donor. However, while the data presented here establish that QueF-L itself utilizes only NH4+, the data does not preclude the potential involvement of a protein partner that might function in generating NH4+ in vivo.
4. Materials and Methods
4.1. General
Buffers, salts and reagents (highest quality grade available), as well as gel filtration molecular weight standards and NTPs, were purchased from Sigma (St. Louis, MO, USA). Dithiothreitol (DTT), isopropyl-β-d-thiogalacto-pyranoside (IPTG), kanamycin sulfate, diethylpyrocarbonate (DEPC), and ampicillin were purchased from RPI Corporation (Chicago, IL, USA). [8-14C]-guanine was obtained from Perkin Elmer (Waltham, MA, USA). Amicon Ultra 15 and 0.5 centrifugal filter units and NovaBlue Singles competent cells were acquired from EMD Millipore (Billerica, MA, USA). Nickel-nitrilotriacetic acid agarose (Ni2+-NTA agarose), silica TLC plates, and Whatman GF-B PVDF syringe filters were purchased from Fisher Scientific (Pittsburgh, PA, USA). GeneJet Plasmid Miniprep kits, Klenow enzyme and PageRuler pre-stained protein ladder were purchased from Fermentas (Glen Burnie, MD, USA). Custom oligonucleotides were obtained from Integrated DNA Technologies (San Diego, CA, USA). Dialysis was carried out in Slide-A-Lyzer cassettes (ThermoFisher, Waltham, MA, USA). All reagents for sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) were purchased from BioRad (Hercules, CA, USA). SDS-PAGE analysis was carried out using 12% gels and visualized with Coomassie Brilliant Blue. Diethylpyrocarbonate-treated water was used in the preparation of all solutions for RNA-related assays. DNA sequencing was carried out at the DNA Services Core at Oregon Health and Sciences University (OHSU), Portland, OR, USA.
4.2. Enzymes
Bacterial alkaline phosphatase, nuclease P1 from Penicillium citrinum, snake venom phophodiesterase I and DNase were purchased as lyophilized powders from Sigma and stored in 50% glycerol with the appropriate buffer at the recommended temperature. PfuUltra DNA polymerase was obtained from Agilent (Santa Clara, CA, USA). Restriction enzymes were purchased from Fermentas (Glen Burnie, MD, USA) and New England Biolabs (Ipswich, MA, USA). Lysozyme was purchased from RPI Corporation (Mount Prospect, IL, USA). The plasmid encoding the Δ(172–173) variant of T7 RNA polymerase [40] was provided by John Perona. Recombinant Methanocaldococcus jannashii TGT was over-produced and purified as described previously [41].
4.3. Instrumentation
PCR was carried out on a 2720 Applied Biosystems cycler (Thermo, San Jose, CA, USA). UV-Vis spectroscopy was performed with a Cary 100 spectrophotometer (Agilent, Santa Clara, CA, USA) equipped with a thermostated cell holder. HPLC was carried out using an Agilent 1100 with photodiode array detector, and controlled via the Agilent Chemstation software, Agilent (Santa Clara, CA, USA). SDS-PAGE was carried out on a mini Protean III system from BioRad (Hercules, CA, USA). Mass spectrometry was performed on a LTQ-Orbitrap mass spectrometer (Thermo Electron, San Jose, CA, USA) equipped with an electrospray ionization (ESI) source in the Department of Chemistry’s core facility at Portland State University. Radioactivity was quantified with a Hidex 300 SL liquid scintillation counter (Turku, Finland) using Econo-Safe liquid scintillation cocktail (RPI).
4.4. PreQ0 Synthesis
PreQ0 was synthesized as previously described by Klepper (yield 12%, purity 98%). The product was purified by HPLC using a Luna C18 semi-preparative column (250 × 10 mm, 5 micron) from Phenomenex (Torrance, CA, USA) using a gradient of ammonium acetate (25 mM, pH 6.5) and acetonitrile (0–50% ACN over 30 min).
4.5. Cloning of P. calidifontis queF-L
The queF-L gene from P. calidifontis was synthesized (GenScript) with codon optimization for E. coli expression (see Supplemental Figure S1) and subcloned via PCR from into the FactorXa/LIC vector (Novagen) with the following primers:
- Sense primer: 5′-GGTATTGAGGGTCGCATGCTGAAAGTCTCAAAAAGCC-3′
- Antisense primer: 5′-AGAGGAGAGTTAGAGCCTTAGATGTAGACCGGCGGC-3′
PCR reactions contained 100 ng of linearized pGP358, 200 μM dNTPs, 50 pmol primers, 1× Pfu Ultra buffer (supplied by the manufacturer), and 2.5 units of Pfu Ultra DNA polymerase in a final volume of 50 μL. A three-step PCR thermocycling protocol was utilized: firstly, 94 °C for 3 min; secondly, 30 cycles of denaturation at 94 °C for 1 min, annealing at 50 °C for 1 min, and extension at 72 °C for 2 min; and thirdly, 72 °C for 3 min. The PCR products were then gel purified (1% agarose), and the DNA isolated (Qiagen PCR purification kit) and inserted into the FactorXa/LIC vector as described by the manufacturer. The primary structure of the resulting construct (pLBI14) was confirmed by capillary electrophoresis DNA sequencing at the OHSU DNA Services Core.
4.6. Over-Production and Purification of Recombinant P. calidifontis QueF-L
Luria-Bertani/kanamycin medium (3 mL) was inoculated with a single colony of E. coli BL21(DE3)/pLBI14 cells, and after 12 h of incubation at 37 °C a 1-mL aliquot was used to inoculate 100 mL of fresh LB/kan medium. The cultures were incubated at 37 °C and 250 rpm for 12 h and a 5-mL aliquot was taken and used to inoculate 500 mL of fresh LB/kan medium. When an optical density (OD)600 of 0.9 was reached, protein over-expression was induced by the addition of IPTG to a final concentration of 0.25 mM. The cell cultures were grown for an additional 4–5 h when the cells were collected by centrifugation at 7500 g for 15 min and frozen with liquid nitrogen. The cells were stored at −80 °C until further use.
The cells were resuspended to a density of 250 mg/mL in lysis buffer (50 mM Tris-HCl (pH 8.0), 300 mM KCl, 2 mM β-mercapto-ethanol (βME), and 1 mM phenylmethylsulfonyl fluoride (PMSF)). Lysozyme was added to a final concentration of 250 µg/mL and the cells incubated at 37 °C for 30 min, followed by three intervals of freeze-thaw cycles. DNase was added to a final concentration of 10 µg/mL and the cells were left at 37 °C for an additional 30 min. The cell lysate was centrifuged at 26,000 g for 30 min, and the cell-free extract (CFE) heated to 80 °C for 15 min followed by centrifugation at 26,000 g for 20 min. The resulting CFE was filtered using a low protein-binding 0.45-µm PVDF syringe filter, then loaded onto 5 mL of Ni2+-NTA agarose resin equilibrated in lysis buffer. The column was washed with five column volumes of lysis buffer followed by five column volumes of lysis buffer with 20 mM imidazole and no PMSF. The recombinant protein was eluted with seven column volumes of lysis buffer (with/out PMSF) containing 200 mM imidazole then concentrated to about 2 mL using the Amicon Ultra YM-10k and dialyzed overnight against 4 L of lysis buffer with no PMSF at 4 °C. Both proteins were cleaved by Factor Xa and purified as previously described. The cleaved protein was stored in 50% glycerol in 100 µL aliquots at −80 °C.
4.7. In Vitro Transcription of tRNA
Duplex DNA templates for in vitro transcription of Methanobacterium thermoautotrophicum tRNAGln were synthesized from two single-stranded oligodeoxynucleotides containing a complementary overlap region as previously described [42]. The oligonucleotide sequences used were:
- 5′-GCAGTAATACGACTCACTATAGGTCCCGTGGGGTAGTGGTAATCCTGCTGGGCTTTG-3′
- 5′-TGGTAGTCCCGAGCGGAGTCGAACCGCTGTCGCCGGGTCCAAAGCCCAGC-3′
The underlined region represents the T7 RNA polymerase promoter sequence. Transcription reactions were performed as previously described using the Δ(172–173) variant of T7 RNA polymerase [40], loaded onto a urea-PAGE gel, and after electrophoresis (80W, 60 min) the band was excised and extracted overnight in 100 mM ammonium acetate (pH 6.5) containing 1 mM EDTA. The gel was discarded and the tRNA precipitated from the remaining solution with three volumes of ethanol followed by cooling at −20 °C for 2 h. The solution was then centrifuged at 20,000 g for 20 min at 4 °C, the supernatant removed, and the RNA pellet washed with 70% cold ethanol. After centrifugation at 20,000 g the supernatant was removed and the tRNA was stored at −80 °C.
4.8. Preparation of preQ0 modified tRNAGln
PreQ0 was inserted into the tRNA transcript using recombinant Methanocaldococcus jannaschii TGT. A solution of tRNA in succinate buffer (100 mM, pH 5.5) was refolded before use [43]. An aliquot of MjTGT (10 µM) was added to a 1-mL solution containing 50 mM succinate (pH 5.5), 20 mM MgCl2, 100 mM KCl, 2 mM DTT, 100 µM tRNA, and 1 mM preQ0. After 45 min at 80 °C, the reaction was terminated by the addition of one-tenth volume of 2 M NaOAc (pH 4.0) followed by one volume of water-saturated phenol and one fifth volume chloroform:isoamyl alcohol (49:1). After vortexing for 20 s, the solution was centrifuged in a fixed angle rotor at 9000 g for 1 min. The aqueous phase was recovered and mixed with an equal volume of chloroform:isoamyl alcohol. After vortexing for 20 s, the solution was centrifuged in a fixed angle rotor for 1 min at 9000 g. The aqueous phase was recovered and concentrated using an Amicon Ultra4 centrifugal concentrator (EMD Millipore, Billerica, MA, USA). Subsequently, the preQ0-tRNAGln was precipitated from the retentate by the addition of three volumes of ethanol and cooling at −20 °C for 2 h. The solution was centrifuged at 20,000 g for 20 min at 4 °C, the supernatant removed, and the RNA pellet washed with 70% cold ethanol. After centrifugation again at 20,000 g the supernatant was removed and the preQ0-tRNA was resuspended in 3 mM sodium citrate (pH 6.3) and stored at −20 °C.
4.9. Guanine Incorporation Controls
To quantify preQ0 incorporation into tRNAGln a control reaction was run in which [8-14C]-guanine (50 mCi/mmol) was incorporated into the tRNA using M. jannaschii TGT and the tRNA isolated as described above. After quantifying the radiochemical specific activity of the [14C]tRNA, preQ0 was incorporated into the tRNA and aliquots of the reaction taken over time to measure the loss of [8-14C]-guanine (and incorporation of preQ0). The tRNA from each aliquot was precipitated and collected on Whatman GF/B glass filters. The filters were washed with cold ethanol in a vacuum filtration system so as to remove any unbound radioactive material. Once dry, the filters were placed in 7 mL scintillation vials with scintillation cocktail and the radioactivity was measured by scintillation counting.
4.10. Substrate Titration Studies
Titrations of QueF (20 µM) with preQ0 (3 mM in dimethylsulfoxide) or preQ0-tRNA were carried out in solutions containing 100 mM phosphate (pH 6.5), 50 mM KCl, 20mM MgCl2, and 1 mM DTT, while monitoring the absorbance from 230 to 450 nm. For the preQ0 titrations the concentrations of preQ0 ranged from 10 to 120 µM and the final concentration of DMSO did not exceed 4% of the total volume. For the preQ0-tRNA titrations the concentrations of preQ0-tRNA ranged from 1.0 to 4.0 µM.
4.11. Amidinotransferase Assays
Assays of amidinotransferase activity were carried out using QueF-L (20 µM) in 100 mM phosphate (pH 6.5), 50 mM KCl, 20 mM MgCl2, and 1 mM DTT, along with 1 mM preQ0 or 10 µM preQ0-tRNA, and incubation for 15 min at 37 °C. For the preQ0-tRNA assays NH4Cl (1 mM), glutamine (1 mM), or asparagine (1 mM) was then added while monitoring the absorption of the covalent thioimide adduct at 376 nm. For the assays with preQ0, the solution was filtered through a centrifugal concentrator (Amicon) in a fixed angle rotor at 8500 rcf for 10 min and the retentate washed with buffer to remove excess preQ0. After 2× washes the retentate was reconstituted with buffer and NH4Cl/glutamine/asparagine was added while monitoring the loss of covalent thioimide adduct at 376 nm.
4.12. LCMS Analysis of QueF-L Assays with preQ0-tRNA
QueF-L (20 µM) was incubated in 100 mM phosphate (pH 6.5), 50 mM KCl, 20 mM MgCl2, 1 mM DTT, 20 µM preQ0-tRNA and 1 mM NH4Cl for 1 h at 37 °C. The tRNA was then precipitated using ethanol and digested using nuclease P1, snake venom phosphodiesterase and alkaline phosphatase and the nucleoside products of the reaction were analyzed by HPLC as previously described [44], using a Gemini C18 column (5 µm, 110 A, 2 × 250 mm; Phenomenex, Torrance CA, USA), with a mobile phase comprised of a linear gradient from 100% 25 mM NH4OAc (pH 6.3) to 85% 25 mM NH4OAc/15% acetonitrile developed over 30 min, then to 50% acetonitrile at 45 min. Flow was maintained at 0.3 mL/min. LCMS analysis of nucleosides was performed on an Orbitrap-LTQ mass spectrometer (Thermo Electron, San Jose, CA, USA) utilizing electrospray ionization (ESI). The ESI interface was operated in the positive mode using the following settings: end plate offset −500 V, capillary voltage −4500 V, nebulizer gas 1.6 bar, dry gas 4 L/min, dry temperature 200 °C, funnel 1 RF 350 Vpp, funnel 2 RF 350 Vpp, hexapole RF 400 Vpp, collision energy 10 eV and collision RF 300 Vpp.
Supplementary Materials
The following are available online at https://www.mdpi.com/2218-273X/7/2/36/s1, Figure S1: Sequence of synthetic gene used for the production of P. calidifontis queF-L.
Acknowledgments
This project was supported by National Science Foundation grant CHE-1309323 to D. Iwata-Reuyl, and National Institutes of Health grant GM70641 to V. de Crécy-Lagard and D. Iwata-Reuyl. We thank Gabriela Phillips for designing the synthetic gene.
Author Contributions
D.I-R. and V. de C-L. conceived and designed the experiments; A.B.R., L.B. and B.T. performed the experiments; A.B.R. and D.I-R. analyzed the data; D.I-R. wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest. The funding sponsors had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, and in the decision to publish the results.
References
- Cantara, W.A.; Crain, P.F.; Rozenski, J.; McCloskey, J.A.; Harris, K.A.; Zhang, X.; Vendeix, F.A.; Fabris, D.; Agris, P.F. The RNA modification database, RNAMDB: 2011 update. Nucleic Acids Res. 2011, 39, D195–D201. [Google Scholar] [CrossRef] [PubMed]
- Grosjean, H. Nucleic acids are not boring long polymers of only four types of nucleosides: A guided tour. In DNA and RNA Modification Enzymes: Structure, Mechanism, Function and Evolution; Grosjean, H., Ed.; Landes Bioscience: Austin, TX, USA, 2009; pp. 1–18. [Google Scholar]
- Iwata-Reuyl, D.; de Crécy Lagard, V. Enzymatic formation of the 7-deazaguanosine hypermodified nucleosides of tRNA. In DNA and RNA Modification Enzymes: Structure, Mechanism, Function and Evolution; Grosjean, H., Ed.; Landes Bioscience: New York, NY, USA, 2009; pp. 379–394. [Google Scholar]
- Gregson, J.M.; Crain, P.F.; Edmonds, C.G.; Gupta, R.; Hashizume, T.; Phillipson, D.W.; McCloskey, J.A. Structure of archaeal transfer RNA nucleoside G*-15 (2-amino-4,7-dihydro-4-oxo-7-b-d-ribofuranosyl-1H-pyrrolo[2,3-d]pyrimidine-5-carboximidamide (archaeosine)). J. Biol. Chem. 1993, 268, 10076–10086. [Google Scholar] [PubMed]
- Kasai, H.; Ohashi, Z.; Harada, F.; Nishimura, S.; Oppenheimer, N.J.; Crain, P.F.; Liehr, J.G.; von Minden, D.L.; McCloskey, J.A. Structure of the modified nucleoside Q isolated from Escherichia coli transfer ribonucleic acid. 7-(4,5-cis-dihydroxy-1-cyclopenten-3-ylaminomethyl)-7-deazaguanosine. Biochemistry 1975, 14, 4198–4208. [Google Scholar] [CrossRef] [PubMed]
- Ohgi, T.; Kondo, T.; Goto, T. Total synthesis of optically pure nucleoside Q. Determination of absolute configuration of natural nucleoside Q. J. Am. Chem. Soc. 1979, 101, 3629–3633. [Google Scholar] [CrossRef]
- Okada, N.; Nishimura, S. Enzymatic synthesis of Q* nucleoside containing mannose in the anticodon of tRNA: Isolation of a novel mannosyltransferase from a cell-free extract of rat liver. Nucleic Acids Res. 1977, 4, 2931–2937. [Google Scholar] [CrossRef] [PubMed]
- Okada, N.; Shindo-Okada, N.; Nishimura, S. Isolation of mammalian tRNAAsp and tRNATyr by lectin-sepharose affinity column chromatography. Nucleic Acids Res. 1977, 4, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Salazar, J.C.; Ambrogelly, A.; Crain, P.F.; McCloskey, J.A.; Soll, D. A truncated aminoacyl-tRNA synthetase modifies RNA. Proc. Natl. Acad. Sci. USA 2004, 101, 7536–7541. [Google Scholar] [CrossRef] [PubMed]
- Dubois, D.Y.; Blaise, M.; Becker, H.D.; Campanacci, V.; Keith, G.; Giege, R.; Cambillau, C.; Lapointe, J.; Kern, D. An aminoacyl-tRNA synthetase-like protein encoded by the Escherichia coli yadB gene glutamylates specifically tRNAAsp. Proc. Natl. Acad. Sci. USA 2004, 101, 7530–7535. [Google Scholar] [CrossRef] [PubMed]
- Campanacci, V.; Dubois, D.Y.; Becker, H.D.; Kern, D.; Spinelli, S.; Valencia, C.; Pagot, F.; Salomoni, A.; Grisel, S.; Vincentelli, R.; et al. The Escherichia coli yadB gene product reveals a novel aminoacyl-tRNA synthetase like activity. J. Mol. Biol. 2004, 337, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Blaise, M.; Becker, H.D.; Keith, G.; Cambillau, C.; Lapointe, J.; Giege, R.; Kern, D. A minimalist glutamyl-tRNA synthetase dedicated to aminoacylation of the tRNAAsp QUC anticodon. Nucleic Acids Res. 2004, 32, 2768–2775. [Google Scholar] [CrossRef] [PubMed]
- Kersten, H. The nutrient factor queuine: Biosynthesis, occurence in transfer RNA and function. BioFactors 1988, 1, 27–29. [Google Scholar] [PubMed]
- Kersten, H.; Kersten, W. Biosynthesis and function of queuine and queuosine tRNAs. In Chromatography and Modification of Nucleosides Part B; Gehrke, C.W., Kuo, K.C.T., Eds.; Elsevier: Amsterdam, The Netherlands, 1990; pp. B69–B108. [Google Scholar]
- Marks, T.; Farkas, W.R. Effects of a diet deficient in tyrosine and queuine on germfree mice. Biochem. Biophys. Res. Commun. 1997, 230, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Carlson, B.A.; Kwon, S.Y.; Chamorro, M.; Oroszlan, S.; Hatfield, D.L.; Lee, B.J. Transfer RNA modification status influences retroviral ribosomal frameshifting. Virology 1999, 255, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Durand, J.; Okada, N.; Tobe, T.; Watarai, M.; Fukuda, I.; Suzuki, T.; Nakata, N.; Komatsu, K.; Yoshikawa, M.; Sasakawa, C. vacC, a virulence-associated chromosomal locus of Shigella flexneri, is homologous to tgt, a gene encoding tRNA-guanine transglycosylase (Tgt) of Escherichia coli K-12. J. Bacteriol. 1994, 176, 4627–4634. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, T.; Hirata, A.; Ohno, S.; Nomura, Y.; Nagano, T.; Nameki, N.; Yokogawa, T.; Hori, H. Multisite-specific archaeosine tRNA-guanine transglycosylase (ArcTGT) from Thermoplasma acidophilum, a thermo-acidophilic archaeon. Nucleic Acids Res. 2016, 44, 1894–1908. [Google Scholar] [CrossRef] [PubMed]
- Sprinzl, M.; Hartmann, T.; Weber, J.; Blank, J.; Zeidler, R. Compilation of tRNA sequences and sequences of tRNA genes. Nuc. Acids Res. 1989, 26, 148–153. [Google Scholar] [CrossRef]
- Oliva, R.; Tramontano, A.; Cavallo, L. Mg2+ binding and archaeosine modification stabilize the G15–C48 Levitt base pair in tRNAs. RNA 2007, 13, 1427–1436. [Google Scholar] [CrossRef] [PubMed]
- Mashhadi, Z.; Xu, H.; White, R.H. An Fe2+-dependent cyclic phosphodiesterase catalyzes the hydrolysis of 7,8-dihydro-d-neopterin 2’,3’-cyclic phosphate in methanopterin biosynthesis. Biochemistry 2009, 48, 9384–9392. [Google Scholar] [CrossRef] [PubMed]
- Grochowski, L.L.; Xu, H.; Leung, K.; White, R.H. Characterization of an Fe2+-dependent archaeal-specific GTP cyclohydrolase, MptA, from Methanocaldococcus jannaschii. Biochemistry 2007, 46, 6658–6667. [Google Scholar] [CrossRef] [PubMed]
- Phillips, G.; El Yacoubi, B.; Lyons, B.; Alvarez, S.; Iwata-Reuyl, D.; de Crecy-Lagard, V. Biosynthesis of 7-deazaguanosine-modified tRNA nucleosides: A new role for GTP cyclohydrolase I. J. Bacteriol. 2008, 190, 7876–7884. [Google Scholar] [CrossRef] [PubMed]
- El Yacoubi, B.; Bonnett, S.; Anderson, J.N.; Swairjo, M.A.; Iwata-Reuyl, D.; de Crecy-Lagard, V. Discovery of a new prokaryotic type I GTP cyclohydrolase family. J. Biol. Chem. 2006, 281, 37586–37593. [Google Scholar] [CrossRef] [PubMed]
- Phillips, G.; Chikwana, V.M.; Maxwell, A.; El-Yacoubi, B.; Swairjo, M.A.; Iwata-Reuyl, D.; de Crecy-Lagard, V. Discovery and characterization of an amidinotransferase involved in the modification of archaeal tRNA. J. Biol. Chem. 2010, 285, 12706–12713. [Google Scholar] [CrossRef] [PubMed]
- Phillips, G.; Swairjo, M.A.; Gaston, K.W.; Bailly, M.; Limbach, P.A.; Iwata-Reuyl, D.; de Crecy-Lagard, V. Diversity of archaeosine synthesis in Crenarchaeota. ACS Chem. Biol. 2012, 7, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Van Lanen, S.G.; Reader, J.S.; Swairjo, M.A.; de Crecy-Lagard, V.; Lee, B.; Iwata-Reuyl, D. From cyclohydrolase to oxidoreductase: Discovery of nitrile reductase activity in a common fold. Proc. Natl. Acad. Sci. USA 2005, 102, 4264–4269. [Google Scholar] [CrossRef] [PubMed]
- Chikwana, V.M.; Stec, B.; Lee, B.W.; de Crecy-Lagard, V.; Iwata-Reuyl, D.; Swairjo, M.A. Structural basis of biological nitrile reduction. J. Biol. Chem. 2012, 287, 30560–30570. [Google Scholar] [CrossRef] [PubMed]
- Colloc’h, N.; Poupon, A.; Mornon, J.P. Sequence and structural features of the T-fold, an original tunnelling building unit. Proteins 2000, 39, 142–154. [Google Scholar] [CrossRef]
- Mei, X.; Alvarez, J.; Bon Ramos, A.; Samanta, U.; Iwata-Reuyl, D.; Swairjo, M.A. Crystal structure of the archaeosine synthase QueF-Like—Insights into amidino transfer and tRNA recognition by the tunnel fold. Proteins 2017, 85, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Matsuo, M.; Tanaka, S.; Akimoto, H.; Asahi, S.; Nishimura, S.; Katz, J.R.; Hashizume, T.; Crain, P.F.; McCloskey, J.A.; et al. Biosynthesis of archaeosine, a novel derivative of 7-deazaguanosine specific to archaeal tRNA, proceeds via a pathway involving base replacement of the tRNA polynucleotide chain. J. Biol. Chem. 1997, 272, 20146–20151. [Google Scholar] [CrossRef] [PubMed]
- Hoops, G.C.; Townsend, L.B.; Garcia, G.A. tRNA-guanine transglycosylase from Escherichia coli: Structure-activity studies investigating the role of the aminomethyl substituent of the heterocyclic substrate PreQ1. Biochemistry 1995, 34, 15381–15387. [Google Scholar] [CrossRef] [PubMed]
- Ishitani, R.; Nureki, O.; Fukai, S.; Kijimoto, T.; Nameki, N.; Watanabe, M.; Kondo, H.; Sekine, M.; Okada, N.; Nishimura, S.; et al. Crystal structure of archaeosine tRNA-guanine transglycosylase. J. Mol. Biol. 2002, 318, 665–677. [Google Scholar] [CrossRef]
- Romier, C.; Meyer, J.E.; Suck, D. Slight sequence variations of a common fold explain the substrate specificities of tRNA-guanine transglycosylases from the three kingdoms. FEBS Lett. 1997, 416, 93–98. [Google Scholar] [CrossRef]
- Lee, B.W.; van Lanen, S.G.; Iwata-Reuyl, D. Mechanistic studies of Bacillus subtilis QueF, the nitrile oxidoreductase involved in queuosine biosynthesis. Biochemistry 2007, 46, 12844–12854. [Google Scholar] [CrossRef] [PubMed]
- Massiere, F.; Badet-Denisot, M.A. The mechanism of glutamine-dependent amidotransferases. Cell. Mol. Life Sci. 1998, 54, 205–222. [Google Scholar] [CrossRef] [PubMed]
- Zalkin, H. The amidotransferases. Adv. Enzymol. Relat. Areas Mol. Biol. 1993, 66, 203–309. [Google Scholar] [PubMed]
- Zallot, R.; Ross, R.; Chen, W.H.; Bruner, S.D.; Limbach, P.A.; De Crecy-Lagard, V. Identification of a novel epoxyqueuosine reductase family by comparative genomics. ACS Chem. Biol. 2017, 12, 844–851. [Google Scholar] [CrossRef] [PubMed]
- Miles, Z.D.; McCarty, R.M.; Molnar, G.; Bandarian, V. Discovery of epoxyqueuosine (oQ) reductase reveals parallels between halorespiration and tRNA modification. Proc. Natl. Acad. Sci. USA 2011, 108, 7368–7372. [Google Scholar] [CrossRef] [PubMed]
- Lyakhov, D.L.; He, B.; Zhang, X.; Studier, F.W.; Dunn, J.J.; McAllister, W.T. Mutant bacteriophage T7 RNA polymerases with altered termination properties. J. Mol. Biol. 1997, 269, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Fox, D.T.; Lacy, J.A.; Van Lanen, S.G.; Iwata-Reuyl, D. Hypermodification of tRNA in Thermophilic archaea. Cloning, overexpression, and characterization of tRNA-guanine transglycosylase from Methanococcus jannaschii. J. Biol. Chem. 2000, 275, 28731–28738. [Google Scholar] [CrossRef] [PubMed]
- Sherlin, L.D.; Bullock, T.L.; Nissan, T.A.; Perona, J.J.; Lariviere, F.J.; Uhlenbeck, O.C.; Scaringe, S.A. Chemical and enzymatic synthesis of tRNAs for high-throughput crystallization. RNA 2001, 7, 1671–1678. [Google Scholar] [PubMed]
- Bhaskaran, H.; Rodriguez-Hernandez, A.; Perona, J.J. Kinetics of tRNA folding monitored by aminoacylation. RNA 2012, 18, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Crain, P.F. Preparation and enzymatic hydrolysis of DNA and RNA for mass spectrometry. Meth. Enzymol. 1990, 193, 782–790. [Google Scholar] [PubMed]
Figure 1.
The biosynthetic pathways to 7-deazaquanosine modified nucleosides of transfer RNA (tRNA). The upper branch occurs in the Archaea and leads to archaeosine (G+), while the lower branch occurs in bacteria and leads to queuosine (Q); the boxed region is common to both. *MptA/B catalyze the initial steps in Archaea [21,22], while GCYH IA or IB catalyze the initial step in bacteria [23,24]. ArcS: Archaeosine Synthase; preQ0: 7-cyano-7-deazaguanine; preQ1: 7-aminomethyl-7-deazaguanine.
Figure 1.
The biosynthetic pathways to 7-deazaquanosine modified nucleosides of transfer RNA (tRNA). The upper branch occurs in the Archaea and leads to archaeosine (G+), while the lower branch occurs in bacteria and leads to queuosine (Q); the boxed region is common to both. *MptA/B catalyze the initial steps in Archaea [21,22], while GCYH IA or IB catalyze the initial step in bacteria [23,24]. ArcS: Archaeosine Synthase; preQ0: 7-cyano-7-deazaguanine; preQ1: 7-aminomethyl-7-deazaguanine.
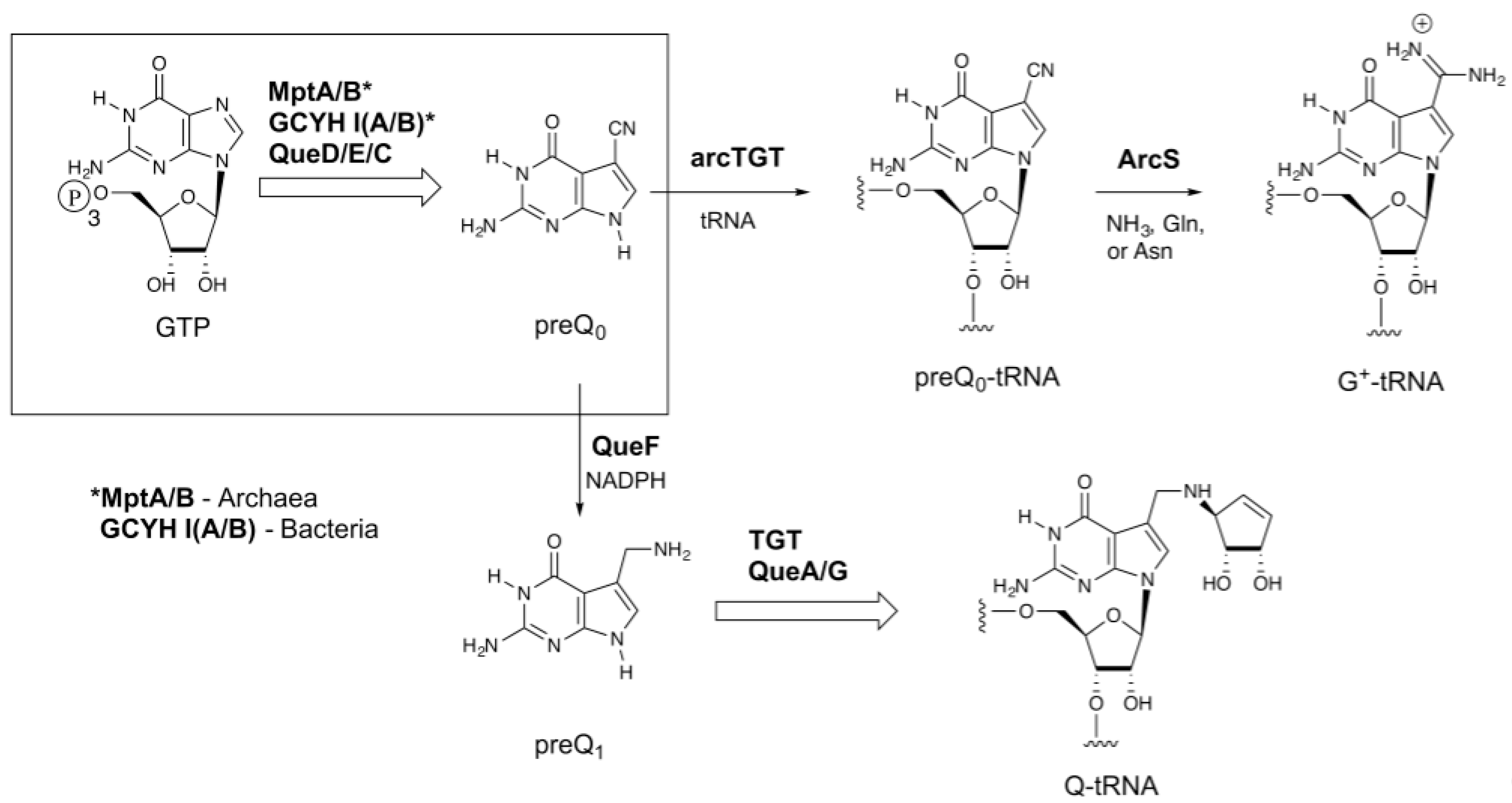
Figure 2.
Possible routes to G+-tRNA in Crenarchaeota possessing QueF-like (QueF-L) enzymes. The top pathway is analogous to the known pathway in Euryarchaeota that utilizes ArcS to carry out the amidation of preQ0-modified tRNA. The bottom pathway is analogous to the known reaction of bacterial QueF, which carries out the four-electron reduction of the nitrile group in preQ0 to give the aminomethyl product preQ1.
Figure 2.
Possible routes to G+-tRNA in Crenarchaeota possessing QueF-like (QueF-L) enzymes. The top pathway is analogous to the known pathway in Euryarchaeota that utilizes ArcS to carry out the amidation of preQ0-modified tRNA. The bottom pathway is analogous to the known reaction of bacterial QueF, which carries out the four-electron reduction of the nitrile group in preQ0 to give the aminomethyl product preQ1.

Figure 3.
Structure and characteristics of the covalent thioimide intermediate formed in the QueF-catalyzed reaction in the biosynthesis of queuosine.
Figure 3.
Structure and characteristics of the covalent thioimide intermediate formed in the QueF-catalyzed reaction in the biosynthesis of queuosine.
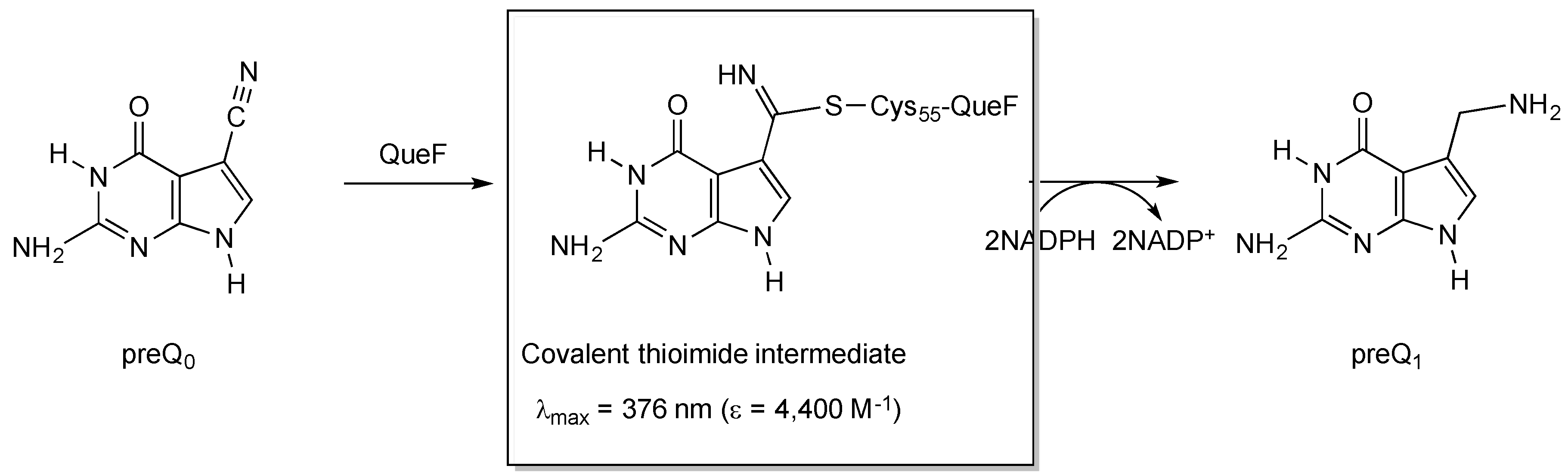
Figure 4.
Ultraviolet-Visible (UV-Vis) spectroscopy of titration of a QueF-like with preQ0. (A) The thicker lower trace corresponds to the protein (20 μM) absorbance prior to preQ0 addition. The finer traces correspond, from bottom to top, to preQ0 concentrations of 10, 20, 40, 60, 80, and 120 μM, respectively. Inset shows the region around 376 nm expanded for clarity; (B) Plot of absorbance at 376 nm vs. [preQ0] exhibits a saturation curve.
Figure 4.
Ultraviolet-Visible (UV-Vis) spectroscopy of titration of a QueF-like with preQ0. (A) The thicker lower trace corresponds to the protein (20 μM) absorbance prior to preQ0 addition. The finer traces correspond, from bottom to top, to preQ0 concentrations of 10, 20, 40, 60, 80, and 120 μM, respectively. Inset shows the region around 376 nm expanded for clarity; (B) Plot of absorbance at 376 nm vs. [preQ0] exhibits a saturation curve.
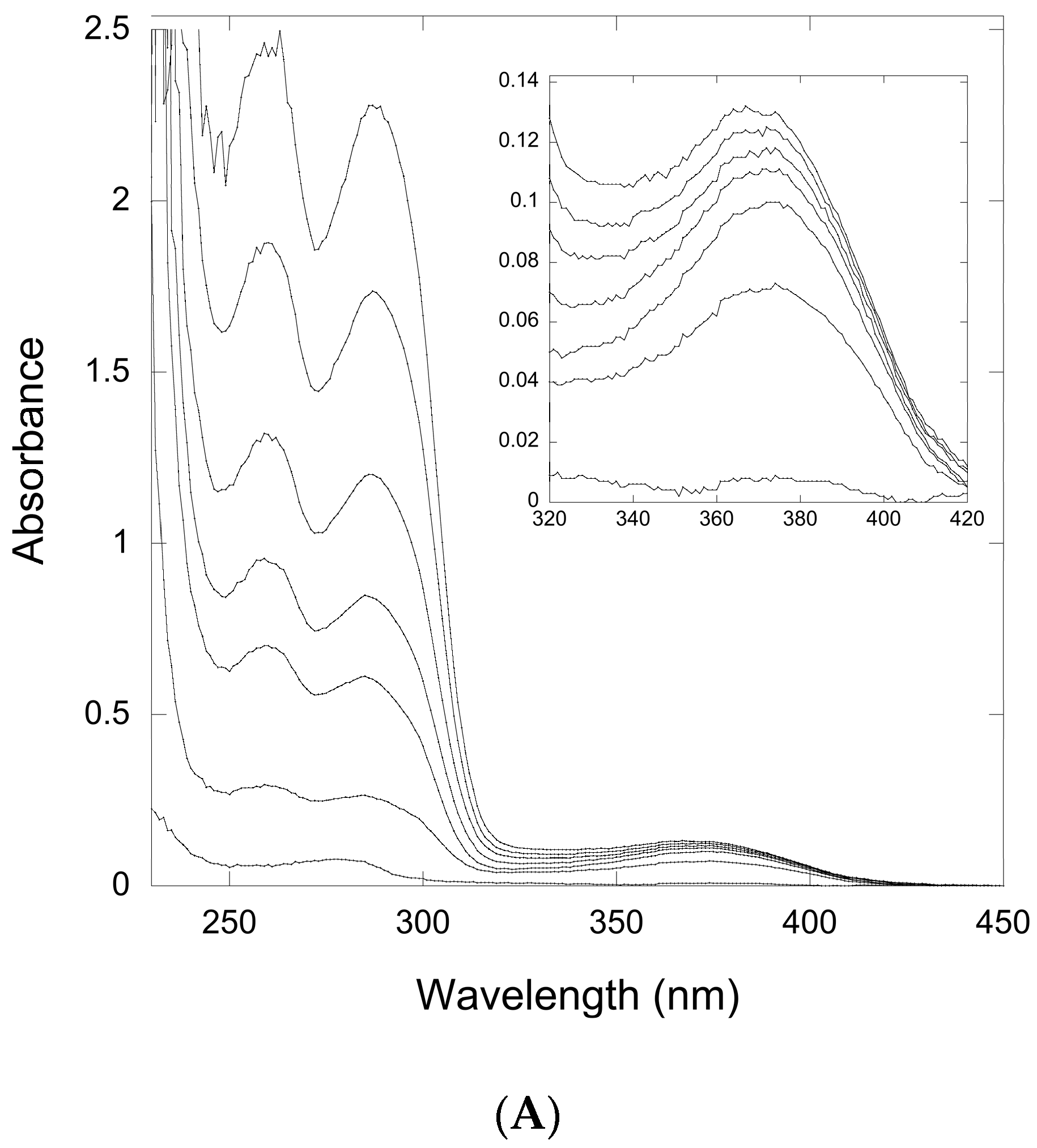
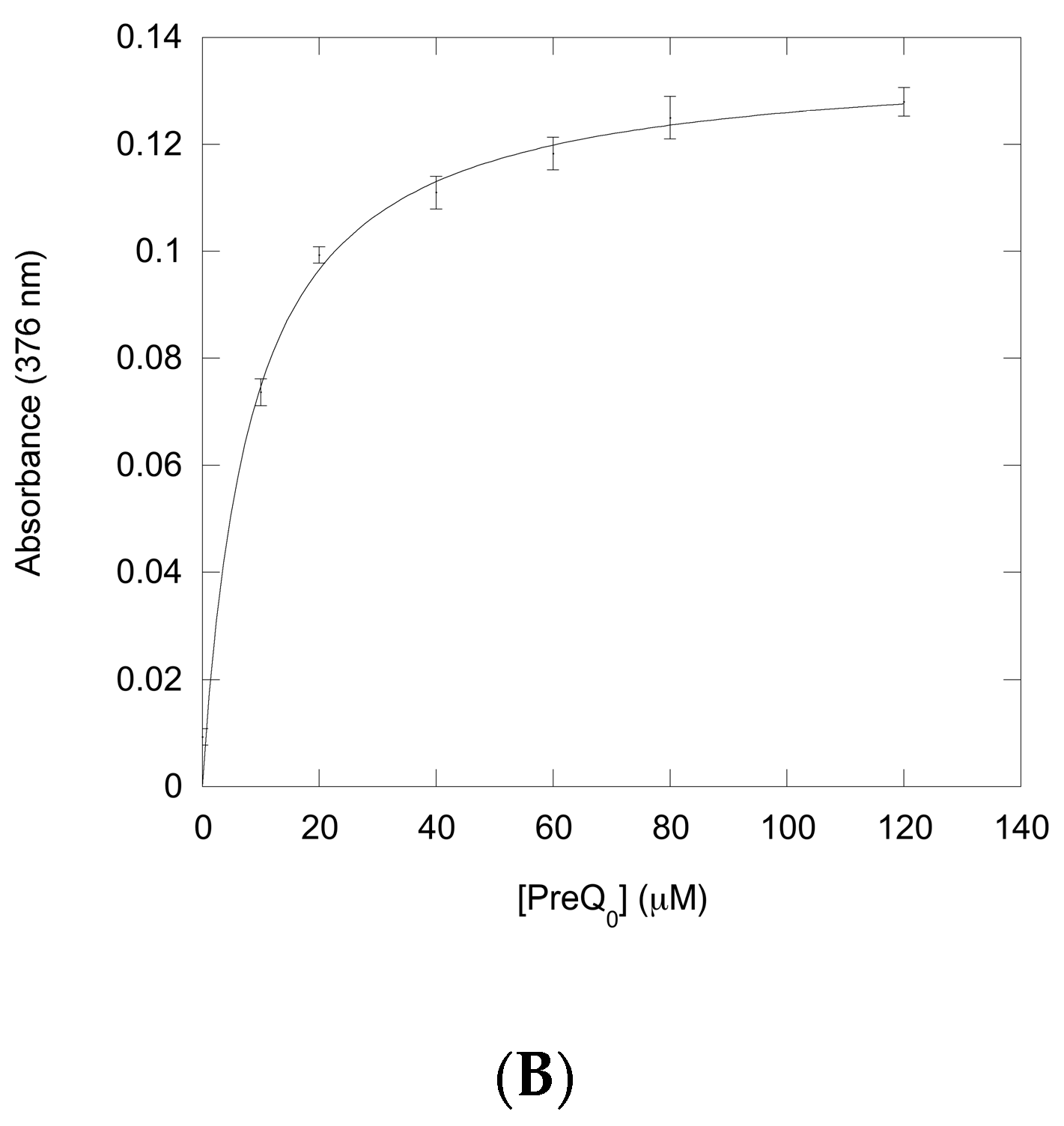
Figure 5.
UV-Vis spectroscopy of a titration of QueF-L with preQ0-tRNA. The traces correspond, from bottom to top, to preQ0-tRNA concentrations of 1, 2, 3, and 4 μM, respectively. QueF-L was present at a concentration of 20 μM. Inset shows the region around 376 nm expanded for clarity.
Figure 5.
UV-Vis spectroscopy of a titration of QueF-L with preQ0-tRNA. The traces correspond, from bottom to top, to preQ0-tRNA concentrations of 1, 2, 3, and 4 μM, respectively. QueF-L was present at a concentration of 20 μM. Inset shows the region around 376 nm expanded for clarity.

Figure 6.
Time-course of the thioimide absorbance (376 nm) from QueF-L/preQ0 and QueF-L/preQ0-tRNA in the presence of various ammonia donors. (A) The absorbance of the QueF-L/preQ0 thioimide adduct in the presence of ammonium chloride (―), glutamine (---), or asparagine (…). Data show an example of a single experiment with each ammonia donor; (B) The absorbance of the QueF-L/preQ0-tRNA thioimide adduct in the presence of ammonium chloride (●,―), glutamine (□,---), or asparagine (▲,…). Data represents the average of three assays, each carried out in replicate with each potential ammonia donor (error bars represent the standard error (SE)).
Figure 6.
Time-course of the thioimide absorbance (376 nm) from QueF-L/preQ0 and QueF-L/preQ0-tRNA in the presence of various ammonia donors. (A) The absorbance of the QueF-L/preQ0 thioimide adduct in the presence of ammonium chloride (―), glutamine (---), or asparagine (…). Data show an example of a single experiment with each ammonia donor; (B) The absorbance of the QueF-L/preQ0-tRNA thioimide adduct in the presence of ammonium chloride (●,―), glutamine (□,---), or asparagine (▲,…). Data represents the average of three assays, each carried out in replicate with each potential ammonia donor (error bars represent the standard error (SE)).
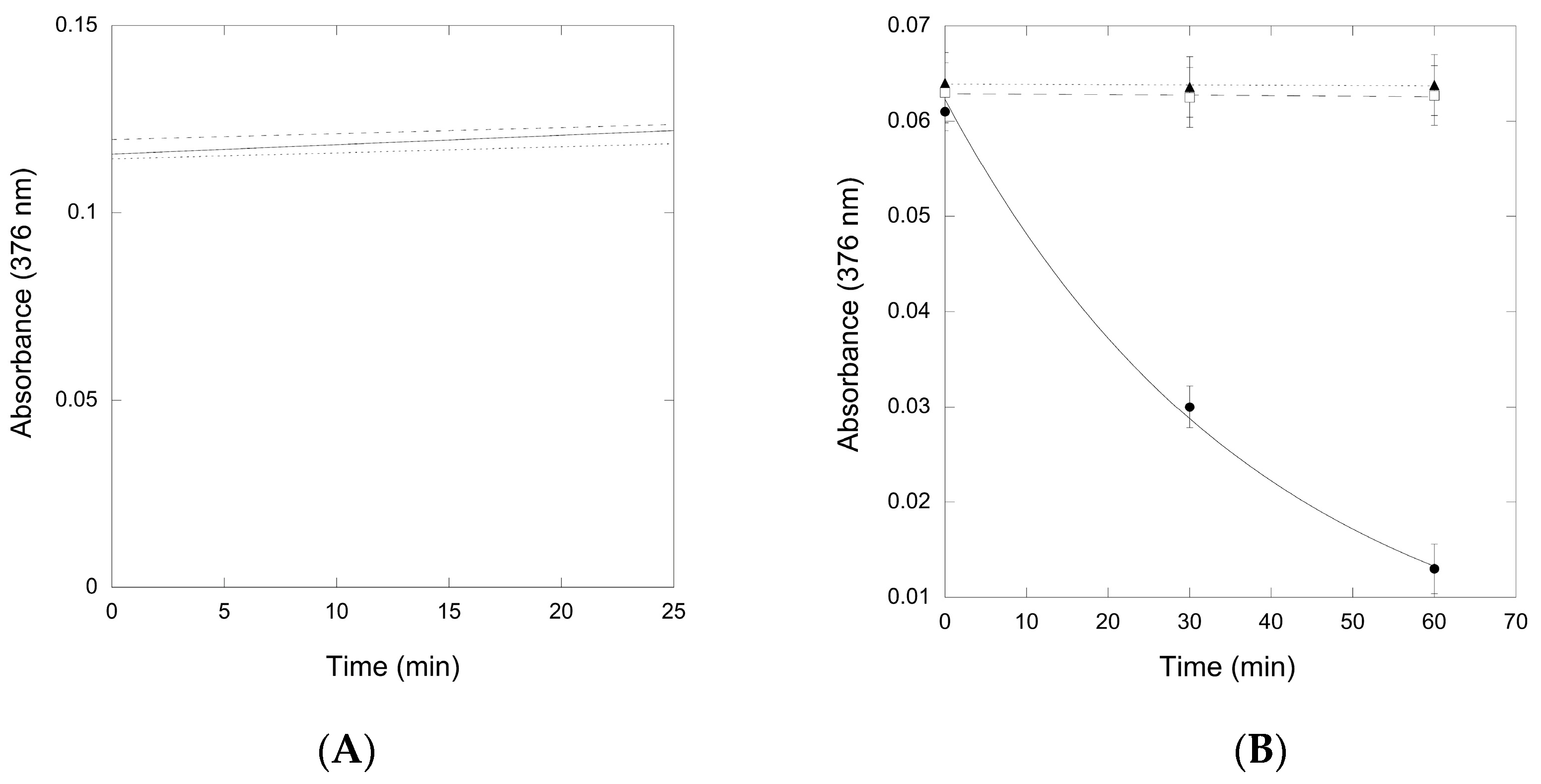
Figure 7.
Confirmation of G+ synthesis by QueF-L. (A) An example of the HPLC chromatogram of nucleoside components of a tRNAGln transcript modified by incorporation of preQ0 with arcTGT, followed by reaction with QueF-L in the presence of NH4Cl. The tRNA was digested and dephosphorylated prior to HPLC analysis. (B) Partial mass spectrum of the component eluting at approximately 32 min and labeled G+ in the chromatogram. The peak at m/z 325.12549 is consistent with G+, and the peak at m/z 326.20742 with the isotopic M+1 ion.
Figure 7.
Confirmation of G+ synthesis by QueF-L. (A) An example of the HPLC chromatogram of nucleoside components of a tRNAGln transcript modified by incorporation of preQ0 with arcTGT, followed by reaction with QueF-L in the presence of NH4Cl. The tRNA was digested and dephosphorylated prior to HPLC analysis. (B) Partial mass spectrum of the component eluting at approximately 32 min and labeled G+ in the chromatogram. The peak at m/z 325.12549 is consistent with G+, and the peak at m/z 326.20742 with the isotopic M+1 ion.
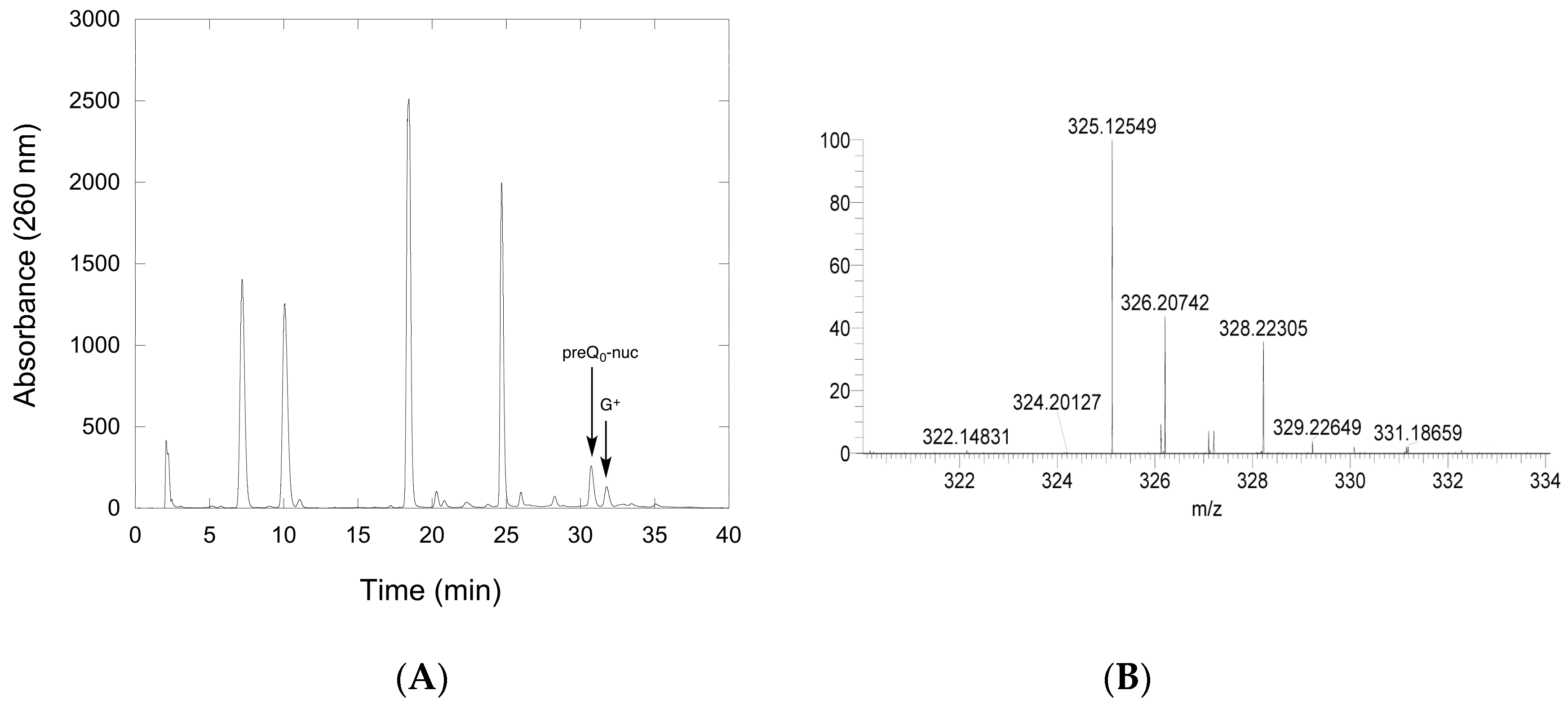
Figure 8.
The common mechanistic steps leading to covalent thioimide intermediates in the reactions catalyzed by QueF and QueF-L.
Figure 8.
The common mechanistic steps leading to covalent thioimide intermediates in the reactions catalyzed by QueF and QueF-L.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bon Ramos, A.; Bao, L.; Turner, B.; De Crécy-Lagard, V.; Iwata-Reuyl, D. QueF-Like, a Non-Homologous Archaeosine Synthase from the Crenarchaeota. Biomolecules 2017, 7, 36. https://doi.org/10.3390/biom7020036
AMA Style
Bon Ramos A, Bao L, Turner B, De Crécy-Lagard V, Iwata-Reuyl D. QueF-Like, a Non-Homologous Archaeosine Synthase from the Crenarchaeota. Biomolecules. 2017; 7(2):36. https://doi.org/10.3390/biom7020036
Chicago/Turabian StyleBon Ramos, Adriana, Lide Bao, Ben Turner, Valérie De Crécy-Lagard, and Dirk Iwata-Reuyl. 2017. "QueF-Like, a Non-Homologous Archaeosine Synthase from the Crenarchaeota" Biomolecules 7, no. 2: 36. https://doi.org/10.3390/biom7020036
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.