Retinoic Acid Signaling during Early Spinal Cord Development

{kind=link}
Abstract
:1. Introduction
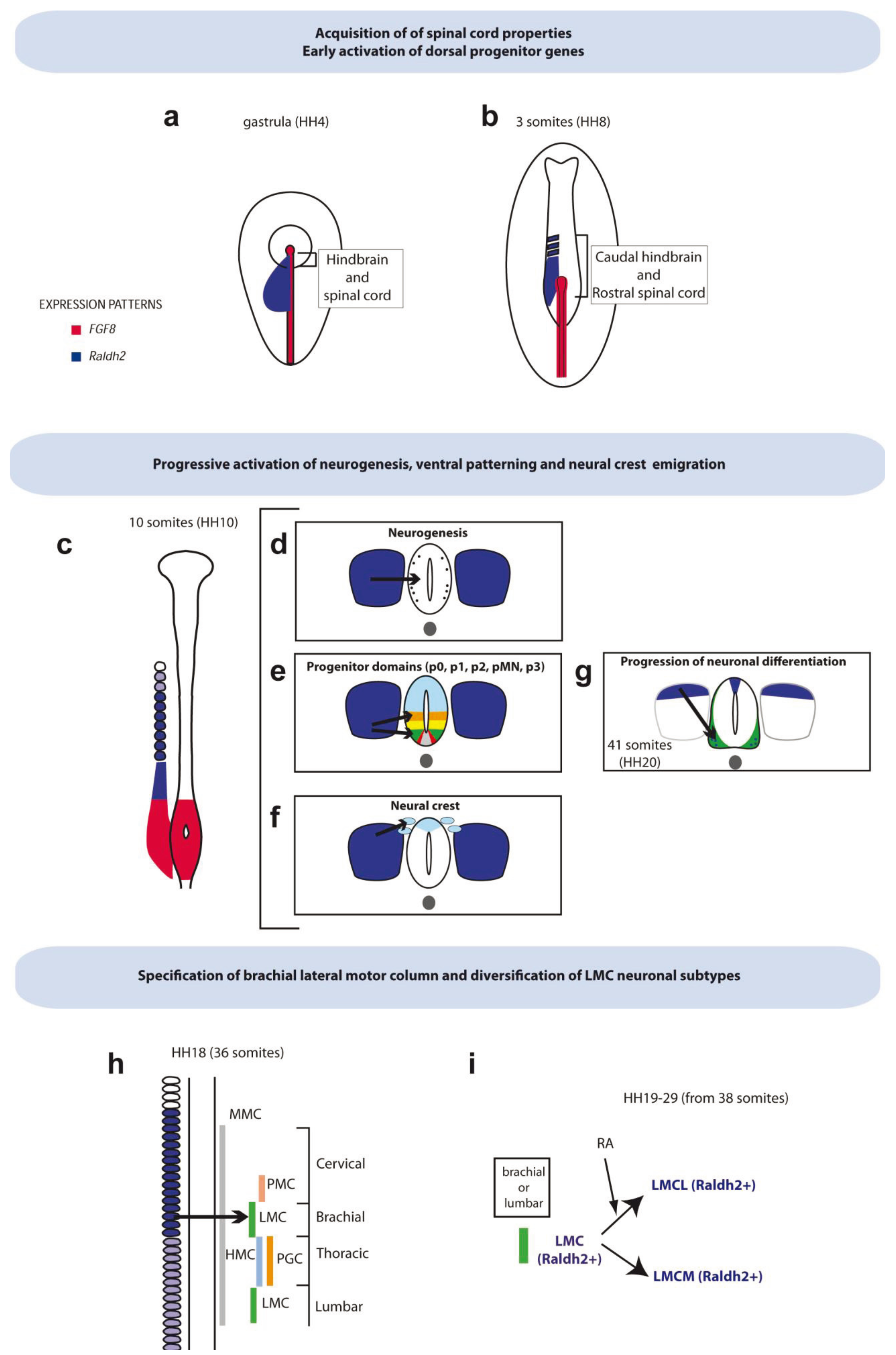
2. RA Signaling Gradients during Axis Elongation
2.1. Sites of Active RA Signaling during Spinal Cord Elongation: Differential Expression of Key Components of the Pathway
2.2. Mechanisms Regulating the Activation of RA Signaling during Axis Elongation: An RA-FGF-Wnt Gene Regulatory Network
2.3. RA in the Control of Trunk-Tail Transition and Axis Termination
3. RA Controlling Early Neurogenesis in Spinal Cord
4. RA in the Acquisition of Dorso-Ventral Identities
4.1. Ventral Patterning of Progenitors and Acquisition of Motor Neuron Identity
4.2. Dorsal Patterning of Progenitors
5. RA in Trunk Neural Crest
6. RA and the Acquisition of Rostro-Caudal Identities in the Spinal Cord
6.1. RA and the Specification of Spinal Cord Progenitor Cells
6.2. RA in the Acquisition of Rostro-Caudal Column Identities of Motor Neurons

7. Conclusions
Acknowledgements
Conflicts of Interest
References
- Stern, C.D. Neural induction: Old problem, new findings, yet more questions. Development 2005, 132, 2007–2021. [Google Scholar] [CrossRef]
- Stern, C.D.; Charite, J.; Deschamps, J.; Duboule, D.; Durston, A.J.; Kmita, M.; Nicolas, J.F.; Palmeirim, I.; Smith, J.C.; Wolpert, L. Head-tail patterning of the vertebrate embryo: One, two or many unresolved problems? Int. J. Dev. Biol. 2006, 50, 3–15. [Google Scholar] [CrossRef]
- Tam, P.P.; Beddington, R.S. The formation of mesodermal tissues in the mouse embryo during gastrulation and early organogenesis. Development 1987, 99, 109–126. [Google Scholar]
- Brown, J.M.; Storey, K.G. A region of the vertebrate neural plate in which neighbouring cells can adopt neural or epidermal cell fates. Curr. Biol. 2000, 10, 869–872. [Google Scholar] [CrossRef]
- Mathis, L.; Kulesa, P.M.; Fraser, S.E. Fgf receptor signalling is required to maintain neural progenitors during Hensen’s node progression. Nat. Cell. Biol. 2001, 3, 559–566. [Google Scholar] [CrossRef]
- Wilson, V.; Olivera-Martinez, I.; Storey, K.G. Stem cells, signals and vertebrate body axis extension. Development 2009, 136, 1591–1604. [Google Scholar] [CrossRef]
- Dersch, H.; Zile, M.H. Induction of normal cardiovascular development in the vitamin A-deprived quail embryo by natural retinoids. Dev. Biol. 1993, 160, 424–433. [Google Scholar] [CrossRef]
- Dong, D.; Zile, M.H. Endogenous retinoids in the early avian embryo. Biochem. Biophys. Res. Commun. 1995, 217, 1026–1031. [Google Scholar] [CrossRef]
- Mic, F.A.; Haselbeck, R.J.; Cuenca, A.E.; Duester, G. Novel retinoic acid generating activities in the neural tube and heart identified by conditional rescue of raldh2 null mutant mice. Development 2002, 129, 2271–2282. [Google Scholar]
- Niederreither, K.; Subbarayan, V.; Dolle, P.; Chambon, P. Embryonic retinoic acid synthesis is essential for early mouse post- implantation development. Nat. Genet. 1999, 21, 444–448. [Google Scholar] [CrossRef]
- Niederreither, K.; Vermot, J.; Fraulob, V.; Chambon, P.; Dolle, P. Retinaldehyde dehydrogenase 2 (raldh2)-independent patterns of retinoic acid synthesis in the mouse embryo. Proc. Natl. Acad. Sci. USA 2002, 99, 16111–16116. [Google Scholar] [CrossRef]
- Vermot, J.; Schuhbaur, B.; Le Mouellic, H.; McCaffery, P.; Garnier, J.M.; Hentsch, D.; Brulet, P.; Niederreither, K.; Chambon, P.; Dolle, P.; et al. Retinaldehyde dehydrogenase 2 and hoxc8 are required in the murine brachial spinal cord for the specification of lim1+ motoneurons and the correct distribution of islet1+ motoneurons. Development 2005, 132, 1611–1621. [Google Scholar] [CrossRef]
- Ji, S.J.; Zhuang, B.; Falco, C.; Schneider, A.; Schuster-Gossler, K.; Gossler, A.; Sockanathan, S. Mesodermal and neuronal retinoids regulate the induction and maintenance of limb innervating spinal motor neurons. Dev. Biol. 2006, 297, 249–261. [Google Scholar] [CrossRef]
- Sockanathan, S.; Perlmann, T.; Jessell, T.M. Retinoid receptor signaling in postmitotic motor neurons regulates rostrocaudal positional identity and axonal projection pattern. Neuron 2003, 40, 97–111. [Google Scholar] [CrossRef]
- Duester, G. Retinoic acid synthesis and signalling during early organogenesis. Cell 2008, 134, 921–931. [Google Scholar] [CrossRef]
- Sandell, L.L.; Lynn, M.L.; Inman, K.E.; McDowell, W.; Trainor, P.A. Rdh10 oxidation of vitamin a is a critical control step in synthesis of retinoic acid during mouse embryogenesis. PLoS ONE 2012, 7, e30698. [Google Scholar]
- Sandell, L.L.; Sanderson, B.W.; Moiseyev, G.; Johnson, T.; Mushegian, A.; Young, K.; Rey, J.P.; Ma, J.X.; Staehling-Hampton, K.; Trainor, P.A. Rdh10 is essential for synthesis of embryonic retinoic acid and is required for limb, craniofacial, and organ development. Genes Dev. 2007, 21, 1113–1124. [Google Scholar] [CrossRef]
- Cammas, L.; Romand, R.; Fraulob, V.; Mura, C.; Dolle, P. Expression of the murine retinol dehydrogenase 10 (rdh10) gene correlates with many sites of retinoid signalling during embryogenesis and organ differentiation. Dev. Dyn. 2007, 236, 2899–2908. [Google Scholar] [CrossRef]
- Reijntjes, S.; Zile, M.H.; Maden, M. The expression of stra6 and rdh10 in the avian embryo and their contribution to the generation of retinoid signatures. Int. J. Dev. Biol. 2010, 54, 1267–1275. [Google Scholar] [CrossRef]
- Rhinn, M.; Schuhbaur, B.; Niederreither, K.; Dolle, P. Involvement of retinol dehydrogenase 10 in embryonic patterning and rescue of its loss of function by maternal retinaldehyde treatment. Proc. Natl. Acad. Sci. USA 2011, 108, 16687–16692. [Google Scholar]
- Belyaeva, O.V.; Lee, S.A.; Adams, M.K.; Chang, C.; Kedishvili, N.Y. Short chain dehydrogenase/reductase rdhe2 is a novel retinol dehydrogenase essential for frog embryonic development. J. Biol. Chem. 2012, 287, 9061–9071. [Google Scholar]
- Chambers, D.; Wilson, L.; Maden, M.; Lumsden, A. Raldh-independent generation of retinoic acid during vertebrate embryogenesis by cyp1b1. Development 2007, 134, 1369–1383. [Google Scholar] [CrossRef]
- Muhr, J.; Graziano, E.; Wilson, S.; Jessell, T.M.; Edlund, T. Convergent inductive signals specify midbrain, hindbrain, and spinal cord identity in gastrula stage chick embryos. Neuron 1999, 23, 689–702. [Google Scholar] [CrossRef]
- Swindell, E.C.; Thaller, C.; Sockanathan, S.; Petkovich, M.; Jessell, T.M.; Eichele, G. Complementary domains of retinoic acid production and degradation in the early chick embryo. Dev. Biol. 1999, 216, 282–296. [Google Scholar] [CrossRef]
- Berggren, K.; McCaffery, P.; Drager, U.; Forehand, C.J. Differential distribution of retinoic acid synthesis in the chicken embryo as determined by immunolocalization of the retinoic acid synthetic enzyme, raldh-2. Dev. Biol. 1999, 210, 288–304. [Google Scholar] [CrossRef]
- Niederreither, K.; McCaffery, P.; Drager, U.C.; Chambon, P.; Dolle, P. Restricted expression and retinoic acid-induced downregulation of the retinaldehyde dehydrogenase type 2 (raldh-2) gene during mouse development. Mech. Dev. 1997, 62, 67–78. [Google Scholar] [CrossRef]
- Niederreither, K.; Vermot, J.; Schuhbaur, B.; Chambon, P.; Dolle, P. Retinoic acid synthesis and hindbrain patterning in the mouse embryo. Development 2000, 127, 75–85. [Google Scholar]
- Ribes, V.; Wang, Z.; Dolle, P.; Niederreither, K. Retinaldehyde dehydrogenase 2 (raldh2)-mediated retinoic acid synthesis regulates early mouse embryonic forebrain development by controlling fgf and sonic hedgehog signaling. Development 2006, 133, 351–361. [Google Scholar]
- Fujii, H.; Sato, T.; Kaneko, S.; Gotoh, O.; Fujii-Kuriyama, Y.; Osawa, K.; Kato, S.; Hamada, H. Metabolic inactivation of retinoic acid by a novel p450 differentially expressed in developing mouse embryos. EMBO J. 1997, 16, 4163–4173. [Google Scholar] [CrossRef]
- Reijntjes, S.; Gale, E.; Maden, M. Generating gradients of retinoic acid in the chick embryo: Cyp26c1 expression and a comparative analysis of the cyp26 enzymes. Dev. Dyn. 2004, 230, 509–517. [Google Scholar] [CrossRef]
- Blentic, A.; Gale, E.; Maden, M. Retinoic acid signalling centres in the avian embryo identified by sites of expression of synthesising and catabolising enzymes. Dev. Dyn. 2003, 227, 114–127. [Google Scholar] [CrossRef]
- Sirbu, I.O.; Gresh, L.; Barra, J.; Duester, G. Shifting boundaries of retinoic acid activity control hindbrain segmental gene expression. Development 2005, 132, 2611–2622. [Google Scholar] [CrossRef]
- Cai, A.Q.; Radtke, K.; Linville, A.; Lander, A.D.; Nie, Q.; Schilling, T.F. Cellular retinoic acid-binding proteins are essential for hindbrain patterning and signal robustness in zebrafish. Development 2012, 139, 2150–2155. [Google Scholar] [CrossRef]
- Lampron, C.; Rochette-Egly, C.; Gorry, P.; Dolle, P.; Mark, M.; Lufkin, T.; LeMeur, M.; Chambon, P. Mice deficient in cellular retinoic acid binding protein ii (crabpii) or in both crabpi and crabpii are essentially normal. Development 1995, 121, 539–548. [Google Scholar]
- Ghyselinck, N.B.; Bavik, C.; Sapin, V.; Mark, M.; Bonnier, D.; Hindelang, C.; Dierich, A.; Nilsson, C.B.; Hakansson, H.; Sauvant, P.; et al. Cellular retinol-binding protein i is essential for vitamin a homeostasis. EMBO J. 1999, 18, 4903–4914. [Google Scholar] [CrossRef]
- Maden, M.; Ong, D.E.; Summerbell, D.; Chytil, F. The role of retinoid-binding proteins in the generation of pattern in the developing limb, the regenerating limb and the nervous system. Development 1989, 107, 109–119. [Google Scholar]
- Perez-Castro, A.V.; Toth-Rogler, L.E.; Wei, L.N.; Nguyen-Huu, M.C. Spatial and temporal pattern of expression of the cellular retinoic acid-binding protein and the cellular retinol-binding protein during mouse embryogenesis. Proc. Natl. Acad. Sci. USA 1989, 86, 8813–8817. [Google Scholar]
- Ruberte, E.; Friederich, V.; Morriss-Kay, G.; Chambon, P. Differential distribution patterns of crabp i and crabp ii transcripts during mouse embryogenesis. Development 1992, 115, 973–987. [Google Scholar]
- Olivera-Martinez, I.; Harada, H.; Halley, P.A.; Storey, K.G. Loss of fgf-dependent mesoderm identity and rise of endogenous retinoid signalling determine cessation of body axis elongation. PLoS Biol. 2012, 10, e1001415. [Google Scholar] [CrossRef]
- Shiga, T.; Gaur, V.P.; Yamaguchi, K.; Oppenheim, R.W. The development of interneurons in the chick embryo spinal cord following in vivo treatment with retinoic acid. J. Comp. Neurol. 1995, 360, 463–474. [Google Scholar] [CrossRef]
- Mark, M.; Ghyselinck, N.B.; Chambon, P. Function of retinoid nuclear receptors: Lessons from genetic and pharmacological dissections of the retinoic acid signaling pathway during mouse embryogenesis. Annu. Rev. Pharmacol. Toxicol. 2006, 46, 451–480. [Google Scholar] [CrossRef]
- Schug, T.T.; Berry, D.C.; Shaw, N.S.; Travis, S.N.; Noy, N. Opposing effects of retinoic acid on cell growth result from alternate activation of two different nuclear receptors. Cell 2007, 129, 723–733. [Google Scholar]
- Dolle, P. Developmental expression of retinoic acid receptors (RARs). Nucl. Recept. Signal. 2009, 7, e006. [Google Scholar]
- Rhinn, M.; Dolle, P. Retinoic acid signalling during development. Development 2012, 139, 843–858. [Google Scholar] [CrossRef]
- Diez del Corral, R.; Olivera-Martinez, I.; Goriely, A.; Gale, E.; Maden, M.; Storey, K.G. Opposing fgf and retinoid pathways control ventral neural pattern, neuronal differentiation, and segmentation during body axis extension. Neuron 2003, 40, 65–79. [Google Scholar] [CrossRef]
- Maden, M.; Sonneveld, E.; van der Saag, P.T.; Gale, E. The distribution of endogenous retinoic acid in the chick embryo: Implications for developmental mechanisms. Development 1998, 125, 4133–4144. [Google Scholar]
- Shimozono, S.; Iimura, T.; Kitaguchi, T.; Higashijima, S.; Miyawaki, A. Visualization of an endogenous retinoic acid gradient across embryonic development. Nature 2013, 496, 363–366. [Google Scholar] [CrossRef]
- Rossant, J.; Zirngibl, R.; Cado, D.; Shago, M.; Giguere, V. Expression of a retinoic acid response element-hsplacz transgene defines specific domains of transcriptional activity during mouse embryogenesis. Genes Dev. 1991, 5, 1333–1344. [Google Scholar] [CrossRef]
- Ribes, V.; Le Roux, I.; Rhinn, M.; Schuhbaur, B.; Dolle, P. Early mouse caudal development relies on crosstalk between retinoic acid, shh and fgf signalling pathways. Development 2009, 136, 665–676. [Google Scholar] [CrossRef]
- Colbert, M.C.; Rubin, W.W.; Linney, E.; LaMantia, A.S. Retinoid signaling and the generation of regional and cellular diversity in the embryonic mouse spinal cord. Dev. Dyn. 1995, 204, 1–12. [Google Scholar] [CrossRef]
- Dubrulle, J.; McGrew, M.J.; Pourquie, O. Fgf signaling controls somite boundary position and regulates segmentation clock control of spatiotemporal hox gene activation. Cell 2001, 106, 219–232. [Google Scholar] [CrossRef]
- Akai, J.; Halley, P.A.; Storey, K.G. Fgf-dependent notch signaling maintains the spinal cord stem zone. Genes Dev. 2005, 19, 2877–2887. [Google Scholar] [CrossRef]
- Diez del Corral, R.; Breitkreuz, D.N.; Storey, K.G. Onset of neuronal differentiation is regulated by paraxial mesoderm and requires attenuation of fgf signalling. Development 2002, 129, 1681–1691. [Google Scholar]
- Olivera-Martinez, I.; Storey, K.G. Wnt signals provide a timing mechanism for the fgf-retinoid differentiation switch during vertebrate body axis extension. Development 2007, 134, 2125–2135. [Google Scholar] [CrossRef]
- Wahl, M.B.; Deng, C.; Lewandoski, M.; Pourquie, O. Fgf signaling acts upstream of the notch and wnt signaling pathways to control segmentation clock oscillations in mouse somitogenesis. Development 2007, 134, 4033–4041. [Google Scholar] [CrossRef]
- Molotkova, N.; Molotkov, A.; Sirbu, I.O.; Duester, G. Requirement of mesodermal retinoic acid generated by raldh2 for posterior neural transformation. Mech. Dev. 2005, 122, 145–155. [Google Scholar] [CrossRef]
- Vermot, J.; Gallego Llamas, J.; Fraulob, V.; Niederreither, K.; Chambon, P.; Dolle, P. Retinoic acid controls the bilateral symmetry of somite formation in the mouse embryo. Science 2005, 308, 563–566. [Google Scholar] [CrossRef]
- Patel, N.S.; Rhinn, M.; Semprich, C.I.; Halley, P.A.; Dolle, P.; Bickmore, W.A.; Storey, K.G. Fgf signalling regulates chromatin organisation during neural differentiation via mechanisms that can be uncoupled from transcription. PLoS Genet. 2013, 9, e1003614. [Google Scholar] [CrossRef]
- Sirbu, I.O.; Duester, G. Retinoic-acid signalling in node ectoderm and posterior neural plate directs left-right patterning of somitic mesoderm. Nat. Cell Biol. 2006, 8, 271–277. [Google Scholar] [CrossRef]
- Afonso, N.D.; Catala, M. Sonic hedgehog and retinoic acid are not sufficient to induce motoneuron generation in the avian caudal neural tube. Dev. Biol. 2005, 279, 356–367. [Google Scholar] [CrossRef]
- Psychoyos, D.; Stern, C.D. Restoration of the organizer after radical ablation of Hensen’s node and the anterior primitive streak in the chick embryo. Development 1996, 122, 3263–3273. [Google Scholar]
- Wilson, V.; Beddington, R.S. Cell fate and morphogenetic movement in the late mouse primitive streak. Mech. Dev. 1996, 55, 79–89. [Google Scholar] [CrossRef]
- Jurberg, A.D.; Aires, R.; Varela-Lasheras, I.; Novoa, A.; Mallo, M. Switching axial progenitors from producing trunk to tail tissues in vertebrate embryos. Dev. Cell 2013, 25, 451–462. [Google Scholar] [CrossRef]
- Kessel, M.; Gruss, P. Homeotic transformations of murine vertebrae and concomitant alteration of hox codes induced by retinoic acid. Cell 1991, 67, 89–104. [Google Scholar] [CrossRef]
- Shum, A.S.; Poon, L.L.; Tang, W.W.; Koide, T.; Chan, B.W.; Leung, Y.C.; Shiroishi, T.; Copp, A.J. Retinoic acid induces down-regulation of wnt-3a, apoptosis and diversion of tail bud cells to a neural fate in the mouse embryo. Mech. Dev. 1999, 84, 17–30. [Google Scholar] [CrossRef]
- Takada, S.; Stark, K.L.; Shea, M.J.; Vassileva, G.; McMahon, J.A.; McMahon, A.P. Wnt-3a regulates somite and tailbud formation in the mouse embryo. Genes Dev. 1994, 8, 174–189. [Google Scholar] [CrossRef]
- Cunningham, T.J.; Zhao, X.; Duester, G. Uncoupling of retinoic acid signaling from tailbud development before termination of body axis extension. Genesis 2011, 49, 776–783. [Google Scholar]
- Brown, J.M.; Robertson, K.E.; Wedden, S.E.; Tickle, C. Alterations in msx 1 and msx 2 expression correlate with inhibition of outgrowth of chick facial primordia induced by retinoic acid. Anat. Embryol. Berl. 1997, 195, 203–207. [Google Scholar] [CrossRef]
- Tickle, C.; Crawley, A.; Farrar, J. Retinoic acid application to chick wing buds leads to a dose-dependent reorganization of the apical ectodermal ridge that is mediated by the mesenchyme. Development 1989, 106, 691–705. [Google Scholar]
- Wichterle, H.; Lieberam, I.; Porter, J.A.; Jessell, T.M. Directed differentiation of embryonic stem cells into motor neurons. Cell 2002, 110, 385–397. [Google Scholar] [CrossRef]
- Wilson, L.; Gale, E.; Maden, M. The role of retinoic acid in the morphogenesis of the neural tube. J. Anat. 2003, 203, 357–368. [Google Scholar] [CrossRef]
- England, S.; Batista, M.F.; Mich, J.K.; Chen, J.K.; Lewis, K.E. Roles of hedgehog pathway components and retinoic acid signalling in specifying zebrafish ventral spinal cord neurons. Development 2011, 138, 5121–5134. [Google Scholar] [CrossRef]
- Sockanathan, S.; Jessell, T.M. Motor neuron-derived retinoid signaling specifies the subtype identity of spinal motor neurons. Cell 1998, 94, 503–514. [Google Scholar] [CrossRef]
- Maden, M.; Gale, E.; Kostetskii, I.; Zile, M. Vitamin a-deficient quail embryos have half a hindbrain and other neural defects. Curr. Biol. 1996, 6, 417–426. [Google Scholar] [CrossRef]
- Stavridis, M.P.; Collins, B.J.; Storey, K.G. Retinoic acid orchestrates fibroblast growth factor signalling to drive embryonic stem cell differentiation. Development 2010, 137, 881–890. [Google Scholar] [CrossRef]
- Guth, S.I.; Wegner, M. Having it both ways: Sox protein function between conservation and innovation. Cell. Mol. Life Sci. 2008, 65, 3000–3018. [Google Scholar] [CrossRef]
- Ribes, V.; Stutzmann, F.; Bianchetti, L.; Guillemot, F.; Dolle, P.; Le Roux, I. Combinatorial signalling controls neurogenin2 expression at the onset of spinal neurogenesis. Dev. Biol. 2008, 321, 470–481. [Google Scholar] [CrossRef]
- Rao, M.; Sockanathan, S. Transmembrane protein gde2 induces motor neuron differentiation in vivo. Science 2005, 309, 2212–2215. [Google Scholar] [CrossRef]
- Sabharwal, P.; Lee, C.; Park, S.; Rao, M.; Sockanathan, S. Gde2 regulates subtype-specific motor neuron generation through inhibition of notch signaling. Neuron 2011, 71, 1058–1070. [Google Scholar] [CrossRef]
- Dessaud, E.; McMahon, A.P.; Briscoe, J. Pattern formation in the vertebrate neural tube: A sonic hedgehog morphogen-regulated transcriptional network. Development 2008, 135, 2489–2503. [Google Scholar] [CrossRef]
- Wilson, L.; Gale, E.; Chambers, D.; Maden, M. Retinoic acid and the control of dorsoventral patterning in the avian spinal cord. Dev. Biol. 2004, 269, 433–446. [Google Scholar] [CrossRef]
- Novitch, B.G.; Wichterle, H.; Jessell, T.M.; Sockanathan, S. A requirement for retinoic acid-mediated transcriptional activation in ventral neural patterning and motor neuron specification. Neuron 2003, 40, 81–95. [Google Scholar] [CrossRef]
- Paschaki, M.; Lin, S.C.; Wong, R.L.; Finnell, R.H.; Dolle, P.; Niederreither, K. Retinoic acid-dependent signaling pathways and lineage events in the developing mouse spinal cord. PLoS ONE 2012, 7, e32447. [Google Scholar]
- Briscoe, J.; Pierani, A.; Jessell, T.M.; Ericson, J. A homeodomain protein code specifies progenitor cell identity and neuronal fate in the ventral neural tube. Cell 2000, 101, 435–445. [Google Scholar] [CrossRef]
- Vallstedt, A.; Muhr, J.; Pattyn, A.; Pierani, A.; Mendelsohn, M.; Sander, M.; Jessell, T.M.; Ericson, J. Different levels of repressor activity assign redundant and specific roles to nkx6 genes in motor neuron and interneuron specification. Neuron 2001, 31, 743–755. [Google Scholar] [CrossRef]
- Pierani, A.; Brenner-Morton, S.; Chiang, C.; Jessell, T.M. A sonic hedgehog-independent, retinoid-activated pathway of neurogenesis in the ventral spinal cord. Cell 1999, 97, 903–915. [Google Scholar] [CrossRef]
- Bertrand, N.; Medevielle, F.; Pituello, F. Fgf signalling controls the timing of pax6 activation in the neural tube. Development 2000, 127, 4837–4843. [Google Scholar]
- Oosterveen, T.; Kurdija, S.; Alekseenko, Z.; Uhde, C.W.; Bergsland, M.; Sandberg, M.; Andersson, E.; Dias, J.M.; Muhr, J.; Ericson, J. Mechanistic differences in the transcriptional interpretation of local and long-range shh morphogen signaling. Dev. Cell 2012, 23, 1006–1019. [Google Scholar] [CrossRef]
- Balaskas, N.; Ribeiro, A.; Panovska, J.; Dessaud, E.; Sasai, N.; Page, K.M.; Briscoe, J.; Ribes, V. Gene regulatory logic for reading the sonic hedgehog signaling gradient in the vertebrate neural tube. Cell 2012, 148, 273–284. [Google Scholar] [CrossRef]
- Lee, S.; Lee, B.; Lee, J.W.; Lee, S.K. Retinoid signaling and neurogenin2 function are coupled for the specification of spinal motor neurons through a chromatin modifier cbp. Neuron 2009, 62, 641–654. [Google Scholar] [CrossRef]
- Jacob, J.; Kong, J.; Moore, S.; Milton, C.; Sasai, N.; Gonzalez-Quevedo, R.; Terriente, J.; Imayoshi, I.; Kageyama, R.; Wilkinson, D.G.; et al. Retinoid acid specifies neuronal identity through graded expression of ascl1. Curr. Biol. 2013, 23, 412–418. [Google Scholar] [CrossRef]
- Le Dreau, G.; Marti, E. Dorsal-ventral patterning of the neural tube: A tale of three signals. Dev. Neurobiol. 2012, 72, 1471–1481. [Google Scholar] [CrossRef]
- Zhuang, B.; Sockanathan, S. Dorsal-ventral patterning: A view from the top. Curr. Opin. Neurobiol. 2006, 16, 20–24. [Google Scholar] [CrossRef]
- Martinez-Morales, P.L.; Diez Del Corral, R.; Olivera-Martinez, I.; Quiroga, A.C.; Das, R.M.; Barbas, J.A.; Storey, K.G.; Morales, A.V. Fgf and retinoic acid activity gradients control the timing of neural crest cell emigration in the trunk. J. Cell. Biol. 2011, 194, 489–503. [Google Scholar] [CrossRef] [Green Version]
- Nordstrom, U.; Jessell, T.M.; Edlund, T. Progressive induction of caudal neural character by graded wnt signaling. Nat. Neurosci. 2002, 5, 525–532. [Google Scholar] [CrossRef]
- Patthey, C.; Gunhaga, L.; Edlund, T. Early development of the central and peripheral nervous systems is coordinated by wnt and bmp signals. PLoS ONE 2008, 3, e1625. [Google Scholar] [CrossRef]
- Villanueva, S.; Glavic, A.; Ruiz, P.; Mayor, R. Posteriorization by fgf, wnt, and retinoic acid is required for neural crest induction. Dev. Biol. 2002, 241, 289–301. [Google Scholar] [CrossRef]
- Patthey, C.; Edlund, T.; Gunhaga, L. Wnt-regulated temporal control of bmp exposure directs the choice between neural plate border and epidermal fate. Development 2009, 136, 73–83. [Google Scholar] [CrossRef]
- Le Douarin, N.; Kalcheim, C. The Neural Crest; Cambridge University Press: Cambridge, UK, 1999; p. 445. [Google Scholar]
- Teillet, M.A.; Kalcheim, C.; Le Douarin, N.M. Formation of the dorsal root ganglia in the avian embryo: Segmental origin and migratory behavior of neural crest progenitor cells. Dev. Biol. 1987, 120, 329–347. [Google Scholar] [CrossRef]
- Sela-Donenfeld, D.; Kalcheim, C. Regulation of the onset of neural crest migration by coordinated activity of bmp4 and noggin in the dorsal neural tube. Development 1999, 126, 4749–4762. [Google Scholar]
- Burstyn-Cohen, T.; Stanleigh, J.; Sela-Donenfeld, D.; Kalcheim, C. Canonical wnt activity regulates trunk neural crest delamination linking bmp/noggin signaling with g1/s transition. Development 2004, 131, 5327–5339. [Google Scholar] [CrossRef]
- Sela-Donenfeld, D.; Kalcheim, C. Inhibition of noggin expression in the dorsal neural tube by somitogenesis: A mechanism for coordinating the timing of neural crest emigration. Development 2000, 127, 4845–4854. [Google Scholar]
- Krispin, S.; Nitzan, E.; Kassem, Y.; Kalcheim, C. Evidence for a dynamic spatiotemporal fate map and early fate restrictions of premigratory avian neural crest. Development 2010, 137, 585–595. [Google Scholar] [CrossRef]
- Aquino, J.B.; Lallemend, F.; Marmigere, F.; Adameyko, I.I.; Golemis, E.A.; Ernfors, P. The retinoic acid inducible Cas-family signaling protein nedd9 regulates neural crest cell migration by modulating adhesion and actin dynamics. Neuroscience 2009, 162, 1106–1119. [Google Scholar] [CrossRef]
- Merrill, R.A.; See, A.W.; Wertheim, M.L.; Clagett-Dame, M. Crk-associated substrate (Cas) family member, nedd9, is regulated in human neuroblastoma cells and in the embryonic hindbrain by all-trans retinoic acid. Dev. Dyn. 2004, 231, 564–575. [Google Scholar] [CrossRef]
- Dickman, E.D.; Thaller, C.; Smith, S.M. Temporally-regulated retinoic acid depletion produces specific neural crest, ocular and nervous system defects. Development 1997, 124, 3111–3121. [Google Scholar]
- Lohnes, D.; Mark, M.; Mendelsohn, C.; Dolle, P.; Decimo, D.; LeMeur, M.; Dierich, A.; Gorry, P.; Chambon, P. Developmental roles of the retinoic acid receptors. J. Steroid Biochem. Mol. Biol. 1995, 53, 475–486. [Google Scholar] [CrossRef]
- Niederreither, K.; Vermot, J.; Le Roux, I.; Schuhbaur, B.; Chambon, P.; Dolle, P. The regional pattern of retinoic acid synthesis by raldh2 is essential for the development of posterior pharyngeal arches and the enteric nervous system. Development 2003, 130, 2525–2534. [Google Scholar] [CrossRef]
- Wright-Jin, E.C.; Grider, J.R.; Duester, G.; Heuckeroth, R.O. Retinaldehyde dehydrogenase enzymes regulate colon enteric nervous system structure and function. Dev. Biol. 2013, 381, 28–37. [Google Scholar] [CrossRef]
- Lohnes, D.; Mark, M.; Mendelsohn, C.; Dolle, P.; Dierich, A.; Gorry, P.; Gansmuller, A.; Chambon, P. Function of the retinoic acid receptors (RARs) during development (I). Craniofacial and skeletal abnormalities in RAR double mutants. Development 1994, 120, 2723–2748. [Google Scholar]
- Ghyselinck, N.B.; Dupe, V.; Dierich, A.; Messaddeq, N.; Garnier, J.M.; Rochette-Egly, C.; Chambon, P.; Mark, M. Role of the retinoic acid receptor beta (RARbeta) during mouse development. Int. J. Dev. Biol. 1997, 41, 425–447. [Google Scholar]
- Mark, M.; Ghyselinck, N.B.; Chambon, P. Function of retinoic acid receptors during embryonic development. Nucl. Recept. Signal. 2009, 7, e002. [Google Scholar]
- Minoux, M.; Rijli, F.M. Molecular mechanisms of cranial neural crest cell migration and patterning in craniofacial development. Development 2010, 137, 2605–2621. [Google Scholar] [CrossRef]
- Halilagic, A.; Ribes, V.; Ghyselinck, N.B.; Zile, M.H.; Dolle, P.; Studer, M. Retinoids control anterior and dorsal properties in the developing forebrain. Dev. Biol. 2007, 303, 362–375. [Google Scholar] [CrossRef]
- Halilagic, A.; Zile, M.H.; Studer, M. A novel role for retinoids in patterning the avian forebrain during presomite stages. Development 2003, 130, 2039–2050. [Google Scholar] [CrossRef]
- Liu, J.P.; Laufer, E.; Jessell, T.M. Assigning the positional identity of spinal motor neurons: Rostrocaudal patterning of hox-c expression by fgfs, gdf11, and retinoids. Neuron 2001, 32, 997–1012. [Google Scholar] [CrossRef]
- Oosterveen, T.; Niederreither, K.; Dolle, P.; Chambon, P.; Meijlink, F.; Deschamps, J. Retinoids regulate the anterior expression boundaries of 5' hoxb genes in posterior hindbrain. Embo J. 2003, 22, 262–269. [Google Scholar] [CrossRef]
- Nordstrom, U.; Maier, E.; Jessell, T.M.; Edlund, T. An early role for wnt signaling in specifying neural patterns of cdx and hox gene expression and motor neuron subtype identity. PLoS Biol. 2006, 4, e252. [Google Scholar] [CrossRef]
- Delfino-Machin, M.; Lunn, J.S.; Breitkreuz, D.N.; Akai, J.; Storey, K.G. Specification and maintenance of the spinal cord stem zone. Development 2005, 132, 4273–4283. [Google Scholar] [CrossRef]
- Houle, M.; Prinos, P.; Iulianella, A.; Bouchard, N.; Lohnes, D. Retinoic acid regulation of cdx1: An indirect mechanism for retinoids and vertebral specification. Mol. Cell. Biol. 2000, 20, 6579–6586. [Google Scholar] [CrossRef]
- Alexander, T.; Nolte, C.; Krumlauf, R. Hox genes and segmentation of the hindbrain and axial skeleton. Annu. Rev. Cell Dev. Biol. 2009, 25, 431–456. [Google Scholar] [CrossRef]
- Shimizu, T.; Bae, Y.K.; Hibi, M. Cdx-hox code controls competence for responding to fgfs and retinoic acid in zebrafish neural tissue. Development 2006, 133, 4709–4719. [Google Scholar] [CrossRef]
- Bel-Vialar, S.; Itasaki, N.; Krumlauf, R. Initiating hox gene expression: In the early chick neural tube differential sensitivity to fgf and ra signaling subdivides the hoxb genes in two distinct groups. Development 2002, 129, 5103–5115. [Google Scholar]
- Philippidou, P.; Dasen, J.S. Hox genes: Choreographers in neural development, architects of circuit organization. Neuron 2013, 80, 12–34. [Google Scholar] [CrossRef]
- Ensini, M.; Tsuchida, T.N.; Belting, H.G.; Jessell, T.M. The control of rostrocaudal pattern in the developing spinal cord: Specification of motor neuron subtype identity is initiated by signals from paraxial mesoderm. Development 1998, 125, 969–982. [Google Scholar]
- Misra, M.; Shah, V.; Carpenter, E.; McCaffery, P.; Lance-Jones, C. Restricted patterns of hoxd10 and hoxd11 set segmental differences in motoneuron subtype complement in the lumbosacral spinal cord. Dev. Biol. 2009, 330, 54–72. [Google Scholar] [CrossRef]
- Kania, A.; Jessell, T.M. Topographic motor projections in the limb imposed by lim homeodomain protein regulation of ephrin-a:Epha interactions. Neuron 2003, 38, 581–596. [Google Scholar] [CrossRef]
- Ji, S.J.; Periz, G.; Sockanathan, S. Nolz1 is induced by retinoid signals and controls motoneuron subtype identity through distinct repressor activities. Development 2009, 136, 231–240. [Google Scholar] [CrossRef]
- Amoroso, M.W.; Croft, G.F.; Williams, D.J.; O’Keeffe, S.; Carrasco, M.A.; Davis, A.R.; Roybon, L.; Oakley, D.H.; Maniatis, T.; Henderson, C.E.; et al. Accelerated high-yield generation of limb-innervating motor neurons from human stem cells. J. Neurosci. 2013, 33, 574–586. [Google Scholar] [CrossRef]
- Peljto, M.; Wichterle, H. Programming embryonic stem cells to neuronal subtypes. Curr. Opin. Neurobiol. 2011, 21, 43–51. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Diez del Corral, R.; Morales, A.V. Retinoic Acid Signaling during Early Spinal Cord Development. J. Dev. Biol. 2014, 2, 174-197. https://doi.org/10.3390/jdb2030174
Diez del Corral R, Morales AV. Retinoic Acid Signaling during Early Spinal Cord Development. Journal of Developmental Biology. 2014; 2(3):174-197. https://doi.org/10.3390/jdb2030174
Chicago/Turabian StyleDiez del Corral, Ruth, and Aixa V. Morales. 2014. "Retinoic Acid Signaling during Early Spinal Cord Development" Journal of Developmental Biology 2, no. 3: 174-197. https://doi.org/10.3390/jdb2030174