Systems Modeling at Multiple Levels of Regulation: Linking Systems and Genetic Networks to Spatially Explicit Plant Populations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Current Tools in Evolutionary Biology, Population Genetic and Landscape Genetic Simulation Models
2.1. Fisherian Population Genetics Models
2.2. Landscape Genetics
2.3. Spatially Explicit Individual-Based Models and Their Use in Simulation Studies
2.3.1. Semi-Spatial Models
2.3.2. Spatially Explicit Models
2.4. Resistance Surfaces
3. GRNs, Network Motifs and Inference
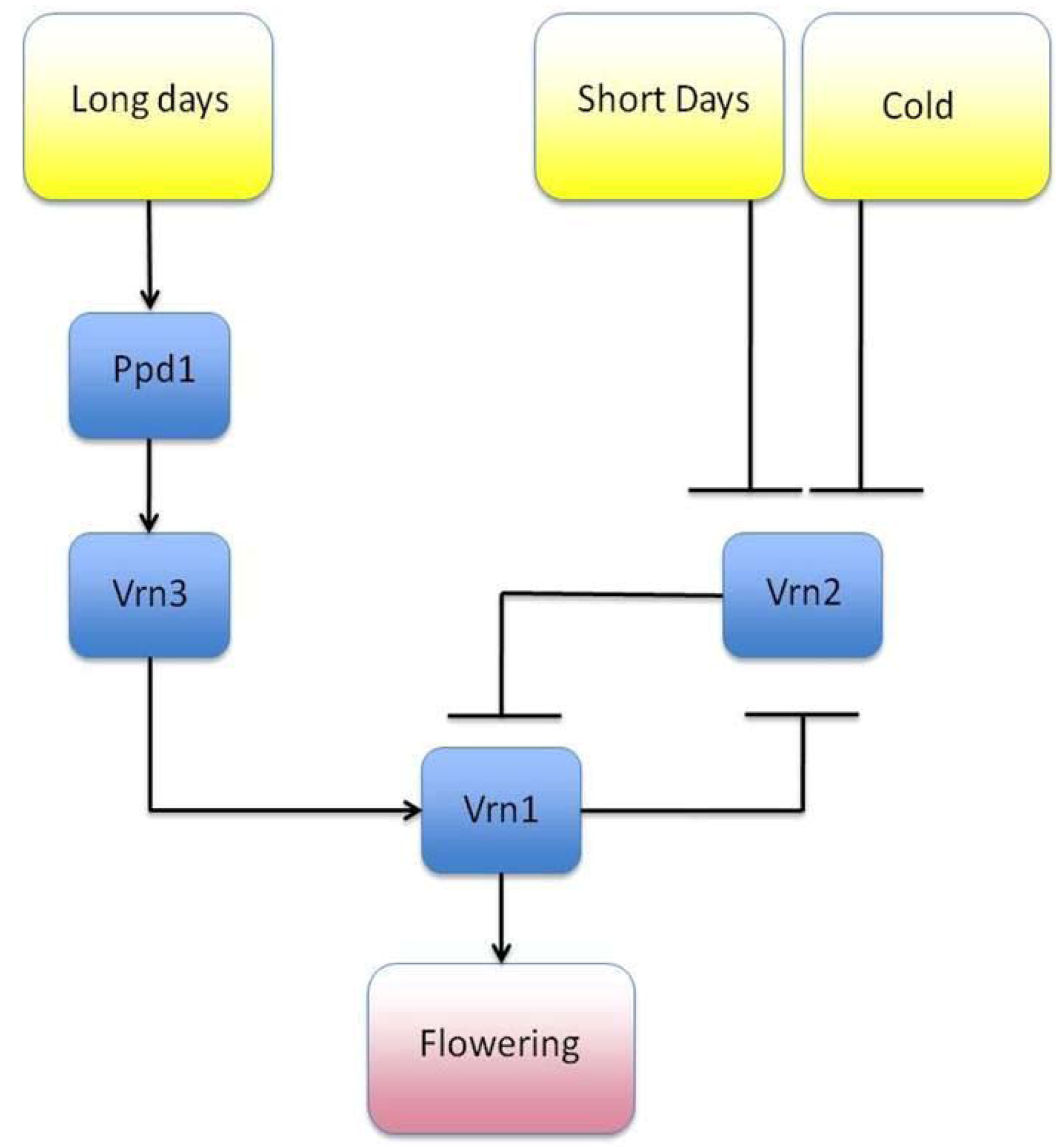
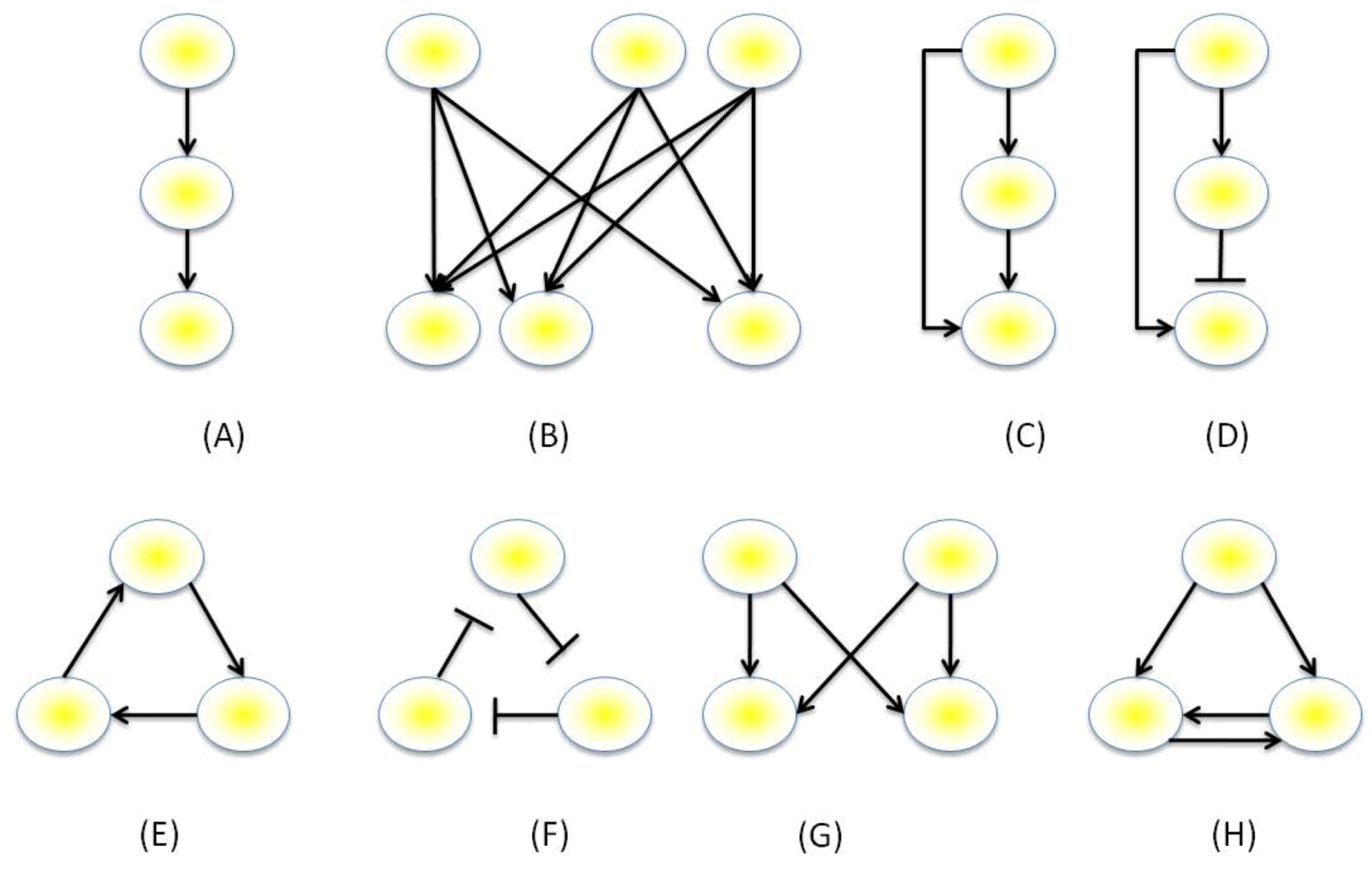
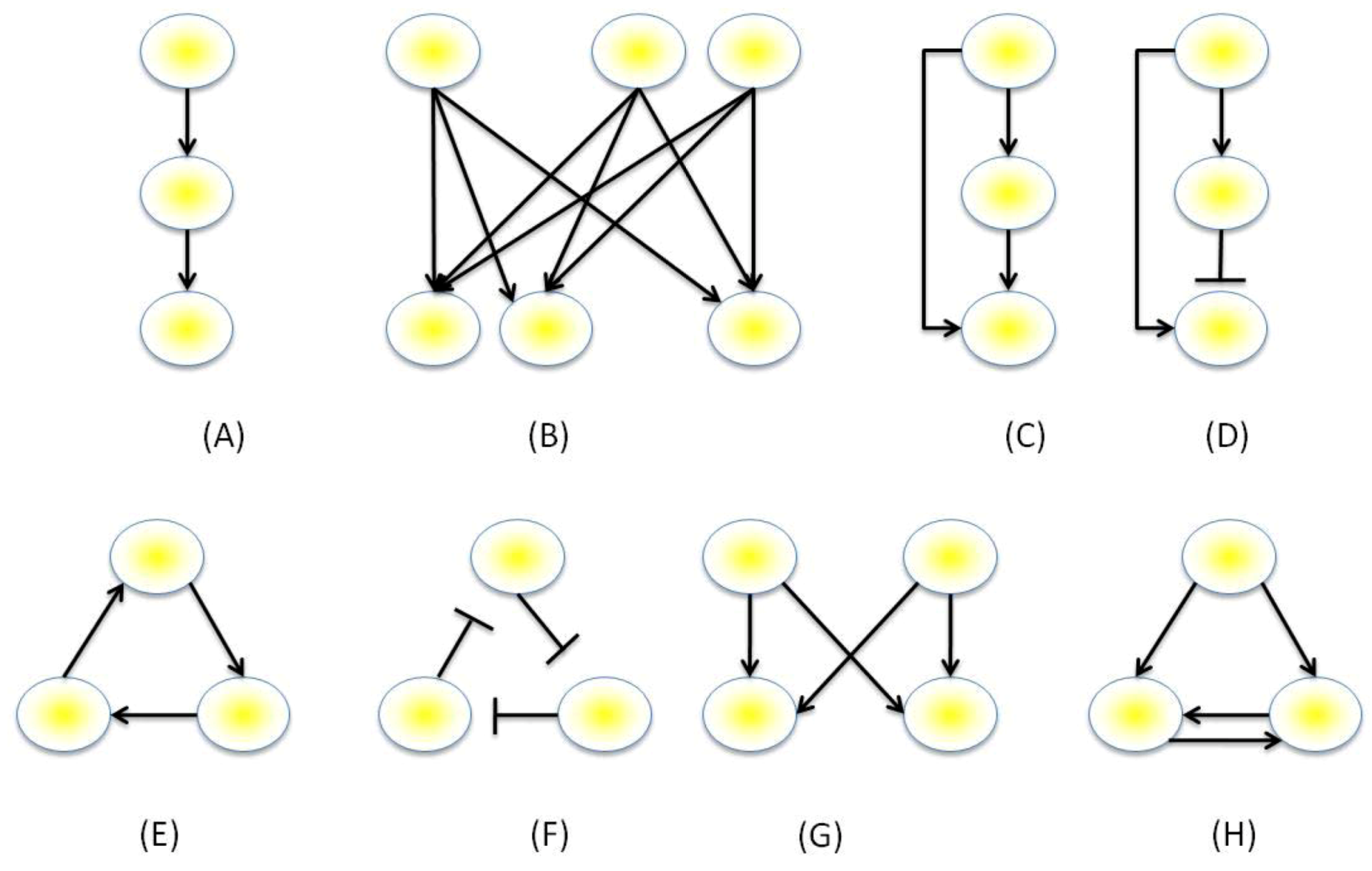
3.1. The GRN Topologies Observed in Nature
3.2. Motif Function
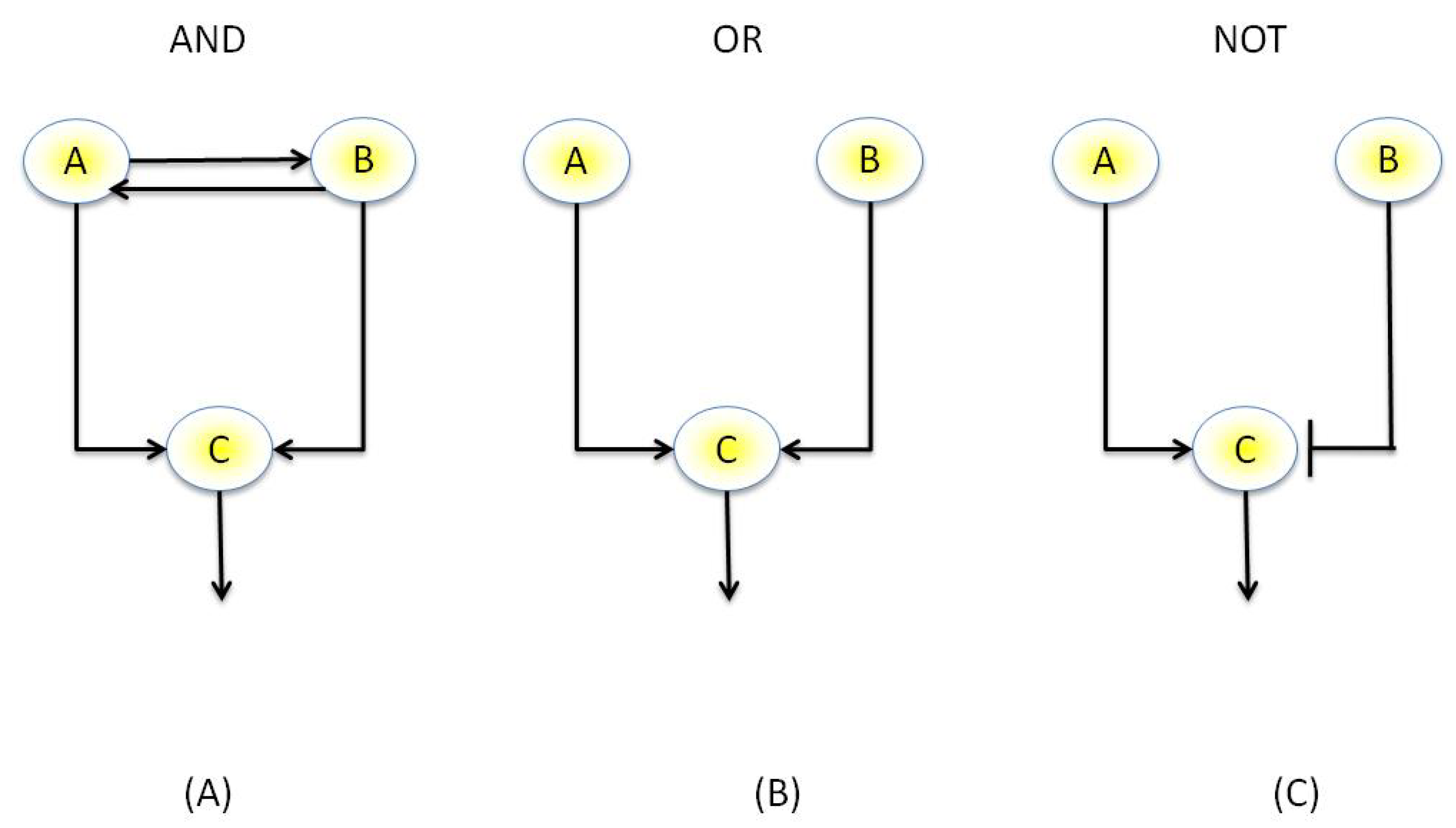
3.3. Mathematical Modeling of GRNs
3.3.1. Boolean Networks
3.3.2. Continuous GRN Models and Bayesian Networks
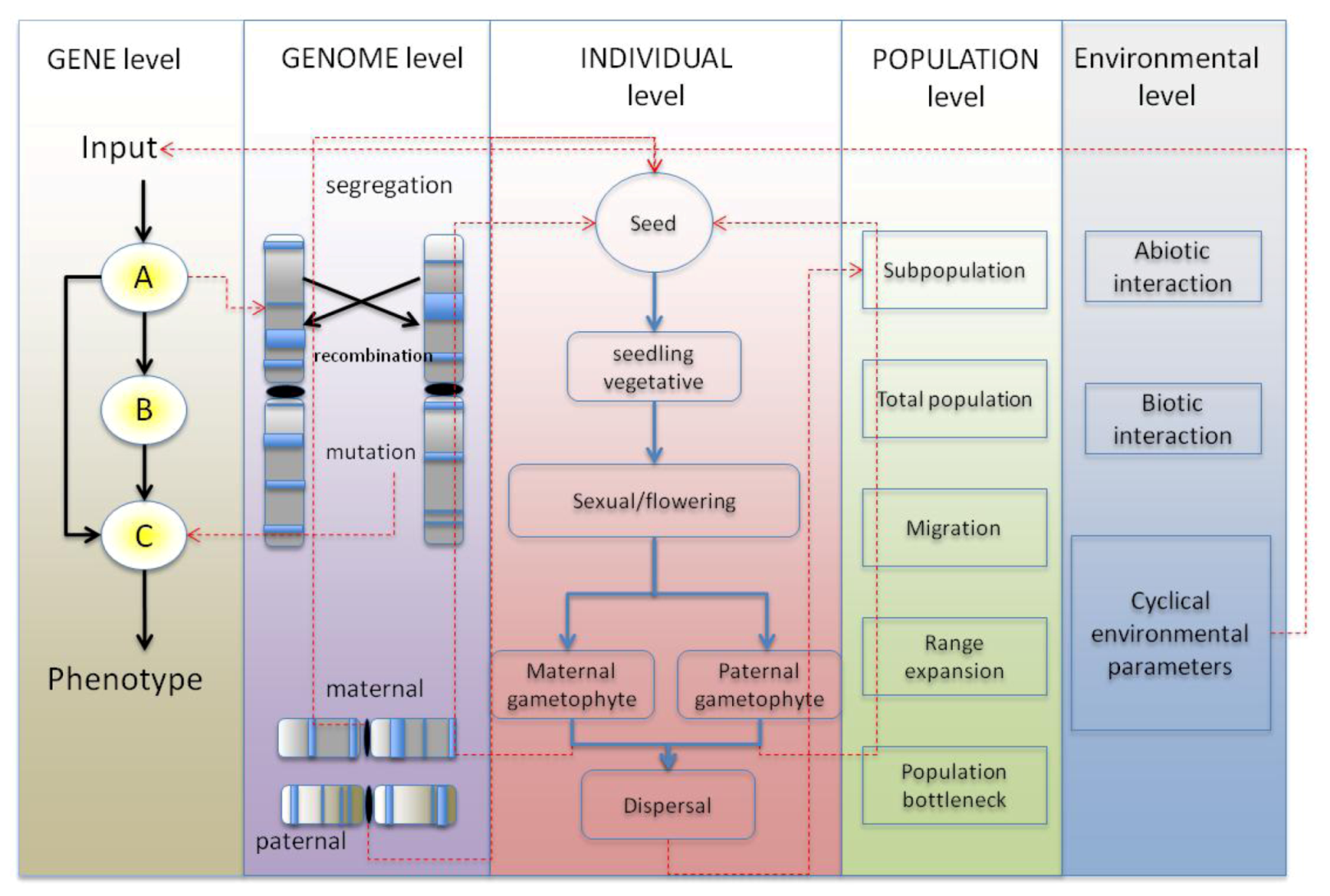
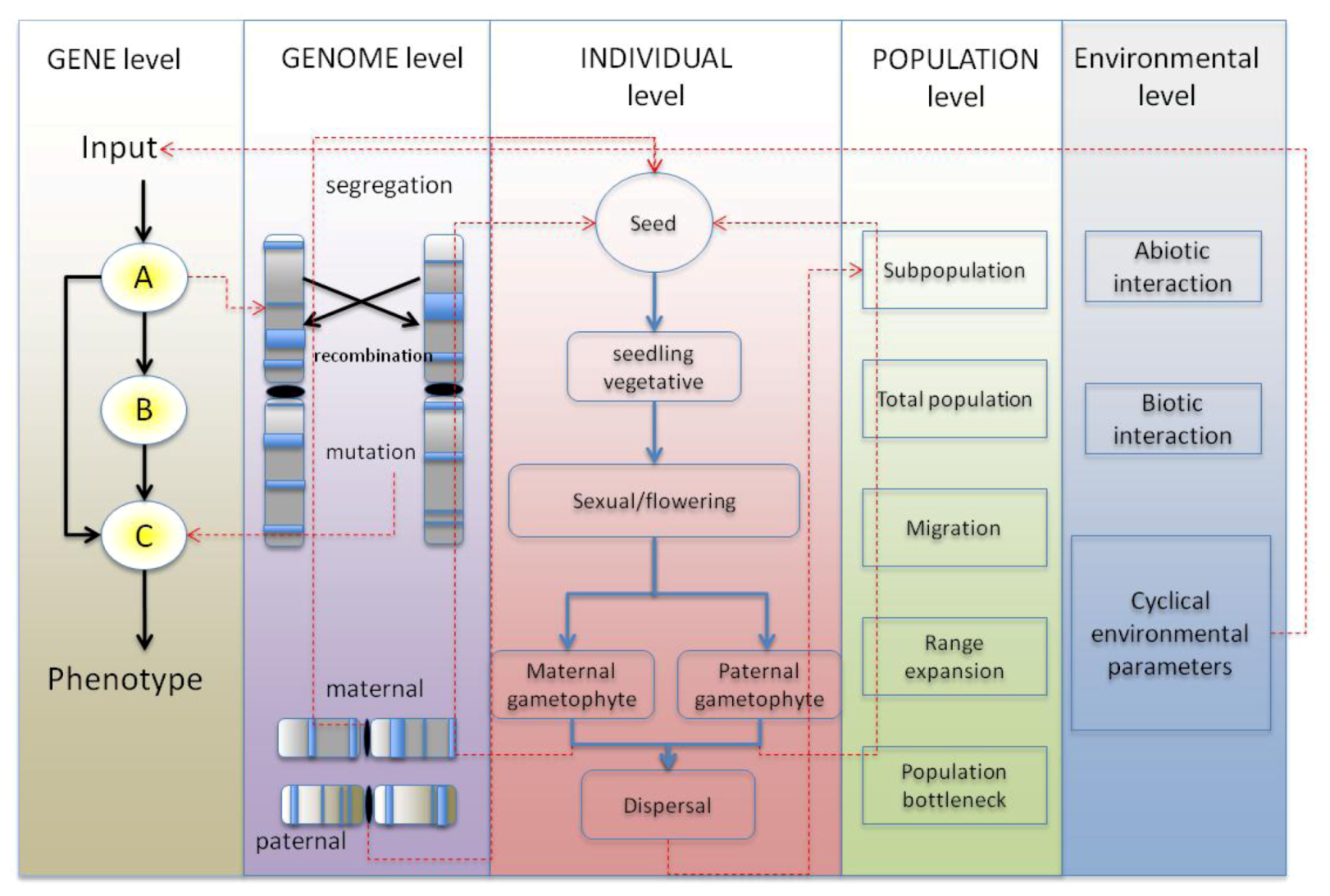
4. Synthesis: Spatial Individual-Based Models with Gene Networks: Approaches, Applications to Plant Science and Potential Pitfalls
- How does a functional (non-neutral) mutation to the sensitivity (as in threshold) or output of a GRN node affect the expressed phenotype or the fitness of an individual? How do the phenotypic effects differ from simulating single non-interacting loci?
- How do perturbations of the edges within a network (such as edge deletion, addition and rewiring) or node duplications impact on the fitness of an individual within different environments?
- How does the conformation of a GRN affect the quantitative trait that is ultimately expressed? Can population models or SIBMs show that certain motifs may be selected for within different environments?
- What role do evolutionary forces such as gene flow and range expansion play on the diversity of GRN topologies?
- Considering the effects of gene flow, can certain environments (i.e., abiotic factors) favor specific GRN topologies? Similarly, can biotic interaction select for certain GRN topologies?
- Which choice of GRN representations (such as static-edge, Boolean, Bayesian, ODE-based networks) is a better fit to the system in question?
4.1. GRN Evolution and the Resulting Phenotypic Effects
4.1.1. Simulating GRNs in Population Models Instead of Quantitative Trait Loci
4.1.2. Simulating Network Evolution
4.1.3. Choice of GRN Model within the Context of a Spatially Explicit Individual-Based Model
4.2. Benefit through Using a Spatially-Explicit System
4.2.1. Biotic Interaction
4.2.2. Analyzing Past and Future Events on Adaptation
4.3. Producing Complex Modeling Systems in a Step-Wise Manner
4.4. Adaptive Dynamics
Simulation of Evolutionary Tradeoffs with GRNs
4.5. Pitfalls
4.5.1. Algorithmic and Programming Complexity
4.5.2. Accurate Representations of GRNs
4.6. Applications to Plant Science
4.6.1. Domestication as a Selection Regime
4.6.2. Simulation Models Accounting for Polyploidy amongst Plants
4.6.3. Functional-Structural Plant Modeling and Efforts in the Simulation of Plant Growth and Morphology
5. Conclusions
Acknowledgments
References
- Fitzgerald, T.L.; Shapter, F.M.; McDonald, S.; Waters, D.L.; Chivers, I.H.; Drenth, A.; Nevo, E.; Henry, R.J. Genome diversity in wild grasses under environmental stress. Proc. Natl. Acad. Sci. USA 2011, 108, 21140–21145. [Google Scholar]
- Nevo, E.; Fu, Y.B.; Pavlicek, T.; Khalifa, S.; Tavasi, M.; Beiles, A. Evolution of wild cereals during 28 years of global warming in Israel. Proc. Natl. Acad. Sci. USA 2012, 109, 3412–3415. [Google Scholar]
- Aschard, H.; Lutz, S.; Maus, B.; Duell, E.J.; Fingerlin, T.E.; Chatterjee, N.; Kraft, P.; van Steen, K. Challenges and opportunities in genome-wide environmental interaction (GWEI) studies. Hum. Genet. 2012, 131, 1591–1613. [Google Scholar] [CrossRef]
- Amato, R.; Pinelli, M.; D’Andrea, D.; Miele, G.; Nicodemi, M.; Raiconi, G.; Cocozza, S. A novel approach to simulate gene-environment interactions in complex diseases. BMC Bioinformatics 2010, 11, 8. [Google Scholar] [CrossRef]
- Pinelli, M.; Scala, G.; Amato, R.; Cocozza, S.; Miele, G. Simulating gene-gene and gene-environment interactions in complex diseases: Gene-Environment iNteraction Simulator 2. BMC Bioinformatics 2012, 13, 132. [Google Scholar] [CrossRef]
- Gunasekera, C.P.; Martin, L.D.; Siddique, K.H.M.; Walton, G.H. Genotype by environment interactions of Indian mustard (Brassica juncea L.) and canola (B. napus L.) in Mediterranean-type environments: 1. Crop growth and seed yield. Eur. J. Agron. 2006, 25, 1–12. [Google Scholar] [CrossRef]
- Helgadottir, A.; Kristjansdottir, T.A. Simple Approach to the Analysis of Gxe Interactions in a Multilocational Spaced Plant Trial with Timothy. Euphytica 1991, 54, 65–73. [Google Scholar] [CrossRef]
- Haji, H.M.; Hunt, L.A. Genotype x environment interactions and underlying environmental factors for winter wheat in Ontario. Can. J. Plant Sci. 1999, 79, 497–505. [Google Scholar] [CrossRef]
- DeLacy, I.H.; Kaul, S.; Rana, B.S.; Cooper, M. Genotypic variation for grain and stover yield of dryland (rabi) sorghum in India: 1. Magnitude of genotype x environment interactions. Field Crops Res. 2010, 118, 228–235. [Google Scholar] [CrossRef]
- Kang, M.S. Using genotype-by-environment interaction for crop cultivar development. Adv. Agron. 1998, 62, 199–252. [Google Scholar] [CrossRef]
- Holderegger, R.; Herrmann, D.; Poncet, B.; Gugerli, F.; Thuiller, W.; Taberlet, P.; Gielly, L.; Rioux, D.; Brodbeck, S.; Aubert, S.; et al. Land ahead: Using genome scans to identify molecular markers of adaptive relevance. Plant Ecol. Div. 2008, 1, 273–283. [Google Scholar] [CrossRef]
- Cox, K.; Broeck, A.V.; van Calster, H.; Mergeay, J. Temperature-related natural selection in a wind-pollinated tree across regional and continental scales. Mol. Ecol. 2011, 20, 2724–2738. [Google Scholar]
- Schuster, S.C. Next-generation sequencing transforms today’s biology. Nat. Methods 2008, 5, 16–18. [Google Scholar] [CrossRef]
- Cannon, C.H.; Kua, C.-S.; Zhang, D.; Harting, J.R. Assembly free comparative genomics of short-read sequence data discovers the needles in the haystack. Mol. Ecol. 2010, 19, 147–161. [Google Scholar] [CrossRef]
- Whittall, J.B.; Syring, J.; Parks, M.; Buenrostro, J.; Dick, C.; Liston, A.; Cronn, R. Finding a (pine) needle in a haystack: Chloroplast genome sequence divergence in rare and widespread pines. Mol. Ecol. 2010, 19, 100–114. [Google Scholar]
- Ferguson, L.; Lee, S.F.; Chamberlain, N.; Nadeau, N.; Joron, M.; Baxter, S.; Wilkinson, P.; Papanicolaou, A.; Kumar, S.; Kee, T.-J.; et al. Characterization of a hotspot for mimicry: Assembly of a butterfly wing transcriptome to genomic sequence at the HmYb/Sb locus. Mol. Ecol. 2010, 19, 240–254. [Google Scholar] [CrossRef]
- Kloch, A.; Babik, W.; Bajer, A.; SiŃSki, E.; Radwan, J. Effects of an MHC-DRB genotype and allele number on the load of gut parasites in the bank vole Myodes glareolus. Mol. Ecol. 2010, 19, 255–265. [Google Scholar] [CrossRef]
- Aparicio, O.; Geisberg, J.V.; Struhl, K. Chromatin Immunoprecipitation for Determining the Association of Proteins with Specific Genomic Sequences in Vivo. Curr. Protoc. Cell Biol. 2004, 23, 17.7.1–17.7.23. [Google Scholar]
- Buck, M.J.; Lieb, J.D. ChIP-chip: Considerations for the design, analysis, and application of genome-wide chromatin immunoprecipitation experiments. Genomics 2004, 83, 349–360. [Google Scholar]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Park, P.J. ChIP-seq: Advantages and challenges of a maturing technology. Nat. Rev. Genet. 2009, 10, 669–680. [Google Scholar] [CrossRef]
- Ferrier, T.; Matus, J.T.; Jin, J.; Riechmann, J.L. Arabidopsis paves the way: Genomic and network analyses in crops. Curr. Opin. Biotechnol. 2011, 22, 260–270. [Google Scholar] [CrossRef]
- Singh, D.; Singh, P.K.; Chaudhary, S.; Mehla, K.; Kumar, S. Chapter Three—Exome Sequencing and Advances in Crop Improvement. In Advances in Genetics; Theodore Friedmann, J.C.D., Stephen, F.G., Eds.; Academic Press: New York, NY, USA, 2012; Volume 79, pp. 87–121. [Google Scholar]
- Jansen, R.C.; Nap, J.P. Genetical genomics: The added value from segregation. Trends Genet. 2001, 17, 388–391. [Google Scholar]
- Hardy, G.H. Mendelian Proportions in a Mixed Population. Science 1908, 28, 49–50. [Google Scholar]
- Barrett, M.D.; Wallace, M.J.; Anthony, J.M. Characterization and Cross Application of Novel Microsatellite Markers for a Rare Sedge, Lepidosperma Gibsonii (Cyperaceae). Am. J. Bot. 2012, 99, E14–E16. [Google Scholar] [CrossRef]
- King, T.L.; Springmann, M.J.; Young, J.A. Tri- and tetra-nucleotide microsatellite DNA markers for assessing genetic diversity, population structure, and demographics in the Holmgren milk-vetch (Astragalus holmgreniorum). Conserv. Genet. Resour. 2012, 4, 39–42. [Google Scholar] [CrossRef]
- Wohrmann, T.; Guicking, D.; Khoshbakht, K.; Weising, K. Genetic variability in wild populations of Prunus divaricata Ledeb. in northern Iran evaluated by EST-SSR and genomic SSR marker analysis. Genet. Resour. Crop Evol. 2011, 58, 1157–1167. [Google Scholar]
- Millar, M.A.; Byrne, M.; Barbour, E. Characterisation of eleven polymorphic microsatellite DNA markers for Australian sandalwood (Santalum spicatum) (R.Br.) A.DC. (Santalaceae). Conserv. Genet. Resour. 2012, 4, 51–53. [Google Scholar] [CrossRef]
- Muir, K.; Byrne, M.; Barbour, E.; Cox, M.C.; Fox, J.E.D. High levels of outcrossing in a family trial of Western Australian sandalwood (Santalum spicatum). Silvae Genetica 2007, 56, 222–230. [Google Scholar]
- Rosenberg, N.A.; Nordborg, M. Genealogical trees, coalescent theory and the analysis of genetic polymorphisms. Nat. Rev. Genet. 2002, 3, 380–390. [Google Scholar]
- O'fallon, B. TreesimJ: A flexible, forward time population genetic simulator. Bioinformatics 2010, 26, 2200–2201. [Google Scholar] [CrossRef]
- Hudson, R.R. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics 2002, 18, 337–338. [Google Scholar] [CrossRef]
- Excoffier, L.; Novembre, J.; Schneider, S. SIMCOAL: A general coalescent program for the simulation of molecular data in interconnected populations with arbitrary demography. J. Hered. 2000, 91, 506–509. [Google Scholar] [CrossRef]
- Laval, G.; Excoffier, L. SIMCOAL 2.0: A program to simulate genomic diversity over large recombining regions in a subdivided population with a complex history. Bioinformatics 2004, 20, 2485–2487. [Google Scholar] [CrossRef]
- Anderson, C.N.K.; Ramakrishnan, U.; Chan, Y.L.; Hadly, E.A. Serial SimCoal: A population genetics model for data from multiple populations and points in time. Bioinformatics 2005, 21, 1733–1734. [Google Scholar] [CrossRef]
- Hellenthal, G.; Stephens, M. msHOT: Modifying Hudson’s ms simulator to incorporate crossover and gene conversion hotspots. Bioinformatics 2007, 23, 520–521. [Google Scholar] [CrossRef]
- Ewing, G.; Hermisson, J. MSMS: A coalescent simulation program including recombination, demographic structure and selection at a single locus. Bioinformatics 2010, 26, 2064–2065. [Google Scholar] [CrossRef]
- Garroway, C.J.; Bowman, J.; Wilson, P.J. Using a genetic network to parameterize a landscape resistance surface for fishers, Martes pennanti. Mol. Ecol. 2011, 20, 3978–3988. [Google Scholar] [CrossRef]
- Manel, S.; Schwartz, M.K.; Luikart, G.; Taberlet, P. Landscape genetics: Combining landscape ecology and population genetics. Trends Ecol. Evol. 2003, 18, 189–197. [Google Scholar] [CrossRef]
- Segelbacher, G.; Cushman, S.A.; Epperson, B.K.; Fortin, M.-J.; Francois, O.; Hardy, O.J.; Holderegger, R.; Taberlet, P.; Waits, L.P.; Manel, S. Applications of landscape genetics in conservation biology: Concepts and challenges. Conserv. Genet. 2010, 11, 375–385. [Google Scholar] [CrossRef]
- Currat, M.; Ray, N.; Excoffier, L. splatche: A program to simulate genetic diversity taking into account environmental heterogeneity. Mol. Ecol. Notes 2004, 4, 139–142. [Google Scholar] [CrossRef]
- Ray, N.; Currat, M.; Foll, M.; Excoffier, L. SPLATCHE2: A spatially explicit simulation framework for complex demography, genetic admixture and recombination. Bioinformatics 2010, 26, 2993–2994. [Google Scholar] [CrossRef]
- Hamilton, G.; Currat, M.; Ray, N.; Heckel, G.; Beaumont, M.A.; Excoffier, L. Bayesian estimation of recent migration rates after a spatial expansion. Genetics 2005, 170, 409–417. [Google Scholar] [CrossRef]
- Klopfstein, S.; Currat, M.; Excoffier, L. The Fate of Mutations Surfing on the Wave of a Range Expansion. Mol. Biol. Evol. 2006, 23, 482–490. [Google Scholar]
- Van Etten, J.; Hijmans, R.J. A geospatial modelling approach integrating archaeobotany and genetics to trace the origin and dispersal of domesticated plants. PLoS One 2010, 5, e12060. [Google Scholar] [CrossRef]
- Itan, Y.; Powell, A.; Beaumont, M.A.; Burger, J.; Thomas, M.G. The Origins of Lactase Persistence in Europe. PLoS Comput. Biol. 2009, 5, e1000491. [Google Scholar] [CrossRef]
- Platt, A.; Horton, M.; Huang, Y.S.; Li, Y.; Anastasio, A.E.; Mulyati, N.W.; Agren, J.; Bossdorf, O.; Byers, D.; Donohue, K.; et al. The scale of population structure in Arabidopsis thaliana. PLoS Genet. 2010, 6, e1000843. [Google Scholar] [CrossRef] [Green Version]
- Jones, H.; Leigh, F.J.; Mackay, I.; Bower, M.A.; Smith, L.M.J.; Charles, M.P.; Jones, G.; Jones, M.K.; Brown, T.A.; Powell, W. Population-Based Resequencing Reveals That the Flowering Time Adaptation of Cultivated Barley Originated East of the Fertile Crescent. Mol. Biol. Evol. 2008, 25, 2211–2219. [Google Scholar] [CrossRef]
- Carvajal-Rodriguez, A. Simulation of genomes: A review. Curr. Genomics 2008, 9, 155–159. [Google Scholar] [CrossRef]
- Carvajal-Rodriguez, A. Simulation of Genes and Genomes Forward in Time. Curr. Genomics 2010, 11, 58–61. [Google Scholar]
- Balloux, F. EASYPOP (version 1.7): A computer program for population genetics simulations. J. Hered. 2001, 92, 301–302. [Google Scholar] [CrossRef]
- Leblois, R.; Estoup, A.; Rousset, F. IBDSim: A computer program to simulate genotypic data under isolation by distance. Mol. Ecol. Resour. 2009, 9, 107–109. [Google Scholar] [CrossRef]
- Neuenschwander, S.; Hospital, F.; Guillaume, F.; Goudet, J. quantiNemo: An individual-based program to simulate quantitative traits with explicit genetic architecture in a dynamic metapopulation. Bioinformatics 2008, 24, 1552–1553. [Google Scholar] [CrossRef]
- Peng, B.; Amos, C.I. Forward-time simulations of non-random mating populations using simuPOP. Bioinformatics 2008, 24, 1408–1409. [Google Scholar] [CrossRef]
- Peng, B.; Kimmel, M. simuPOP: A forward-time population genetics simulation environment. Bioinformatics 2005, 21, 3686–3687. [Google Scholar] [CrossRef]
- Carvajal-Rodriguez, A. GENOMEPOP: A program to simulate genomes in populations. BMC Bioinformatics 2008, 9, 223. [Google Scholar] [CrossRef]
- Epperson, B.K.; McRae, B.H.; Scribner, K.; Cushman, S.A.; Rosenberg, M.S.; Fortin, M.J.; James, P.M.; Murphy, M.; Manel, S.; Legendre, P.; et al. Utility of computer simulations in landscape genetics. Mol. Ecol. 2010, 19, 3549–3564. [Google Scholar] [CrossRef]
- Doligez, A.; Baril, C.; Joly, H.I. Fine-scale spatial genetic structure with nonuniform distribution of individuals. Genetics 1998, 148, 905–919. [Google Scholar]
- Kitchen, J.L.; Allaby, R.G. The Limits of Mean-Field Heterozygosity Estimates under Spatial Extension in Simulated Plant Populations. PLoS One 2012, 7, e43254. [Google Scholar] [CrossRef]
- Kuparinen, A.; Schurr, F.M. A flexible modelling framework linking the spatio-temporal dynamics of plant genotypes and populations: Application to gene flow from transgenic forests. Ecol. Modell. 2007, 202, 476–486. [Google Scholar] [CrossRef]
- Beckie, H.J.; Hall, L.M. Simple to complex: Modelling crop pollen-mediated gene flow. Plant Sci. 2008, 175, 615–628. [Google Scholar] [CrossRef]
- McRae, B.H. Isolation by resistance. Evolution 2006, 60, 1551–1561. [Google Scholar]
- Spear, S.F.; Balkenhol, N.; Fortin, M.J.; McRae, B.H.; Scribner, K. Use of resistance surfaces for landscape genetic studies: Considerations for parameterization and analysis. Mol. Ecol. 2010, 19, 3576–3591. [Google Scholar] [CrossRef]
- Landguth, E.L.; Cushman, S.A. cdpop: A spatially explicit cost distance population genetics program. Mol. Ecol. Resour. 2010, 10, 156–161. [Google Scholar] [CrossRef]
- Landguth, E.L.; Cushman, S.A.; Johnson, N.A. Simulating natural selection in landscape genetics. Mol. Ecol. Resour. 2012, 12, 363–368. [Google Scholar] [CrossRef]
- Landguth, E.; Balkenhol, N. Relative sensitivity of neutral versus adaptive genetic data for assessing population differentiation. Conserv. Genet. 2012, 13, 1421–1426. [Google Scholar] [CrossRef]
- McRae, B.H.; Dickson, B.G.; Keitt, T.H.; Shah, V.B. Using circuit theory to model connectivity in ecology, evolution, and conservation. Ecology 2008, 89, 2712–2724. [Google Scholar] [CrossRef]
- Shah, V.; McRae, B. Circuitscape: A Tool for Landscape Ecology. In Proceedings of the 7th Python in Science Conference (SciPy), Pasadena, CA, USA, 19–24 August 2008; Varoquaux, G., Millman, J., Vaught, T., Eds.; pp. 62–65.
- Pigliucci, M. Genotype-phenotype mapping and the end of the “genes as blueprint” metaphor. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2010, 365, 557–566. [Google Scholar] [CrossRef]
- Fuller, D.Q.; Allaby, R. Seed Dispersal and Crop Domestication: Shattering, Germination and Seasonality in Evolution under Cultivation. In Annual Plant Reviews; Fruit Development and Seed Dispersal, Østergaard, L., Ed.; Wiley-Blackwell: Oxford, UK, 2009; Volume 38, pp. 238–295. [Google Scholar]
- Jeong, H.; Tombor, B.; Albert, R.; Oltvai, Z.N.; Barabasi, A.L. The large-scale organization of metabolic networks. Nature 2000, 407, 651–654. [Google Scholar] [CrossRef]
- Bork, P.; Jensen, L.J.; von Mering, C.; Ramani, A.K.; Lee, I.; Marcotte, E.M. Protein interaction networks from yeast to human. Curr. Opin. Struct. Biol. 2004, 14, 292–299. [Google Scholar] [CrossRef]
- Hecker, M.; Lambeck, S.; Toepfer, S.; van Someren, E.; Guthke, R. Gene regulatory network inference: Data integration in dynamic models—A review. Biosystems 2009, 96, 86–103. [Google Scholar] [CrossRef]
- Smolen, P.; Baxter, D.A.; Byrne, J.H. Mathematical modeling of gene networks. Neuron 2000, 26, 567–580. [Google Scholar] [CrossRef]
- Milo, R.; Shen-Orr, S.; Itzkovitz, S.; Kashtan, N.; Chklovskii, D.; Alon, U. Network motifs: Simple building blocks of complex networks. Science 2002, 298, 824–827. [Google Scholar] [CrossRef]
- Milo, R.; Itzkovitz, S.; Kashtan, N.; Levitt, R.; Shen-Orr, S.; Ayzenshtat, I.; Sheffer, M.; Alon, U. Superfamilies of evolved and designed networks. Science 2004, 303, 1538–1542. [Google Scholar] [CrossRef]
- Artzy-Randrup, Y.; Fleishman, S.J.; Ben-Tal, N.; Stone, L. Comment on “Network motifs: Simple building blocks of complex networks” and “Superfamilies of evolved and designed networks”. Science 2004, 305, 1107. [Google Scholar]
- Mangan, S.; Zaslaver, A.; Alon, U. The Coherent Feedforward Loop Serves as a Sign-sensitive Delay Element in Transcription Networks. J. Mol. Biol. 2003, 334, 197–204. [Google Scholar] [CrossRef]
- Kalir, S.; Mangan, S.; Alon, U. A coherent feed-forward loop with a SUM input function prolongs flagella expression in Escherichia coli. Mol. Syst. Biol. 2005. [Google Scholar] [CrossRef]
- Basu, S.; Mehreja, R.; Thiberge, S.; Chen, M.T.; Weiss, R. Spatiotemporal control of gene expression with pulse-generating networks. Proc. Natl. Acad. Sci. USA 2004, 101, 6355–6360. [Google Scholar] [CrossRef]
- Prill, R.J.; Iglesias, P.A.; Levchenko, A. Dynamic properties of network motifs contribute to biological network organization. PLoS Biol. 2005, 3, e343. [Google Scholar] [CrossRef]
- Widder, S.; Sole, R.; Macia, J. Evolvability of feed-forward loop architecture biases its abundance in transcription networks. BMC Syst. Biol. 2012, 6, 7. [Google Scholar] [CrossRef]
- Ingram, P.J.; Stumpf, M.P.; Stark, J. Network motifs: Structure does not determine function. BMC Genomics 2006, 7, 108. [Google Scholar]
- Konagurthu, A.S.; Lesk, A.M. On the origin of distribution patterns of motifs in biological networks. BMC Syst. Biol. 2008, 2, 73. [Google Scholar] [CrossRef]
- Kauffman, S.A. Metabolic stability and epigenesis in randomly constructed genetic nets. J. Theor. Biol. 1969, 22, 437–467. [Google Scholar] [CrossRef]
- Liang, S.; Fuhrman, S.; Somogyi, R. Reveal, a general reverse engineering algorithm for inference of genetic network architectures. Pac. Symp. Biocomput. 1998, 1998, 18–29. [Google Scholar]
- Thomas, R. Regulatory networks seen as asynchronous automata: A logical description. J. Theor. Biol. 1991, 153, 1–23. [Google Scholar] [CrossRef]
- Shmulevich, I.; Dougherty, E.R.; Kim, S.; Zhang, W. Probabilistic Boolean networks: A rule-based uncertainty model for gene regulatory networks. Bioinformatics 2002, 18, 261–274. [Google Scholar] [CrossRef]
- Liang, J.; Han, J. Stochastic Boolean networks: An efficient approach to modeling gene regulatory networks. BMC Syst. Biol. 2012, 6, 113. [Google Scholar] [CrossRef]
- Deng, X.; Geng, H.; Ali, H. EXAMINE: A computational approach to reconstructing gene regulatory networks. Biosystems 2005, 81, 125–136. [Google Scholar] [CrossRef]
- Friedman, N.; Linial, M.; Nachman, I.; Pe'er, D. Using Bayesian networks to analyze expression data. In Proceedings of the Fourth Annual International Conference on Computational Molecular Biology; ACM: Tokyo, Japan, 2000; pp. 127–135. [Google Scholar]
- Van Berlo, R.J.P.; van Someren, E.P.; Reinders, M.J.T. Studying the Conditions for Learning Dynamic Bayesian Networks to Discover Genetic Regulatory Networks. Simulation 2003, 79, 689–702. [Google Scholar]
- Hartemink, A.; Gifford, D.; Jaakkola, T.; Young, R. Using Graphical Models and Genomic Expression Data to Statistically Validate Models of Genetic Regulatory Networks, Pacific Symposium on Biocomputing; Altman, R., Dunker, K., Hunker, L., Eds.; World Scientific Publishing: Stanford, CA, USA, 2001; pp. 422–433. [Google Scholar]
- Prud'homme, B.; Gompel, N.; Carroll, S.B. Emerging principles of regulatory evolution. Proc. Natl. Acad. Sci. USA 2007, 104, 8605–8612. [Google Scholar] [CrossRef]
- Stumpf, M.P.H.; Kelly, W.P.; Thorne, T.; Wiuf, C. Evolution at the system level: The natural history of protein interaction networks. Trends Ecol. Evol. 2007, 22, 366–373. [Google Scholar] [CrossRef]
- Chouard, T. Darwin 200: Beneath the surface. Nature 2008, 456, 300–303. [Google Scholar] [CrossRef]
- Stern, D.L.; Orgogozo, V. Is genetic evolution predictable? Science 2009, 323, 746–751. [Google Scholar] [CrossRef]
- Lander, E.S. Initial impact of the sequencing of the human genome. Nature 2011, 470, 187–197. [Google Scholar] [CrossRef]
- Allaby, R. Integrating the processes in the evolutionary system of domestication. J. Exp. Bot. 2010, 61, 935–944. [Google Scholar] [CrossRef]
- Knight, C.G.; Pinney, J.W. Making the right connections: Biological networks in the light of evolution. Bioessays 2009, 31, 1080–1090. [Google Scholar] [CrossRef]
- Fischer, A.H.; Smith, J. Evo-devo in the era of gene regulatory networks. Integr. Comp. Biol. 2012, 52, 842–849. [Google Scholar] [CrossRef]
- Muller, G.B. Evo-devo: Extending the evolutionary synthesis. Nat. Rev. Genet. 2007, 8, 943–949. [Google Scholar]
- Flannick, J.; Novak, A.; Do, C.B.; Srinivasan, B.S.; Batzoglou, S. Automatic parameter learning for multiple local network alignment. J. Comput. Biol. 2009, 16, 1001–1022. [Google Scholar] [CrossRef]
- Kolar, M.; Meier, J.; Mustonen, V.; Lassig, M.; Berg, J. GraphAlignment: Bayesian pairwise alignment of biological networks. BMC Syst. Biol. 2012, 6, 144. [Google Scholar] [CrossRef]
- Knight, C.G.; Zitzmann, N.; Prabhakar, S.; Antrobus, R.; Dwek, R.; Hebestreit, H.; Rainey, P.B. Unraveling adaptive evolution: How a single point mutation affects the protein coregulation network. Nat. Genet. 2006, 38, 1015–1022. [Google Scholar] [CrossRef]
- Ohno, S. Evolution by Gene Duplication; Springer: New York, NY, USA, 1970. [Google Scholar]
- Farid, N.; Christensen, K. Evolving networks through deletion and duplication. New J. Phys. 2006, 8, 212. [Google Scholar] [CrossRef]
- Evlampiev, K.; Isambert, H. Modeling protein network evolution under genome duplication and domain shuffling. BMC Syst. Biol. 2007, 1, 49. [Google Scholar]
- Tsai, T.Y.C.; Choi, Y.S.; Ma, W.Z.; Pomerening, J.R.; Tang, C.; Ferrell, J.E. Robust, tunable biological oscillations from interlinked positive and negative feedback loops. Science 2008, 321, 126–129. [Google Scholar] [CrossRef]
- Zhang, Z.H.; Qian, W.F.; Zhang, J.Z. Positive selection for elevated gene expression noise in yeast. Mol. Syst. Biol. 2009, 5, 299. [Google Scholar]
- Schumacher, M.A.; Piro, K.M.; Xu, W.; Hansen, S.; Lewis, K.; Brennan, R.G. Molecular Mechanisms of HipA-Mediated Multidrug Tolerance and Its Neutralization by HipB. Science 2009, 323, 396–401. [Google Scholar] [CrossRef]
- Koh, R.; Dunlop, M. Modeling suggests that gene circuit architecture controls phenotypic variability in a bacterial persistence network. BMC Syst. Biol. 2012, 6, 47. [Google Scholar]
- Tsong, A.E.; Tuch, B.B.; Li, H.; Johnson, A.D. Evolution of alternative transcriptional circuits with identical logic. Nature 2006, 443, 415–420. [Google Scholar]
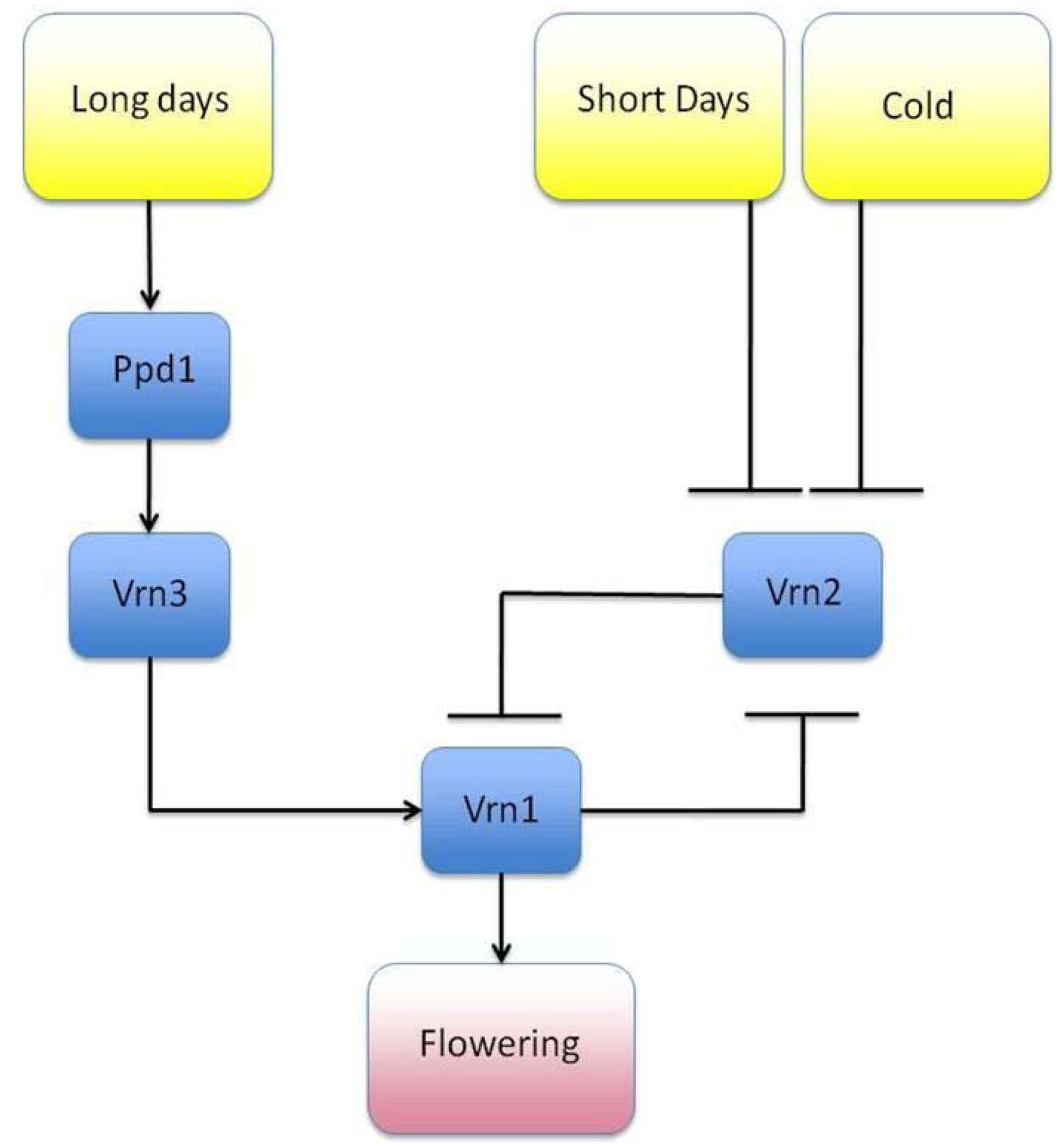
- Song, Y.H.; Ito, S.; Imaizumi, T. Similarities in the circadian clock and photoperiodism in plants. Curr. Opin. Plant Biol. 2010, 13, 594–603. [Google Scholar] [CrossRef]
- Fischer, A.G. Latitudinal Variations in Organic Diversity. Evolution 1960, 14, 64–81. [Google Scholar] [CrossRef]
- Schemske, D.W.; Mittelbach, G.G.; Cornell, H.V.; Sobel, J.M.; Roy, K. Is There a Latitudinal Gradient in the Importance of Biotic Interactions? Annu. Rev. Ecol. Evol. Syst. 2009, 40, 245–269. [Google Scholar] [CrossRef]
- Connel, J.H. On the role of natural enemies in preventing competitive exclusion in some marine animals and in rain forest trees. In Dynamics of Population; den Boer, P.J., Gradwell, G.R., Eds.; Cent. Agric.: Wageningen, The Netherlands, 1971; pp. 298–312. [Google Scholar]
- Janzen, D.H. Herbivores and the Number of Tree Species in Tropical Forests. Am. Nat. 1970, 104, 501–528. [Google Scholar]
- Beaumont, M.A.; Zhang, W.Y.; Balding, D.J. Approximate Bayesian computation in population genetics. Genetics 2002, 162, 2025–2035. [Google Scholar]
- Estoup, A.; Lombaert, E.; Marin, J.M.; Guillemaud, T.; Pudlo, P.; Robert, C.P.; Cornuet, J.M. Estimation of demo-genetic model probabilities with Approximate Bayesian Computation using linear discriminant analysis on summary statistics. Mol. Ecol. Resour. 2012, 12, 846–855. [Google Scholar]
- Itan, Y.; Powell, A.; Beaumont, M.A.; Burger, J.; Thomas, M.G. The Origins of Lactase Persistence in Europe. PLoS Comput. Biol. 2009, 5, e1000491. [Google Scholar] [CrossRef] [Green Version]
- Williams, G.C. Pleiotropy, Natural-Selection, and the Evolution of Senescence. Evolution 1957, 11, 398–411. [Google Scholar] [CrossRef]
- Cheverud, J.M. Developmental integration and the evolution of pleiotropy. Am. Zool. 1996, 36, 44–50. [Google Scholar]
- Elena, S.F.; Sanjuan, R. Climb every mountain? Science 2003, 302, 2074–2075. [Google Scholar] [CrossRef]
- Van Drunen, W.E.; Dorken, M.E. Trade-offs between clonal and sexual reproduction in Sagittaria latifolia (Alismataceae) scale up to affect the fitness of entire clones. New Phytol. 2012, 196, 606–616. [Google Scholar] [CrossRef]
- 127 Kalske, A.; Muola, A.; Laukkanen, L.; Mutikainen, P.; Leimu, R. Variation and constraints of local adaptation of a long-lived plant, its pollinators and specialist herbivores. J. Ecol. 2012, 100, 1359–1372. [Google Scholar]
- Freitak, D.; Wheat, C.W.; Heckel, D.G.; Vogel, H. Immune system responses and fitness costs associated with consumption of bacteria in larvae of Trichoplusia ni. BMC Biol. 2007, 5, 56. [Google Scholar] [CrossRef]
- Hollister, J.D.; Gaut, B.S. Epigenetic silencing of transposable elements: A trade-off between reduced transposition and deleterious effects on neighboring gene expression. Genome Res. 2009, 19, 1419–1428. [Google Scholar] [CrossRef]
- Jacobs, M.W.; Sherrard, K.M. Bigger is not always better: Offspring size does not predict growth or survival for seven ascidian species. Ecology 2010, 91, 3598–3608. [Google Scholar]
- Denison, R.F. Past evolutionary tradeoffs represent opportunities for crop genetic improvement and increased human lifespan. Evol. Appl. 2011, 4, 216–224. [Google Scholar] [CrossRef]
- Sanchez-Humanes, B.; Sork, V.L.; Espelta, J.M. Trade-offs between vegetative growth and acorn production in Quercus lobata during a mast year: The relevance of crop size and hierarchical level within the canopy. Oecologia 2011, 166, 101–110. [Google Scholar] [CrossRef]
- Friesen, M.L. Widespread fitness alignment in the legume-rhizobium symbiosis. New Phytol. 2012, 194, 1096–1111. [Google Scholar] [CrossRef]
- Moon, Y.H.; Chen, L.J.; Pan, R.L.; Chang, H.S.; Zhu, T.; Maffeo, D.M.; Sung, Z.R. EMF genes maintain vegetative development by repressing the flower program in Arabidopsis (vol 15, pg 681, 2003). Plant Cell 2003, 15, 1257–1257. [Google Scholar] [CrossRef]
- Dieckmann, U. Can adaptive dynamics invade? Trends Ecol. Evol. 1997, 12, 128–131. [Google Scholar] [CrossRef]
- Geritz, S.A.H.; Gyllenberg, M. Seven answers from adaptive dynamics. J. Evol. Biol. 2005, 18, 1174–1177. [Google Scholar] [CrossRef]
- Geritz, S.A.H.; van der Meijden, E.; Metz, J.A.J. Evolutionary dynamics of seed size and seedling competitive ability. Theor. Popul. Biol. 1999, 55, 324–343. [Google Scholar]
- Boudsocq, S.; Barot, S.; Loeuille, N. Evolution of nutrient acquisition: When adaptation fills the gap between contrasting ecological theories. Proc. R. Soc. B Biol. Sci. 2011, 278, 449–457. [Google Scholar] [CrossRef]
- Grimm, V.; Berger, U.; Bastiansen, F.; Eliassen, S.; Ginot, V.; Giske, J.; Goss-Custard, J.; Grand, T.; Heinz, S.K.; Huse, G.; et al. A standard protocol for describing individual-based and agent-based models. Ecol. Modell. 2006, 198, 115–126. [Google Scholar] [CrossRef]
- Finkel, R.A.; Bentley, J.L. Quad trees: A data structure for retrieval on composite keys. Acta Informatica 1974, 4, 1–9. [Google Scholar] [CrossRef]
- Tischendorf, L. Modelling individual movements in heterogeneous landscapes: Potentials of a new approach. Ecol. Modell. 1997, 103, 33–42. [Google Scholar] [CrossRef]
- Sommerville, I. Software Engineering, 6th ed; Addison Wesley: Harlow, UK, 2007. [Google Scholar]
- Kool, J.T. An object-oriented, individual-based approach for simulating the dynamics of genes in subdivided populations. Ecol. Inform. 2009, 4, 136–146. [Google Scholar] [CrossRef]
- Bian, L. Object-Oriented Representation of Environmental Phenomena: Is Everything Best Represented as an Object? Ann. Assoc. Am. Geogr. 2007, 97, 267–281. [Google Scholar] [CrossRef]
- Barnes, D.J.; Hopkins, T.R. The impact of programming paradigms on the efficiency of an individual-based simulation model. Simul. Modell. Pract. Theory 2003, 11, 557–569. [Google Scholar] [CrossRef]
- Abbo, S.; Lev-Yadun, S.; Gopher, A. Origin of Near Eastern plant domestication: Homage to Claude Levi-Strauss and “La Pensee Sauvage”. Genet. Resour. Crop Evol. 2011, 58, 175–179. [Google Scholar] [CrossRef]
- Ross-Ibarra, J.; Morrell, P.L.; Gaut, B.S. Plant domestication, a unique opportunity to identify the genetic basis of adaptation. Proc. Natl. Acad. Sci. USA 2007, 104, 8641–8648. [Google Scholar] [CrossRef]
- Fuller, D.Q.; Allaby, R.G.; Stevens, C. Domestication as innovation: The entanglement of techniques, technology and chance in the domestication of cereal crops. World Archaeol. 2010, 42, 13–28. [Google Scholar]
- Brown, T.A.; Jones, M.K.; Powell, W.; Allaby, R.G. The complex origins of domesticated crops in the Fertile Crescent. Trends Ecol. Evol. 2009, 24, 103–109. [Google Scholar] [CrossRef]
- Munguía-Rosas, M.A.; Ollerton, J.; Parra-Tabla, V.; de-Nova, J.A. Meta-analysis of phenotypic selection on flowering phenology suggests that early flowering plants are favoured. Ecol. Lett. 2011, 14, 511–521. [Google Scholar] [CrossRef]
- Leinonen, P.H.; Remington, D.L.; Leppala, J.; Savolainen, O. Genetic basis of local adaptation and flowering time variation in Arabidopsis lyrata. Mol. Ecol. 2013, 22, 709–723. [Google Scholar] [CrossRef]
- Streck, N.A. A vernalization model in onion (Allium cepa L.). Revista Brasileira de Agrociência 2003, 10, 99–105. [Google Scholar]
- Finch-Savage, W.E.; Leubner-Metzger, G. Seed dormancy and the control of germination. New Phytol. 2006, 171, 501–523. [Google Scholar]
- White, C.N.; Proebsting, W.M.; Hedden, P.; Rivin, C.J. Gibberellins and seed development in maize. I. Evidence that gibberellin/abscisic acid balance governs germination versus maturation pathways. Plant Physiol. 2000, 122, 1081–1088. [Google Scholar] [CrossRef]
- Watt, M.S.; Bloomberg, M.; Finch-Savage, W.E. Development of a hydrothermal time model that accurately characterises how thermoinhibition regulates seed germination. Plant Cell Environ. 2011, 34, 870–876. [Google Scholar]
- Meyers, L.A.; Levin, D.A. On the abundance of polyploids in flowering plants. Evolution 2006, 60, 1198–1206. [Google Scholar]
- Lysak, M.A.; Cheung, K.; Kitschke, M.; Bures, P. Ancestral chromosomal blocks are triplicated in Brassiceae species with varying chromosome number and genome size. Plant Physiol. 2007, 145, 402–410. [Google Scholar] [CrossRef]
- Voorrips, R.; Maliepaard, C. The simulation of meiosis in diploid and tetraploid organisms using various genetic models. BMC Bioinformatics 2012, 13, 248. [Google Scholar] [CrossRef]
- Godin, C.; Sinoquet, H. Functional-structural plant modelling. New Phytol. 2005, 166, 705–708. [Google Scholar] [CrossRef]
- Fourcaud, T.; Zhang, X.; Stokes, A.; Lambers, H.; Körner, C. Plant Growth Modelling and Applications: The Increasing Importance of Plant Architecture in Growth Models. Ann. Bot. 2008, 101, 1053–1063. [Google Scholar]
- Vos, J.; Evers, J.B.; Buck-Sorlin, G.H.; Andrieu, B.; Chelle, M.; de Visser, P.H.B. Functional-structural plant modelling: A new versatile tool in crop science. J. Exp. Bot. 2010, 61, 2101–2115. [Google Scholar] [CrossRef] [Green Version]
- Prusinkiewicz, P.; Lindenmayer, A. The Algorithmic Beauty of Plants; Springer-Verlag: New York, NY, USA, 1990; p. 228. [Google Scholar]
- Chenu, K.; Franck, N.; Lecoeur, J. Simulations of virtual plants reveal a role for SERRATE in the response of leaf development to light in Arabidopsis thaliana. New Phytol. 2007, 175, 472–481. [Google Scholar]
- Qu, H.; Wang, Y.; Cai, L.; Wang, T.; Lu, Z. Orange tree simulation under heterogeneous environment using agent-based model ORASIM. Simul. Modell. Pract. Theory 2012, 23, 19–35. [Google Scholar] [CrossRef]
- Drouet, J.-L.; Pagès, L. GRAAL-CN: A model of GRowth, Architecture and ALlocation for Carbon and Nitrogen dynamics within whole plants formalised at the organ level. Ecol. Modell. 2007, 206, 231–249. [Google Scholar] [CrossRef]
- Clark, B.; Bullock, S. Shedding light on plant competition: Modelling the influence of plant morphology on light capture (and vice versa). J. Theor. Biol. 2007, 244, 208–217. [Google Scholar] [CrossRef]
- Buck-Sorlin, G.; Kniemeyer, O.; Kurth, W. A Model of Poplar (Populus sp.) Physiology and Morphology Based on Relational Growth Grammars Mathematical Modeling of Biological Systems; Deutsch, A., Parra, R.B.D.L., Boer, R.J.D., Diekmann, O., Jagers, P., Kisdi, E., Kretzschmar, M., Lansky, P., Metz, H., Eds.; Birkhäuser: Boston, MA, USA, 2008; Volume II, pp. 313–322. [Google Scholar]
- Kurth, W.; Kniemeyer, O.; Buck-Sorlin, G. Relational Growth Grammars—A Graph Rewriting Approach to Dynamical Systems with a Dynamical Structure Unconventional Programming Paradigms. In Unconventional Programming Paradigms; Banâtre, J.-P., Fradet, P., Giavitto, J.-L., Michel, O., Eds.; Springer: Berlin/Heidelberg, Germany, 2005; Volume 3566, p. 97. [Google Scholar]
- Buck-Sorlin, G.H.; Kniemeyer, O.; Kurth, W. Barley morphology, genetics and hormonal regulation of internode elongation modelled by a relational growth grammar. New Phytol. 2005, 166, 859–867. [Google Scholar] [CrossRef]
- Xu, L.; Henke, M.; Zhu, J.; Kurth, W.; Buck-Sorlin, G. A functional-structural model of rice linking quantitative genetic information with morphological development and physiological processes. Ann. Bot. 2011. [Google Scholar] [CrossRef] [Green Version]
- Luquet, D.; Soulié, J.C.; Rebolledo, M.C.; Rouan, L.; Clément-Vidal, A.; Dingkuhn, M. Developmental Dynamics and Early Growth Vigour in Rice 2. Modelling Genetic Diversity Using Ecomeristem. J. Agron. Crop Sci. 2012, 198, 385–398. [Google Scholar] [CrossRef]
- Bornhofen, S.; Barot, S.; Lattaud, C. The evolution of CSR life-history strategies in a plant model with explicit physiology and architecture. Ecol. Modell. 2011, 222, 1–10. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kitchen, J.L.; Allaby, R.G. Systems Modeling at Multiple Levels of Regulation: Linking Systems and Genetic Networks to Spatially Explicit Plant Populations. Plants 2013, 2, 16-49. https://doi.org/10.3390/plants2010016
Kitchen JL, Allaby RG. Systems Modeling at Multiple Levels of Regulation: Linking Systems and Genetic Networks to Spatially Explicit Plant Populations. Plants. 2013; 2(1):16-49. https://doi.org/10.3390/plants2010016
Chicago/Turabian StyleKitchen, James L., and Robin G. Allaby. 2013. "Systems Modeling at Multiple Levels of Regulation: Linking Systems and Genetic Networks to Spatially Explicit Plant Populations" Plants 2, no. 1: 16-49. https://doi.org/10.3390/plants2010016