Development of pGEMINI, a Plant Gateway Destination Vector Allowing the Simultaneous Integration of Two cDNA via a Single LR-Clonase Reaction

 , and
, and 
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods and Materials
2.1. Generation of pGEMINI Constitutive Double Expression Vector
2.2. Generation of pGEM-RED/YELLOW Expression Clones
2.3. Plant Cultivation and Agrobacterium-Mediated Transient Transformation of Nicotina Benthamiana
3. Results
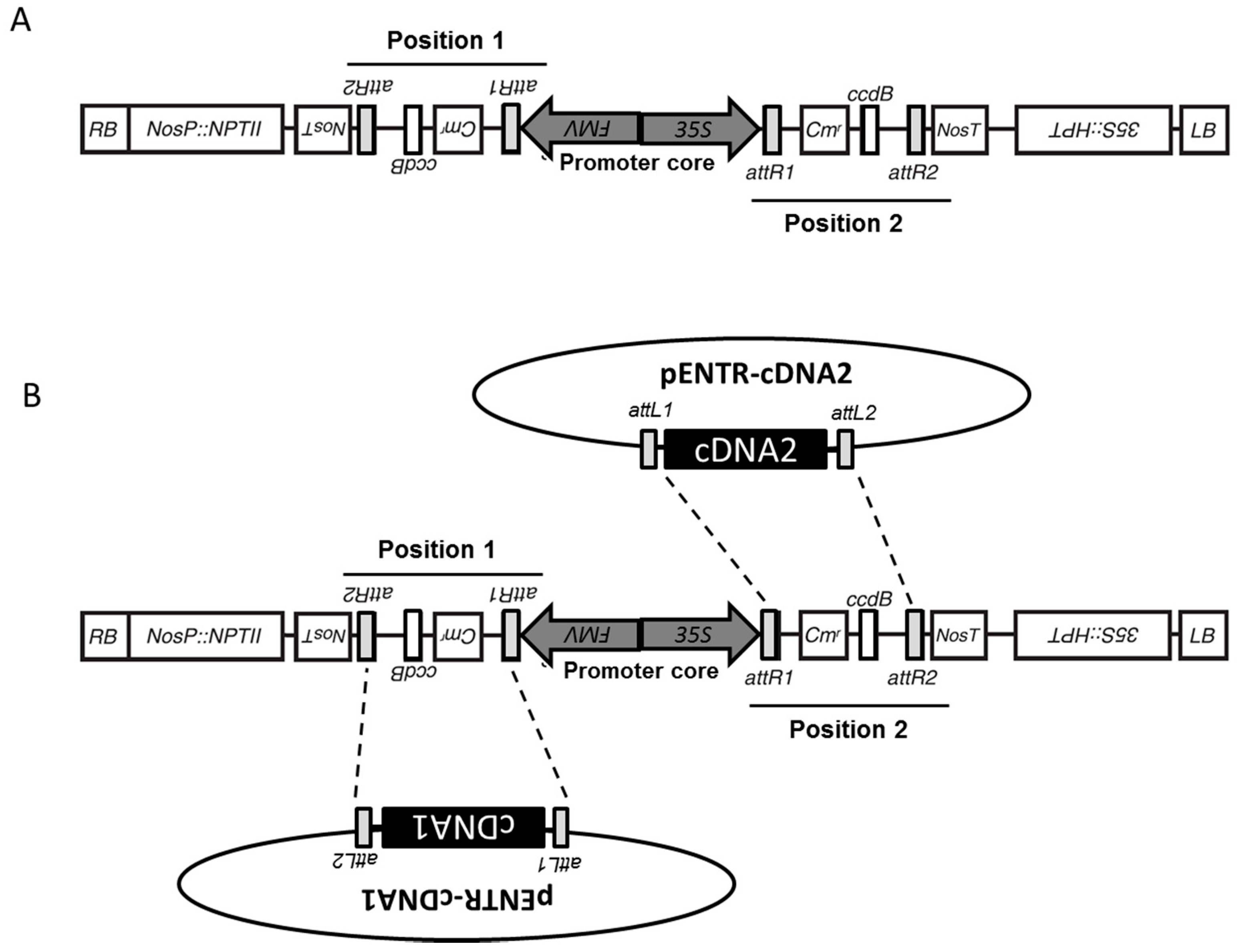
3.1. Structure of pGEMINI
3.2. Integration of cDNA from pENTR/D via an LR Clonase Reaction
3.3. Agrobacterium-Mediated Transient Transformation of Nicotiana Benthamiana and Fluorescent Protein Imaging
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Earley, K.W.; Haag, J.R.; Pontes, O.; Opper, K.; Juehne, T.; Song, K.; Pikaard, C.S. Gateway-compatible vectors for plant functional genomics and proteomics. Plant J. Cell Mol. Biol. 2006, 45, 616–629. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Kurose, T.; Hino, T.; Tanaka, K.; Kawamukai, M.; Niwa, Y.; Toyooka, K.; Matsuoka, K.; Jinbo, T.; Kimura, T. Development of series of gateway binary vectors, pgwbs, for realizing efficient construction of fusion genes for plant transformation. J. Biosci. Bioeng. 2007, 104, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Zarzycki, J.; Axen, S.D.; Kinney, J.N.; Kerfeld, C.A. Cyanobacterial-based approaches to improving photosynthesis in plants. J. Exp. Bot. 2013, 64, 787–798. [Google Scholar] [CrossRef] [PubMed]
- Hanson, D.T.; Stutz, S.S.; Boyer, J.S. Why small fluxes matter: The case and approaches for improving measurements of photosynthesis and (photo)respiration. J. Exp. Bot. 2016, 67, 3027–3039. [Google Scholar] [CrossRef] [PubMed]
- Snowden, K.C.; Simkin, A.J.; Janssen, B.J.; Templeton, K.R.; Loucas, H.M.; Simons, J.L.; Karunairetnam, S.; Gleave, A.P.; Clark, D.G.; Klee, H.J. The decreased apical dominance1/petunia hybrida carotenoid cleavage dioxygenase8 gene affects branch production and plays a role in leaf senescence, root growth, and flower development. Plant Cell 2005, 17, 746–759. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Suzuki, T.; Murata, S.; Nakamura, S.; Hino, T.; Maeo, K.; Tabata, R.; Kawai, T.; Tanaka, K.; Niwa, Y.; et al. Improved gateway binary vectors: High performance vectors for creation of fusion constructs in transgenic analysis of plants. Biosci. Biotechnol. Biochem. 2007, 71, 2095–2100. [Google Scholar] [CrossRef] [PubMed]
- Simkin, A.J.; McAusland, L.; Headland, L.R.; Lawson, T.; Raines, C.A. Multigene manipulation of photosynthetic carbon assimilation increases co2 fixation and biomass yield in tobacco. J. Exp. Bot. 2015, 66, 4075–4090. [Google Scholar] [CrossRef] [PubMed]
- Simkin, A.J.; Lopez-Calcagno, P.E.; Davey, P.A.; Headland, L.R.; Lawson, T.; Timm, S.; Bauwe, H.; Raines, C.A. Simultaneous stimulation of sedoheptulose 1,7-bisphosphatase, fructose 1,6-bisphophate aldolase and the photorespiratory glycine decarboxylase h-protein increases co2 assimilation, vegetative biomass and seed yield in arabidopsis. Plant Biotechnol. J. 2017, 15, 805–816. [Google Scholar] [CrossRef] [PubMed]
- Que, Q.; Chilton, M.M.; de Fontes, C.M.; He, C.; Nuccio, M.; Zhu, T.; Wu, Y.; Chen, J.S.; Shi, L. Trait stacking in transgenic crops. Challenges and opportunities. GM Crops 2010, 1, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Tindamanyire, J.M.; Townsley, B.; Kiggundu, A.; Tushemereirwe, W.; Sinha, N. Building a bi-directional promoter binary vector from the intergenic region of arabidopsis thaliana cab1 and cab2 divergent genes useful for plant transformation. Afr. J. Biotechnol. 2013, 12, 1203–1208. [Google Scholar]
- Hiyama, T. Isolation of photosystem i particles from spinach. Methods Mol. Biol. 2004, 274, 11–17. [Google Scholar] [PubMed]
- Engler, C.; Kandzia, R.; Marillonnet, S. A one pot, one step, precision cloning method with high throughput capability. PLoS ONE 2008, 3, e3647. [Google Scholar] [CrossRef] [PubMed]
- Engler, C.; Gruetzner, R.; Kandzia, R.; Marillonnet, S. Golden gate shuffling: A one-pot DNA shuffling method based on type iis restriction enzymes. PLoS ONE 2009, 4, e5553. [Google Scholar] [CrossRef] [PubMed]
- Booker, J.; Auldridge, M.; Wills, S.; McCarty, D.; Klee, H.; Leyser, O. Max3/ccd7 is a carotenoid cleavage dioxygenase required for the synthesis of a novel plant signaling molecule. Curr. Biol. 2004, 14, 1232–1238. [Google Scholar] [CrossRef] [PubMed]
- Richins, R.D.; Scholthof, H.B.; Shepherd, R.J. Sequence of figwort mosaic-virus DNA (caulimovirus group). Nucleic Acids Res. 1987, 15, 8451–8466. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Dey, N.; Maiti, I.B. Analysis of cis-sequence of subgenomic transcript promoter from the Figwort mosaic virus and comparison of promoter activity with the cauliflower mosaic virus promoters in monocot and dicot cells. Virus Res. 2002, 90, 47–62. [Google Scholar] [PubMed]
- Wesley, S.V.; Helliwell, C.A.; Smith, N.A.; Wang, M.B.; Rouse, D.T.; Liu, Q.; Gooding, P.S.; Singh, S.P.; Abbott, D.; Stoutjesdijk, P.A.; et al. Construct design for efficient, effective and high-throughput gene silencing in plants. Plant J. Cell Mol. Biol. 2001, 27, 581–590. [Google Scholar] [CrossRef]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Exposito-Rodriguez, M.; Laissue, P.P.; López-Calcagno, P.E.; Mullineaux, P.M.; Raines, C.A.; Simkin, A.J. Development of pGEMINI, a Plant Gateway Destination Vector Allowing the Simultaneous Integration of Two cDNA via a Single LR-Clonase Reaction. Plants 2017, 6, 55. https://doi.org/10.3390/plants6040055
Exposito-Rodriguez M, Laissue PP, López-Calcagno PE, Mullineaux PM, Raines CA, Simkin AJ. Development of pGEMINI, a Plant Gateway Destination Vector Allowing the Simultaneous Integration of Two cDNA via a Single LR-Clonase Reaction. Plants. 2017; 6(4):55. https://doi.org/10.3390/plants6040055
Chicago/Turabian StyleExposito-Rodriguez, Marino, Philippe P. Laissue, Patricia E. López-Calcagno, Philip M. Mullineaux, Christine A. Raines, and Andrew J. Simkin. 2017. "Development of pGEMINI, a Plant Gateway Destination Vector Allowing the Simultaneous Integration of Two cDNA via a Single LR-Clonase Reaction" Plants 6, no. 4: 55. https://doi.org/10.3390/plants6040055