The Role of Translational Regulation in Survival after Radiation Damage; an Opportunity for Proteomics Analysis

{kind=link}
{kind=link}
Abstract
:1. The Effect of Ionizing Radiation on Cells and Tissues
1.1. DNA Damage Responses: Cell Cycle Checkpoints, DNA Repair and Cell Death
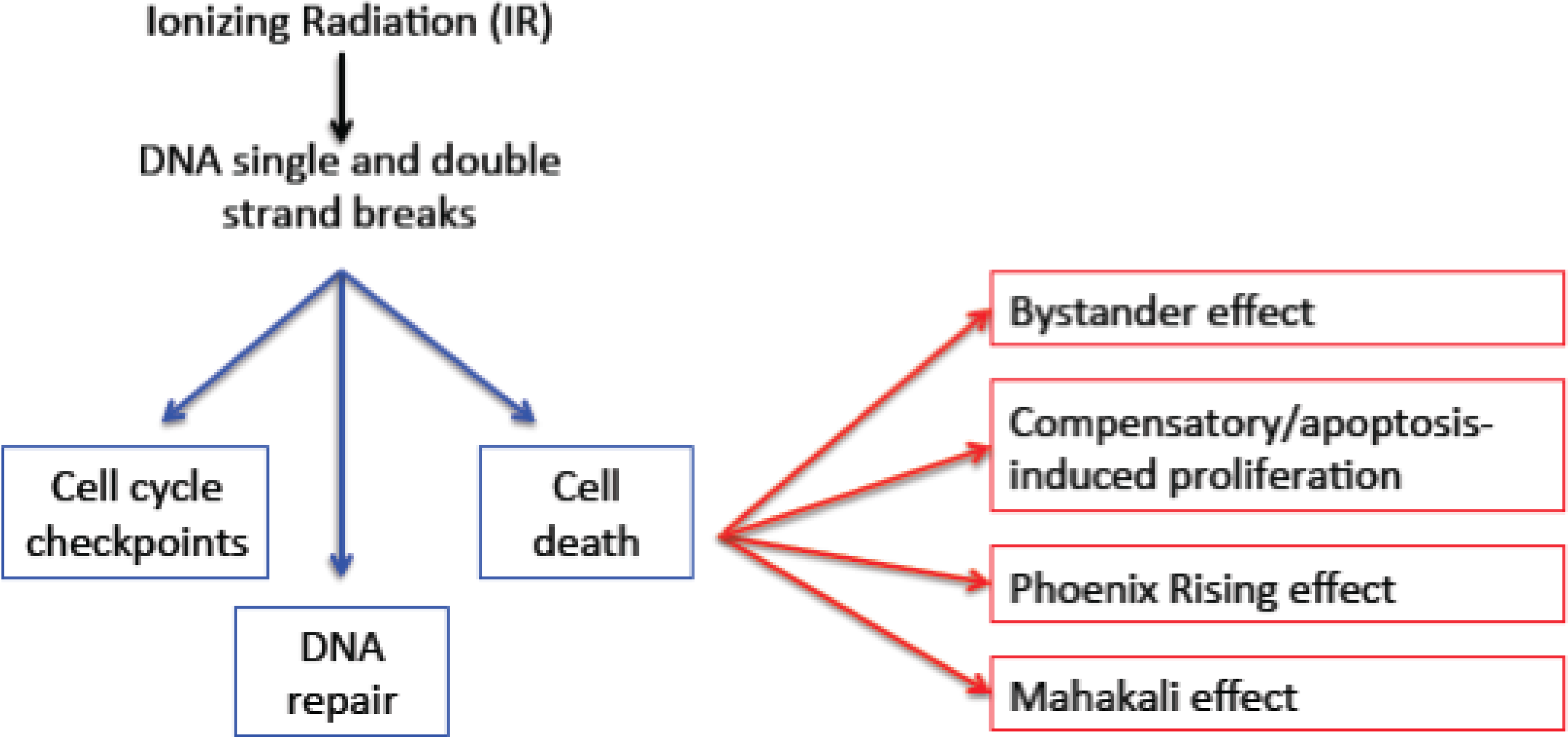
1.2. Secondary Consequences of Cell Death
1.3. The Effect on Macromolecules
2. Translational Regulation in Response to IR
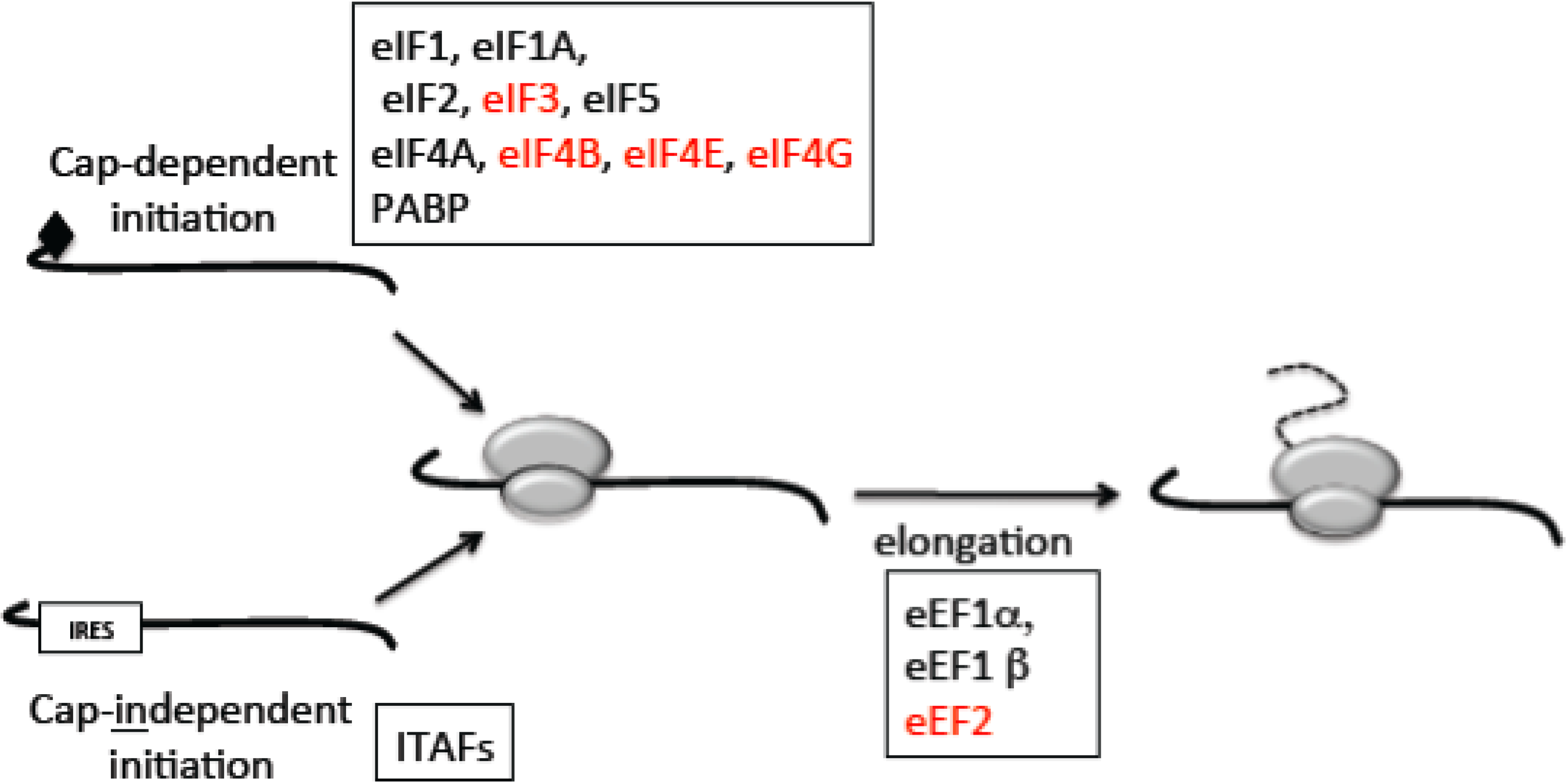
2.1. Mechanisms of Translation
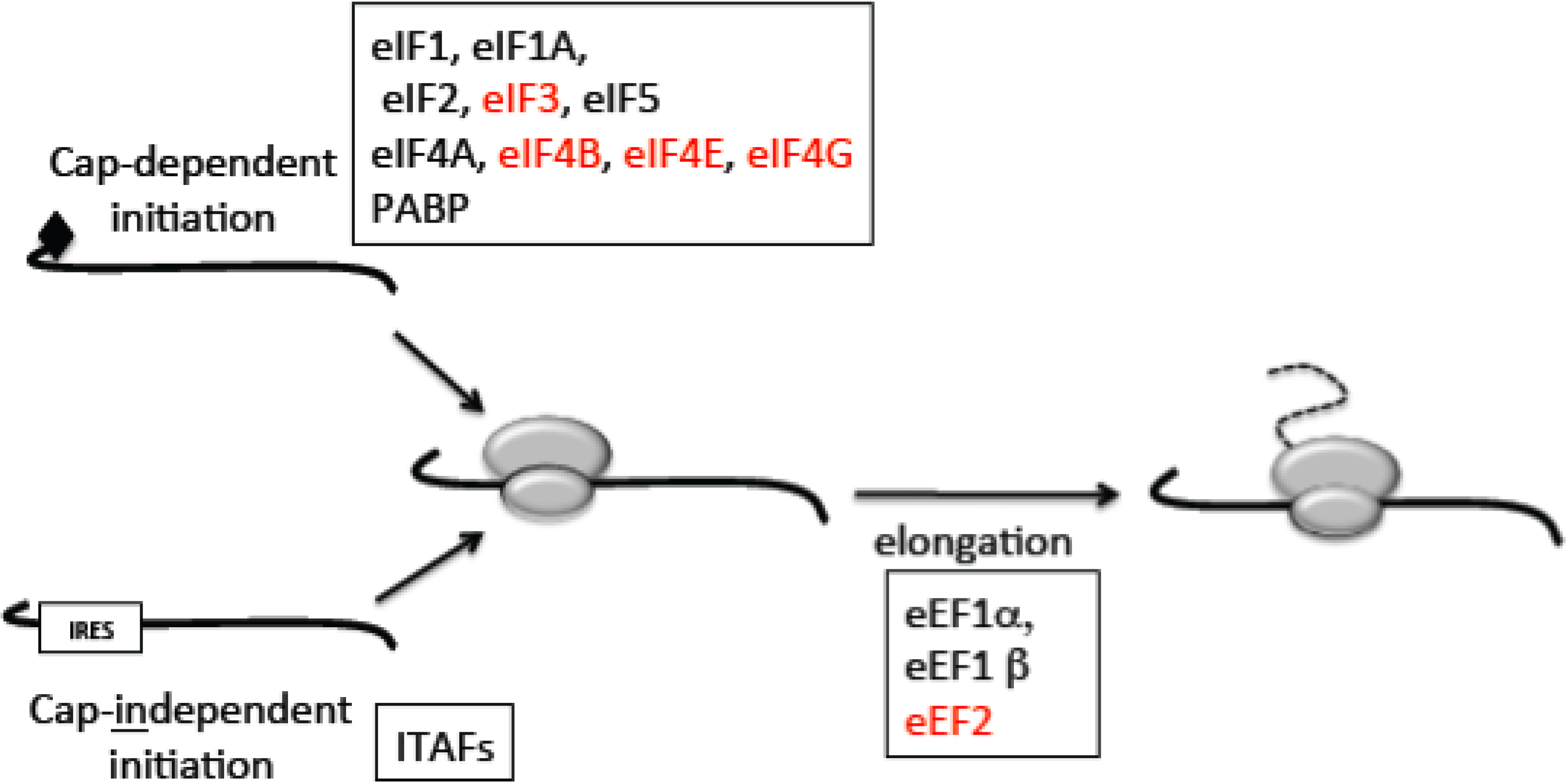
2.2. The Switch to Cap-Independent Initiation under Stress
2.3. Mechanisms for Regulation of Translation
2.4. The Evidence for Translational Regulation after IR: Changes in Transcriptome vs. Ribosome-Associated Transcriptome
2.5. Potential Mechanisms for Altered Polysome Profiles: The Effect of IR on Translation Factors
2.6. Interfering with Translation Compromises Radiation Responses
3. Prior Studies Using Proteomic Analysis after IR Exposure
3.1. Proteomic Studies of DDR Responses
3.2. Searches for Biomarkers for Radiotherapy Pharmacodynamics and Side Effects
4. Opportunities: Unanswered Questions that Will Benefit from Proteomics Analysis
5. Conclusions
Supplementary Materials
Supplementary File 1Abbreviations
| 4E-BP1 | 4E binding protein 1 |
| ATM | ataxia telangiectasia mutated |
| CAF | cancer associated fibroblast |
| DDR | DNA damage responses |
| DSB | double strand break |
| eEF | eukaryotic elongation factor |
| EGFR | epidermal growth factor receptor |
| eIF | eukaryotic Initiation Factor |
| ERLIC | electrostatic repulsion-hydrophilic interaction chromatography, a method for enriching phosphor-peptides |
| ESI | Electron Spray Ionization, a technique to ionize molecules for mass spectrometry, especially useful with macromolecules |
| GEF | guanine nucleotide exchange factor |
| HuR | Hu antigen R, also known as ELAVL1 |
| ICPL | isotope coded protein labeling, typically applied to tissue lysates |
| IR | ionizing radiation |
| IRES | internal ribosome entry site |
| ITAF | IRES transacting factors |
| JNK | Jun N-terminal kinase |
| LC | liquid chromatography for physical separation of molecules |
| LC-MS | a combination of LC with mass spectrometry |
| MALDI-TOF | matrix assisted laser desorption/ionization-time of flight, a mass spectrometry technique |
| MDM2 | mouse double minute 2 |
| PABP | poly A binding protein |
| PDGF | platelet-derived growth factor |
| PI3K | phosphatidylinositol 3-kinase |
| PIC | pre-initiation complex |
| SILAC | stable isotope labeling by/with amino acids in cell culture |
| TOR | target of rapamycin |
| UTR | untranslated region |
| VEGF | vascular endothelial growth factor |
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hall, E.; Giaccia, A.J. Radiobiology for the Radiologist, 6th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2006. [Google Scholar]
- Su, T.T. Cellular responses to DNA damage: One signal, multiple choices. Annu. Rev. Genet. 2006, 40, 187–208. [Google Scholar] [CrossRef]
- Jackson, S.P. Sensing and repairing DNA double-strand breaks. Carcinogenesis 2002, 23, 687–696. [Google Scholar] [CrossRef]
- Belli, M.; Sapora, O.; Tabocchini, M.A. Molecular targets in cellular response to ionizing radiation and implications in space radiation protection. J. Radiat. Res. 2002, 43, S13–S19. [Google Scholar] [CrossRef]
- Shiloh, Y.; Ziv, Y. The ATM protein kinase: Regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 2013, 14, 197–210. [Google Scholar] [CrossRef]
- Sirbu, B.M.; Cortez, D. DNA damage response: Three levels of DNA repair regulation. Cold Spring Harb. Perspect. Biol. 2013, 5, a012724. [Google Scholar]
- Surova, O.; Zhivotovsky, B. Various modes of cell death induced by DNA damage. Oncogene 2013, 32, 3789–3797. [Google Scholar] [CrossRef]
- Fan, Y.; Bergmann, A. Distinct mechanisms of apoptosis-induced compensatory proliferation in proliferating and differentiating tissues in the Drosophila eye. Dev. Cell 2008, 14, 399–410. [Google Scholar] [CrossRef]
- Grusche, F.A.; Degoutin, J.L.; Richardson, H.E.; Harvey, K.F. The salvador/warts/hippo pathway controls regenerative tissue growth in Drosophila melanogaster. Dev. Biol. 2011, 350, 255–266. [Google Scholar] [CrossRef]
- Ryoo, H.D.; Gorenc, T.; Steller, H. Apoptotic cells can induce compensatory cell proliferation through the jnk and the wingless signaling pathways. Dev. Cell 2004, 7, 491–501. [Google Scholar] [CrossRef]
- Sun, G.; Irvine, K.D. Regulation of hippo signaling by jun kinase signaling during compensatory cell proliferation and regeneration, and in neoplastic tumors. Dev. Biol. 2011, 350, 139–151. [Google Scholar] [CrossRef]
- Li, F.; Huang, Q.; Chen, J.; Peng, Y.; Roop, D.R.; Bedford, J.S.; Li, C.Y. Apoptotic cells activate the “phoenix rising” pathway to promote wound healing and tissue regeneration. Sci. Signal. 2010, 3, ra13. [Google Scholar]
- Huang, Q.; Li, F.; Liu, X.; Li, W.; Shi, W.; Liu, F.F.; O’Sullivan, B.; He, Z.; Peng, Y.; Tan, A.C.; et al. Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat. Med. 2011, 17, 860–866. [Google Scholar] [CrossRef]
- Marcu, L.G.; Bezak, E. Radiobiological modeling of interplay between accelerated repopulation and altered fractionation schedules in head and neck cancer. J. Med. Phys. 2009, 34, 206–211. [Google Scholar]
- Szczepanski, L.; Trott, K.R. Post-irradiation proliferation kinetics of a serially transplanted murine adenocarcinoma. Br. J. Radiol. 1975, 48, 200–208. [Google Scholar] [CrossRef]
- Chen, C.P.; Weinberg, V.K.; Jahan, T.M.; Jablons, D.M.; Yom, S.S. Implications of delayed initiation of radiotherapy: Accelerated repopulation after induction chemotherapy for stage III non-small cell lung cancer. J. Thorac. Oncol. 2011, 6, 1857–1864. [Google Scholar] [CrossRef]
- Huang, Z.; Mayr, N.A.; Gao, M.; Lo, S.S.; Wang, J.Z.; Jia, G.; Yuh, W.T. Onset time of tumor repopulation for cervical cancer: First evidence from clinical data. Int. J. Radiat. Oncol. Biol. Phys. 2012, 84, 478–484. [Google Scholar] [CrossRef]
- Kim, J.J.; Tannock, I.F. Repopulation of cancer cells during therapy: An important cause of treatment failure. Nat. Rev. Cancer 2005, 5, 516–525. [Google Scholar] [CrossRef]
- Robertson, K.L.; Mostaghim, A.; Cuomo, C.A.; Soto, C.M.; Lebedev, N.; Bailey, R.F.; Wang, Z. Adaptation of the black yeast wangiella dermatitidis to ionizing radiation: Molecular and cellular mechanisms. PLoS One 2012, 7, e48674. [Google Scholar]
- Bilak, A.; Uyetake, L.; Su, T.T. Dying cells protect survivors from radiation-induced cell death in Drosophila. PLoS Genet. 2014, 10, e1004220. [Google Scholar] [CrossRef]
- Jaklevic, B.; Uyetake, L.; Wichmann, A.; Bilak, A.; English, C.N.; Su, T.T. Modulation of ionizing radiation-induced apoptosis by bantam microrna in Drosophila. Dev. Biol. 2008, 320, 122–130. [Google Scholar]
- Mothersill, C.; Seymour, C. Radiation-induced bystander effects: Evidence for an adaptive response to low dose exposures? Dose-Response 2006, 4, 283–290. [Google Scholar] [CrossRef]
- Mothersill, C.; Seymour, C.B. Radiation-induced bystander effects and the DNA paradigm: An “out of field” perspective. Mutat. Res. 2006, 597, 5–10. [Google Scholar] [CrossRef]
- Singh, H.; Saroya, R.; Smith, R.; Mantha, R.; Guindon, L.; Mitchel, R.E.; Seymour, C.; Mothersill, C. Radiation induced bystander effects in mice given low doses of radiation in vivo. Dose-Response 2011, 9, 225–242. [Google Scholar]
- Mothersill, C.; Stamato, T.D.; Perez, M.L.; Cummins, R.; Mooney, R.; Seymour, C.B. Involvement of energy metabolism in the production of ‘bystander effects’ by radiation. Br. J. Cancer 2000, 82, 1740–1746. [Google Scholar] [CrossRef]
- Cheng, A.; Caffrey, M. Free radical mediated x-ray damage of model membranes. Biophys. J. 1996, 70, 2212–2222. [Google Scholar] [CrossRef]
- Corre, I.; Niaudet, C.; Paris, F. Plasma membrane signaling induced by ionizing radiation. Mutat. Res. 2010, 704, 61–67. [Google Scholar] [CrossRef]
- Krisko, A.; Radman, M. Protein damage and death by radiation in Escherichia coli and deinococcus radiodurans. Proc. Natl. Acad. Sci. USA 2010, 107, 14373–14377. [Google Scholar] [CrossRef]
- Daly, M.J. Death by protein damage in irradiated cells. DNA Repair (Amst) 2012, 11, 12–21. [Google Scholar] [CrossRef]
- Pavitt, G.D.; Ashe, M.P. Translation controlled. Genome Biol. 2008, 9, e323. [Google Scholar]
- Shahbazian, D.; Parsyan, A.; Petroulakis, E.; Topisirovic, I.; Martineau, Y.; Gibbs, B.F.; Svitkin, Y.; Sonenberg, N. Control of cell survival and proliferation by mammalian eukaryotic initiation factor 4b. Mol. Cell. Biol. 2010, 30, 1478–1485. [Google Scholar] [CrossRef]
- Gandin, V.; Miluzio, A.; Barbieri, A.M.; Beugnet, A.; Kiyokawa, H.; Marchisio, P.C.; Biffo, S. Eukaryotic initiation factor 6 is rate-limiting in translation, growth and transformation. Nature 2008, 455, 684–688. [Google Scholar] [CrossRef]
- Svitkin, Y.V.; Herdy, B.; Costa-Mattioli, M.; Gingras, A.C.; Raught, B.; Sonenberg, N. Eukaryotic translation initiation factor 4e availability controls the switch between cap-dependent and internal ribosomal entry site-mediated translation. Mol. Cell Biol. 2005, 25, 10556–10565. [Google Scholar] [CrossRef]
- Braunstein, S.; Badura, M.L.; Xi, Q.; Formenti, S.C.; Schneider, R.J. Regulation of protein synthesis by ionizing radiation. Mol. Cell Biol. 2009, 29, 5645–5656. [Google Scholar] [CrossRef]
- Aitken, C.E.; Lorsch, J.R. A mechanistic overview of translation initiation in eukaryotes. Nat. Struct. Mol. Biol. 2012, 19, 568–576. [Google Scholar]
- Fraser, C.S.; Doudna, J.A. Structural and mechanistic insights into hepatitis C viral translation initiation. Nat. Rev. Microbiol. 2007, 5, 29–38. [Google Scholar] [CrossRef]
- Trivigno, D.; Bornes, L.; Huber, S.M.; Rudner, J. Regulation of protein translation initiation in response to ionizing radiation. Radiat. Oncol. 2013, 8, e35. [Google Scholar] [CrossRef]
- Komar, A.A.; Hatzoglou, M. Cellular IRES-mediated translation: The war of ITAFs in pathophysiological states. Cell Cycle 2011, 10, 229–240. [Google Scholar] [CrossRef]
- Kapp, L.D.; Lorsch, J.R. The molecular mechanics of eukaryotic translation. Annu. Rev. Biochem. 2004, 73, 657–704. [Google Scholar] [CrossRef]
- Buttgereit, F.; Brand, M.D. A hierarchy of ATP-consuming processes in mammalian cells. Biochem. J. 1995, 312, 163–167. [Google Scholar]
- Holcik, M.; Sonenberg, N. Translational control in stress and apoptosis. Nat. Rev. Mol. Cell Biol. 2005, 6, 318–327. [Google Scholar] [CrossRef]
- Hay, N.; Sonenberg, N. Upstream and downstream of mTOR. Genes Dev. 2004, 18, 1926–1945. [Google Scholar] [CrossRef]
- Mamane, Y.; Petroulakis, E.; LeBacquer, O.; Sonenberg, N. mTOR, translation initiation and cancer. Oncogene 2006, 25, 6416–6422. [Google Scholar] [CrossRef]
- Petroulakis, E.; Mamane, Y.; le Bacquer, O.; Shahbazian, D.; Sonenberg, N. mTOR signaling: Implications for cancer and anticancer therapy. Br. J. Cancer 2006, 94, 195–199. [Google Scholar] [CrossRef]
- Wang, X.; Proud, C.G. The mtor pathway in the control of protein synthesis. Physiology (Bethesda) 2006, 21, 362–369. [Google Scholar] [CrossRef]
- Harris, T.E.; Chi, A.; Shabanowitz, J.; Hunt, D.F.; Rhoads, R.E.; Lawrence, J.C., Jr. mTOR-dependent stimulation of the association of eIF4G and eIF3 by insulin. EMBO J. 2006, 25, 1659–1668. [Google Scholar] [CrossRef]
- Kruiswijk, F.; Yuniati, L.; Magliozzi, R.; Low, T.Y.; Lim, R.; Bolder, R.; Mohammed, S.; Proud, C.G.; Heck, A.J.; Pagano, M.; et al. Coupled activation and degradation of eEF2K regulates protein synthesis in response to genotoxic stress. Sci. Signal. 2012, 5, ra40. [Google Scholar]
- Browne, G.J.; Proud, C.G. A novel mTOR-regulated phosphorylation site in elongation factor 2 kinase modulates the activity of the kinase and its binding to calmodulin. Mol. Cell. Biol. 2004, 24, 2986–2997. [Google Scholar] [CrossRef]
- Wang, X.; Li, W.; Williams, M.; Terada, N.; Alessi, D.R.; Proud, C.G. Regulation of elongation factor 2 kinase by p90(RSK1) and p70 S6 kinase. EMBO J. 2001, 20, 4370–4379. [Google Scholar] [CrossRef]
- Lu, X.; de la Pena, L.; Barker, C.; Camphausen, K.; Tofilon, P.J. Radiation-induced changes in gene expression involve recruitment of existing messenger RNAs to and away from polysomes. Cancer Res. 2006, 66, 1052–1061. [Google Scholar] [CrossRef]
- Kumaraswamy, S.; Chinnaiyan, P.; Shankavaram, U.T.; Lu, X.; Camphausen, K.; Tofilon, P.J. Radiation-induced gene translation profiles reveal tumor type and cancer-specific components. Cancer Res. 2008, 68, 3819–3826. [Google Scholar] [CrossRef]
- Kumar, V.; Sabatini, D.; Pandey, P.; Gingras, A.C.; Majumder, P.K.; Kumar, M.; Yuan, Z.M.; Carmichael, G.; Weichselbaum, R.; Sonenberg, N.; et al. Regulation of the rapamycin and FKBP-target 1/mammalian target of rapamycin and cap-dependent initiation of translation by the c-Abl protein-tyrosine kinase. J. Biol. Chem. 2000, 275, 10779–10787. [Google Scholar] [CrossRef]
- Roig, J.; Traugh, J.A. p21-activated protein kinase gamma-PAK is activated by ionizing radiation and other DNA-damaging agents. Similarities and differences to alpha-PAK. J. Biol. Chem. 1999, 274, 31119–31122. [Google Scholar] [CrossRef]
- Ling, J.; Morley, S.J.; Traugh, J.A. Inhibition of cap-dependent translation via phosphorylation of eIF4G by protein kinase Pak2. EMBO J. 2005, 24, 4094–4105. [Google Scholar] [CrossRef]
- Paglin, S.; Lee, N.Y.; Nakar, C.; Fitzgerald, M.; Plotkin, J.; Deuel, B.; Hackett, N.; McMahill, M.; Sphicas, E.; Lampen, N.; et al. Rapamycin-sensitive pathway regulates mitochondrial membrane potential, autophagy, and survival in irradiated mcf-7 cells. Cancer Res. 2005, 65, 11061–11070. [Google Scholar] [CrossRef]
- Mazan-Mamczarz, K.; Hagner, P.R.; Zhang, Y.; Dai, B.; Lehrmann, E.; Becker, K.G.; Keene, J.D.; Gorospe, M.; Liu, Z.; Gartenhaus, R.B. ATM regulates a DNA damage response posttranscriptional RNA operon in lymphocytes. Blood 2011, 117, 2441–2450. [Google Scholar] [CrossRef]
- Gu, L.; Zhu, N.; Zhang, H.; Durden, D.L.; Feng, Y.; Zhou, M. Regulation of XIAP translation and induction by MDM2 following irradiation. Cancer Cell 2009, 15, 363–375. [Google Scholar]
- Hashimoto, Y.; Hosoda, N.; Datta, P.; Alnemri, E.S.; Hoshino, S. Translation termination factor ERF3 is targeted for caspase-mediated proteolytic cleavage and degradation during DNA damage-induced apoptosis. Apoptosis 2012, 17, 1287–1299. [Google Scholar]
- Djordjevic, B.; Kim, J.H. Modification of radiation response in synchronized hela cells by metabolic inhibitors: Effects of inhibitors of DNA and protein synthesis. Radiat. Res. 1969, 37, 435–450. [Google Scholar]
- Youngblom, J.H.; Wiencke, J.K.; Wolff, S. Inhibition of the adaptive response of human lymphocytes to very low doses of ionizing radiation by the protein synthesis inhibitor cycloheximide. Mutat. Res. 1989, 227, 257–261. [Google Scholar] [CrossRef]
- Taylor, M.H.; Buckwalter, M.R.; Stephenson, A.C.; Hart, J.L.; Taylor, B.J.; O’Neill, K.L. Radiation-induced apoptosis in molt-4 cells requires de novo protein synthesis independent of de novo RNA synthesis. FEBS Lett. 2002, 514, 199–203. [Google Scholar] [CrossRef]
- Gobe, G.C.; Harmon, B.; Leighton, J.; Allan, D.J. Radiation-induced apoptosis and gene expression in neonatal kidney and testis with and without protein synthesis inhibition. Int. J. Radiat. Biol. 1999, 75, 973–983. [Google Scholar] [CrossRef]
- Weissberg, J.B.; Fischer, J.J. Protective effect of cycloheximide on the response of rat hind limbs to X irradiation. Radiat. Res. 1981, 88, 597–606. [Google Scholar] [CrossRef]
- Yentrapalli, R.; Azimzadeh, O.; Sriharshan, A.; Malinowsky, K.; Merl, J.; Wojcik, A.; Harms-Ringdahl, M.; Atkinson, M.J.; Becker, K.F.; Haghdoost, S.; et al. The PI3K/Akt/mTOR pathway is implicated in the premature senescence of primary human endothelial cells exposed to chronic radiation. PLoS One 2013, 8, e70024. [Google Scholar]
- Wiita, A.P.; Ziv, E.; Wiita, P.J.; Urisman, A.; Julien, O.; Burlingame, A.L.; Weissman, J.S.; Wells, J.A. Global cellular response to chemotherapy-induced apoptosis. Elife 2013, 2, e01236. [Google Scholar] [CrossRef]
- Kronja, I.; Yuan, B.; Eichhorn, S.; Dzeyk, K.; Krijgsveld, J.; Bartel, D.P.; Orr-Weaver, T.L. Widespread changes in the posttranscriptional landscape at the Drosophila oocyte-to-embryo transition. Cell Rep. 2014. [Google Scholar] [CrossRef]
- Azimzadeh, O.; Atkinson, M.J.; Tapio, S. Proteomics in radiation research: Present status and future perspectives. Radiat. Environ. Biophys. 2014, 53, 31–38. [Google Scholar] [CrossRef]
- Leszczynski, D. Radiation proteomics: A brief overview. Proteomics 2014, 14, 481–488. [Google Scholar] [CrossRef]
- Cheema, A.K.; Varghese, R.S.; Timofeeva, O.; Zhang, L.; Kirilyuk, A.; Zandkarimi, F.; Kaur, P.; Ressom, H.W.; Jung, M.; Dritschilo, A. Functional proteomics analysis to study ATM dependent signaling in response to ionizing radiation. Radiat. Res. 2013, 179, 674–683. [Google Scholar] [CrossRef]
- Bennetzen, M.V.; Larsen, D.H.; Bunkenborg, J.; Bartek, J.; Lukas, J.; Andersen, J.S. Site-specific phosphorylation dynamics of the nuclear proteome during the DNA damage response. Mol. Cell. Proteomics 2010, 9, 1314–1323. [Google Scholar] [CrossRef]
- Tang, Y.; Zhao, W.; Chen, Y.; Zhao, Y.; Gu, W. Acetylation is indispensable for p53 activation. Cell 2008, 133, 612–626. [Google Scholar] [CrossRef]
- Bennetzen, M.V.; Larsen, D.H.; Dinant, C.; Watanabe, S.; Bartek, J.; Lukas, J.; Andersen, J.S. Acetylation dynamics of human nuclear proteins during the ionizing radiation-induced DNA damage response. Cell Cycle 2013, 12, 1688–1695. [Google Scholar] [CrossRef]
- Lacombe, J.; Mange, A.; Azria, D.; Solassol, J. Identification of predictive biomarkers to radiotherapy outcome through proteomics approaches. Cancer Radiother. 2013, 17, 62–69, quiz 70, 72. [Google Scholar] [CrossRef]
- Walpurgis, K.; Kohler, M.; Thomas, A.; Wenzel, F.; Geyer, H.; Schanzer, W.; Thevis, M. Effects of gamma irradiation and 15 days of subsequent ex vivo storage on the cytosolic red blood cell proteome analyzed by 2D-DIGE and Orbitrap MS. Proteomics Clin. Appl. 2013, 7, 561–570. [Google Scholar] [CrossRef]
- Lukkahatai, N.; Patel, S.; Gucek, M.; Hsiao, C.P.; Saligan, L.N. Proteomic serum profile of fatigued men receiving localized external beam radiation therapy for non-metastatic prostate cancer. J. Pain Symptom Manag. 2013, 47, 748–756. [Google Scholar]
- Li, H.Y.; Zhang, H. Proteome analysis for profiling infertility markers in male mouse sperm after carbon ion radiation. Toxicology 2013, 306, 85–92. [Google Scholar] [CrossRef]
- Azimzadeh, O.; Sievert, W.; Sarioglu, H.; Yentrapalli, R.; Barjaktarovic, Z.; Sriharshan, A.; Ueffing, M.; Janik, D.; Aichler, M.; Atkinson, M.J.; et al. PPAR alpha: A novel radiation target in locally exposed Mus musculus heart revealed by quantitative proteomics. J. Proteome Res. 2013, 12, 2700–2714. [Google Scholar] [CrossRef]
- Hellevik, T.; Pettersen, I.; Berg, V.; Bruun, J.; Bartnes, K.; Busund, L.T.; Chalmers, A.; Bremnes, R.; Martinez-Zubiaurre, I. Changes in the secretory profile of NSCLC-associated fibroblasts after ablative radiotherapy: Potential impact on angiogenesis and tumor growth. Transl. Oncol. 2013, 6, 66–74. [Google Scholar]
- Ahlemann, M.; Zeidler, R.; Lang, S.; Mack, B.; Munz, M.; Gires, O. Carcinoma-associated eIF3i overexpression facilitates mTOR-dependent growth transformation. Mol. Carcinog. 2006, 45, 957–967. [Google Scholar] [CrossRef]
- Avdulov, S.; Li, S.; Michalek, V.; Burrichter, D.; Peterson, M.; Perlman, D.M.; Manivel, J.C.; Sonenberg, N.; Yee, D.; Bitterman, P.B.; et al. Activation of translation complex eIF4F is essential for the genesis and maintenance of the malignant phenotype in human mammary epithelial cells. Cancer Cell 2004, 5, 553–563. [Google Scholar]
- Fukuchi-Shimogori, T.; Ishii, I.; Kashiwagi, K.; Mashiba, H.; Ekimoto, H.; Igarashi, K. Malignant transformation by overproduction of translation initiation factor eIF4G. Cancer Res. 1997, 57, 5041–5044. [Google Scholar]
- Lazaris-Karatzas, A.; Montine, K.S.; Sonenberg, N. Malignant transformation by a eukaryotic initiation factor subunit that binds to mRNA 5' cap. Nature 1990, 345, 544–547. [Google Scholar] [CrossRef]
- Savinainen, K.J.; Helenius, M.A.; Lehtonen, H.J.; Visakorpi, T. Overexpression of EIF3S3 promotes cancer cell growth. Prostate 2006, 66, 1144–1150. [Google Scholar]
- Zhang, L.; Pan, X.; Hershey, J.W. Individual overexpression of five subunits of human translation initiation factor eIF3 promotes malignant transformation of immortal fibroblast cells. J. Biol. Chem. 2007, 282, 5790–5800. [Google Scholar] [CrossRef]
- De Benedetti, A.; Graff, J.R. eIF-4E expression and its role in malignancies and metastases. Oncogene 2004, 23, 3189–3199. [Google Scholar] [CrossRef]
- Graff, J.R.; Konicek, B.W.; Vincent, T.M.; Lynch, R.L.; Monteith, D.; Weir, S.N.; Schwier, P.; Capen, A.; Goode, R.L.; Dowless, M.S.; et al. Therapeutic suppression of translation initiation factor eIF4E expression reduces tumor growth without toxicity. J. Clin. Investig. 2007, 117, 2638–2648. [Google Scholar] [CrossRef]
- Mamane, Y.; Petroulakis, E.; Rong, L.; Yoshida, K.; Ler, L.W.; Sonenberg, N. eIF4E—From translation to transformation. Oncogene 2004, 23, 3172–3179. [Google Scholar] [CrossRef]
- Rosenwald, I.B.; Kaspar, R.; Rousseau, D.; Gehrke, L.; Leboulch, P.; Chen, J.J.; Schmidt, E.V.; Sonenberg, N.; London, I.M. Eukaryotic translation initiation factor 4E regulates expression of cyclin D1 at transcriptional and post-transcriptional levels. J. Biol. Chem. 1995, 270, 21176–21180. [Google Scholar]
- Anand, N.; Murthy, S.; Amann, G.; Wernick, M.; Porter, L.A.; Cukier, I.H.; Collins, C.; Gray, J.W.; Diebold, J.; Demetrick, D.J.; et al. Protein elongation factor eEF1A2 is a putative oncogene in ovarian cancer. Nat. Genet. 2002, 31, 301–305. [Google Scholar]
- DeFatta, R.J.; Nathan, C.O.; de Benedetti, A. Antisense RNA to eIF4E suppresses oncogenic properties of a head and neck squamous cell carcinoma cell line. Laryngoscope 2000, 110, 928–933. [Google Scholar] [CrossRef]
- Mazan-Mamczarz, K.; Zhao, X.F.; Dai, B.; Steinhardt, J.J.; Peroutka, R.J.; Berk, K.L.; Landon, A.L.; Sadowska, M.; Zhang, Y.; Lehrmann, E.; et al. Down-regulation of eIF4GII by miR-5 20c-3p represses diffuse large b cell lymphoma development. PLoS Genet. 2014, 10, e1004105. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Stickel, S.; Gomes, N.; Su, T.T. The Role of Translational Regulation in Survival after Radiation Damage; an Opportunity for Proteomics Analysis. Proteomes 2014, 2, 272-290. https://doi.org/10.3390/proteomes2020272
Stickel S, Gomes N, Su TT. The Role of Translational Regulation in Survival after Radiation Damage; an Opportunity for Proteomics Analysis. Proteomes. 2014; 2(2):272-290. https://doi.org/10.3390/proteomes2020272
Chicago/Turabian StyleStickel, Stefanie, Nathan Gomes, and Tin Tin Su. 2014. "The Role of Translational Regulation in Survival after Radiation Damage; an Opportunity for Proteomics Analysis" Proteomes 2, no. 2: 272-290. https://doi.org/10.3390/proteomes2020272