
Proteomic Profiling of the Microsomal Root Fraction: Discrimination of Pisum sativum L. Cultivars and Identification of Putative Root Growth Markers


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. Microsomal Enrichment
2.3. Washing Membranes
2.4. Sample Preparation for Mass Spectrometry
2.5. Mass Spectrometry
2.6. Targeted Protein Marker Evaluation
3. Results
3.1. Morphological Changes
3.2. Proteome Analysis
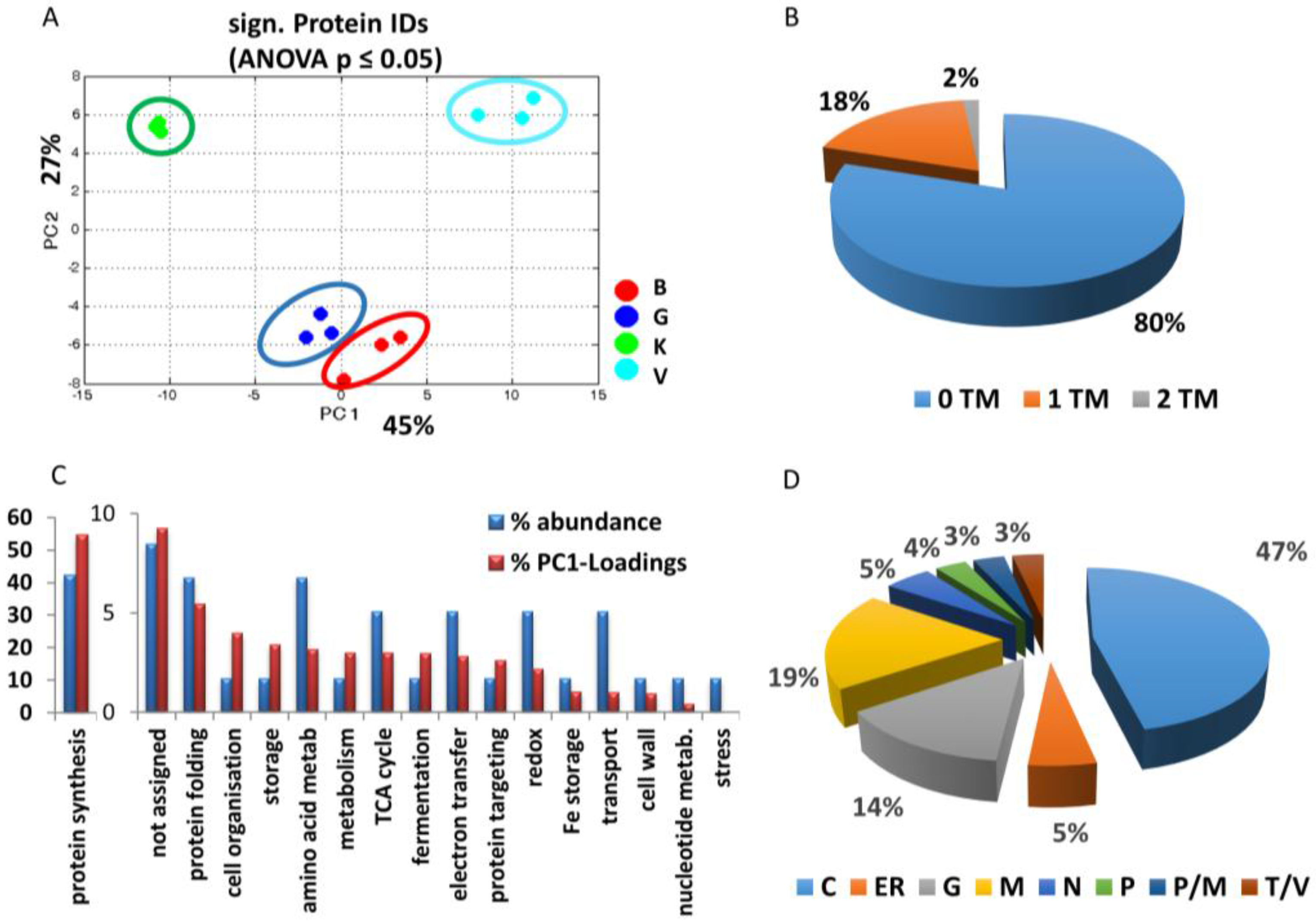
3.2.1. General Overview
3.2.2. Cultivar Specific Differences—Proteins with the Highest Separation Impact on PC1
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Smýkal, P.; Aubert, G.; Burstin, J.; Coyne, C.J.; Ellis, N.T.H.; Flavell, A.J.; Ford, R.; Hýbl, M.; Macas, J.; Neumann, P.; et al. Pea (Pisum sativum L.) in the Genomic Era. Agronomy 2012, 2, 74–115. [Google Scholar] [CrossRef]
- FAOSTAT. Available online: http://faostat3.fao.org/home/E (accessed on 1 September 2016).
- Meisrimler, C.-N.; Menckhoff, L.; Kukavica, B.M.; Lüthje, S. Pre-fractionation strategies to resolve pea (Pisum sativum) sub-proteomes. Front Plant Sci. 2015, 6, 849. [Google Scholar] [CrossRef] [PubMed]
- Cool Season Food Legume Database. Available online: https://www.coolseasonfoodlegume.org/ (accessed on 1 September 2016).
- Wang, J.; Meng, Y.; Li, B.; Ma, X.; Lai, Y.; Si, E.; Yang, K.; Xu, X.; Shang, X.; Wang, H.; et al. Physiological and proteomic analyses of salt stress response in the halophyte Halogeton glomeratus. Plant Cell Environ. 2015, 38, 655–669. [Google Scholar] [CrossRef] [PubMed]
- Meisrimler, C.N.; Planchon, S.; Renaut, J.; Sergeant, K.; Lüthje, S. Alteration of plasma membrane-bound redox systems of Fe deficient pea roots by chitosan. J. Proteom. 2011, 74, 1437–1449. [Google Scholar] [CrossRef] [PubMed]
- Vincent, D.; Ergül, A.; Bohlman, M.C.; Tattersall, E.A.; Tillett, R.L.; Wheatley, M.D.; Woolsey, D.R.; Quilici, D.R.; Joets, J.; Schlauch, K.; et al. Proteomic analysis reveals differences between Vitis vinifera L. cv. Chardonnay and cv. Cabernet Sauvignon and their responses to water deficit and salinity. J. Exp. Bot. 2011, 58, 1873–1892. [Google Scholar] [CrossRef] [PubMed]
- Stark, A.L.; Hause, R.J., Jr.; Gorsic, L.K.; Antao, N.N.; Wong, S.S.; Chung, S.H.; Daniel, F.G.; Im, H.K.; Myers, J.L.; White, K.P.; et al. Protein quantitative trait loci identify novel candidates modulating cellular response to chemotherapy. PLoS Genet. 2014, 10, e1004192. [Google Scholar] [CrossRef] [PubMed]
- Castillejo, M.Á.; Iglesias-García, R.; Wienkoop, S.; Rubiales, D. Label-free quantitative proteomic analysis of tolerance to drought in Pisum sativum. Proteomics 2016, 16, 2776–2787. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Singh, A.; Singh, S.; Singh, V.; Singh, H.B. Comparative proteomic analysis in pea treated with microbial consortia of beneficial microbes reveals changes in the protein network to enhance resistance against Sclerotinia sclerotiorum. J. Plant Physiol. 2015, 182, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.Q.; Møller, I.M.; Song, S.Q. Proteomic analysis of embryonic axis of Pisum sativum seeds during germination and identification of proteins associated with loss of desiccation tolerance. J. Protoc. 2012, 77, 68–86. [Google Scholar] [CrossRef] [PubMed]
- Badowiec, A.; Swigonska, S.; Weidner, S. Changes in the protein patterns in pea (Pisum sativum L.) roots under the influence of long- and short-term chilling stress and post-stress recovery. Plant Phys. Biochem. 2013, 71, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Nacry, P.; Canivenc, G.; Muller, B.; Azmi, A.; Van Onckelen, H.; Rossignol, M.; Doumas, P. A role for auxin redistribution in the responses of the root system architecture to phosphate starvation in Arabidopsis. Plant Physiol. 2005, 138, 2061–2074. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Lee, E.J.; Yang, E.J.; Lee, J.E.; Park, A.R.; Song, W.H.; Park, O.K. Proteomic identification of annexins, calcium-dependent membrane binding proteins that mediate osmotic stress and abscisic acid signal transduction in Arabidopsis. Plant Cell 2004, 16, 1378–1391. [Google Scholar] [CrossRef] [PubMed]
- McPhee, K. Variation for seedling root architecture in the core collection of pea germplasm. Crop Sci. 2005, 45, 1758–1763. [Google Scholar] [CrossRef]
- Shabalina, S.A.; Spiridonov, N.A.; Kashina, A. Sounds of silence: synonymous nucleotides as a key to biological regulation and complexity. Nucleic Acids Res. 2013, 41, 2073–2094. [Google Scholar] [CrossRef] [PubMed]
- Meisrimler, C.N.; Wienkoop, S.; Lyon, D.; Geilfus, C.M.; Lüthje, S. Long-term iron deficiency: Tracing changes in the proteome of different pea (Pisum sativum L.) cultivars. J. Proteom. 2016, 140, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Staudinger, C.; Mehmeti, V.; Turetschek, R.; Lyon, D.; Egelhofer, V.; Wienkoop, S. Possible Role of Nutritional Priming for Early Salt and Drought Stress Responses in Medicago truncatula. Front Plant Sci. 2012, 3, 285. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Fleming, R.M.T.; Thiele, I.; Saunders, M.A. Robust flux balance analysis of multiscale biochemical reaction networks. BMC Bioinf. 2013, 14, 240. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.; Nagel, A.; Herter, T.; May, P.; Schroda, M.; Zrenner, R.; Tohge, T.; Fernie, A.R.; Stitt, M.; Usadel, B. Mercator: A fast and simple web server for genome scale functional annotation of plant sequence data. Plant Cell Environ. 2014, 37, 1250–1258. [Google Scholar] [CrossRef] [PubMed]
- Turetschek, R.; Lyon, D.; Desalegn, G.; Kaul, H.P.; Wienkoop, S. A Proteomic Workflow Using High-Throughput De Novo Sequencing Towards Complementation of Genome Information for Improved Comparative Crop Science. In Proteomis in Systems Biology SE - 17 Methods in Molecular Biology; Reinders, J., Ed.; Springer: New York, NY, USA, 2016; pp. 233–243. [Google Scholar]
- Egelhofer, V.; Hoehenwarter, W.; Lyon, D.; Weckwerth, W.; Wienkoop, S. Using ProtMAX to Create High-Mass-Accuracy Precursor Alignments from Label-Free Quantitative Mass Spectrometry Data Generated in Shotgun Proteomics Experiments. Nat. Protoc. 2013, 8, 595–601. [Google Scholar] [CrossRef] [PubMed]
- TMHMM. Available online: http://www.cbs.dtu.dk/services/TMHMM/ (accessed on 1 September 2016).
- HMMTOP. Available online: http://www.enzim.hu/hmmtop/ (accessed on 1 January 2017).
- Chou, K.C.; Shen, H.B. Plant-mPLoc: A top-down strategy to augment the power for predicting plant protein subcellular localization. PLoS ONE 2010, 5, e11335. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.C.; Shen, H.B. Cell-PLoc: A package of web-servers for predicting subcellular localization of proteins in various organisms. Nat. Protoc. 2008, 3, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.C.; Shen, H.B. Large-Scale Plant Protein Subcellular Location Prediction. J. Cell. Biochem. 2007, 100, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Freedman, S.D.; Jamieson, J.D. Hormone-induced protein phosphorylation. II. Localization to the ribosomal fraction from rat exocrine pancreas and parotid of a 29,000-dalton protein phosphorylated in situ in response to secretagogues. J. Cell. Biol. 1982, 95, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Koldamova, R.P.; Lefterov, I.M.; DiSabella, M.T.; Almonte, C.; Watkins, S.C.; Lazo, J.S. Human bleomycin hydrolase binds ribosomal proteins. Biochemistry 1999, 38, 7111–7117. [Google Scholar] [CrossRef] [PubMed]
- Zargar, S.M.; Kurata, R.; Inaba, S.; Oikawa, A.; Fukui, R.; Ogata, Y.; Agrawal, G.K.; Rakwal, R.; Fukao, Y. Quantitative proteomics of Arabidopsis shoot microsomal proteins reveals a cross-talk between excess zinc and iron deficiency. Proteomics 2015, 15, 1196–1201. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lan, P.; Gao, H.; Zheng, L.; Li, W.; Schmidt, W. Expression changes of ribosomal proteins in phosphate-and iron-deficient Arabidopsis roots predict stress-specific alterations in ribosome composition. BMC Genom. 2013, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Lan, P.; Schmidt, W. The enigma of eIF5A in the iron deficiency response of Arabidopsis. Plant Sign. Behav. 2011, 6, 528–530. [Google Scholar] [CrossRef]
- Jelali, N.; Moez, S.; Dhifi, W.; Mnif, W.; Abdelly, C.; Gharsalli, M. Secondary metabolism responses in two Pisum sativum L. cultivars cultivated under Fe deficiency conditions. Afr. J. Biotech. 2012, 11, 14828–14836. [Google Scholar]
- Rahman, M.F.; Islam, M.; Begum, M.C.; Kabir, A.H.; Alam, M.F. Genetic variation in cadmium tolerance is related to transport and antioxidant activities in field peas (Pisum sativum L.). Arch. Agron. Soil Sci. 2016, 1–8. [Google Scholar] [CrossRef]
- Comas, L.H.; Becker, S.R.; von Mark, V.C.; Byrne, P.F.; Dierig, D. Root traits contributing to plant productivity under drought. Front. Plant Sci. 2013, 4, 442. [Google Scholar] [CrossRef] [PubMed]
- Chimungu, J.G.; Brown, K.M.; Lynch, J.P. Large root cortical cell size improves drought tolerance in maize. Plant Physiol. 2014, 166, 2166–2178. [Google Scholar] [CrossRef] [PubMed]
- Nie, L.; Feng, J.; Fan, P.; Chen, X.; Guo, J.; Lv, S.; Bao, H.; Jia, W.; Tai1, F.; Jiang, P.; et al. Comparative proteomics of root plasma membrane proteins reveals the involvement of calcium signalling in NaCl-facilitated nitrate uptake in Salicornia europaea. J. Exp. Bot. 2015, erv216. [Google Scholar] [CrossRef]
- Wienkoop, S.; Saalbach, G. Proteome analysis. Novel proteins identified at the peribacteroid membrane from Lotus japonicus root nodules. Plant Physiol. 2003, 131, 1080–1090. [Google Scholar] [CrossRef] [PubMed]
- Talukdar, T.; Gorecka, K.M.; de Carvalho-Niebel, F.; Downie, J.A.; Cullimore, J.; Pikula, S. Annexins—Calcium- and membrane-binding proteins in the plant kingdom. Potential role in nodulation and mycorrhization in Medicago truncatula. Acta Biochim. Pol. 2009, 56, 199–210. [Google Scholar] [PubMed]
- Lino, B.; Chagolla, A.; de la Vara, L.G. Membrane proteins involved in transport, vesicle traffic and Ca2+ signaling increase in beetroots grown in saline soils. Planta 2016, 244, 87–101. [Google Scholar] [CrossRef] [PubMed]
- Baucher, M.; Lowe, Y.O.; Vandeputte, O.M.; Bopopi, J.M.; Moussawi, J.; Vermeersch, M.; Mol, A.; El Jaziri, M.; Homblé, F.; Pérez-Morga, D. Ntann12 annexin expression is induced by auxin in tobacco roots. J. Exp. Bot. 2011, 62, 4055–4065. [Google Scholar] [CrossRef] [PubMed]
- Overvoorde, P.; Fukaki, H.; Beeckman, T. Auxin control of root development. Cold Spring Harb. Perspect. Biol. 2010, 2, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Saini, S.; Sharma, I.; Kaur, N.; Pati, P.K. Auxin: A master regulator in plant root development. Plant Cell Rep. 2013, 32, 741–757. [Google Scholar] [CrossRef] [PubMed]
- James, B.; Koepfli, J.B. The Inhibition of Root Growth by Auxins. Am. J. Bot. 1939, 26, 557–566. [Google Scholar]
- Croy, R.R.; Gatehouse, J.A.; Evans, I.M.; Boulter, D. Characterisation of the storage protein subunits synthesised in vitro by polyribosomes and RNA from developing pea (Pisum sativum L.). Planta 1980, 148, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Klein, E.M.; Mascheroni, L.; Pompa, A.; Ragni, L.; Weimar, T.; Lilley, K.S.; Dupree, P.; Vitale, A. Plant endoplasmin supports the protein secretory pathway and has a role in proliferating tissues. Plant J. 2006, 48, 657–673. [Google Scholar] [CrossRef] [PubMed]
- Qian, D.; Tian, L.; Qu, L. Proteomic analysis of endoplasmic reticulum stress responses in rice seeds. Sci. Rep. 2015, 5, 14255. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wei, Z.; Qiao, Z.; Wu, Z.; Cheng, L.; Wang, Y. Proteomics Analysis of Alfalfa Response to Heat Stress. PLoS ONE 2013, 8, e82725. [Google Scholar] [CrossRef] [PubMed]
- Hill, K.; Model, K.; Ryan, M.T.; Dietmeier, K.; Martin, F.; Wagner, R.; Pfanner, N. Tom40 forms the hydrophilic channel of the mitochondrial import pore for preproteins. Nature 1998, 395, 516–521. [Google Scholar] [PubMed]
- Hsieh, W.Y.; Liao, J.C.; Hsieh, M.H. Dysfunctional mitochondria regulate the size of root apical meristem and leaf development in Arabidopsis. Plant Signal. Behav. 2015, 10, e1071002. [Google Scholar] [CrossRef] [PubMed]
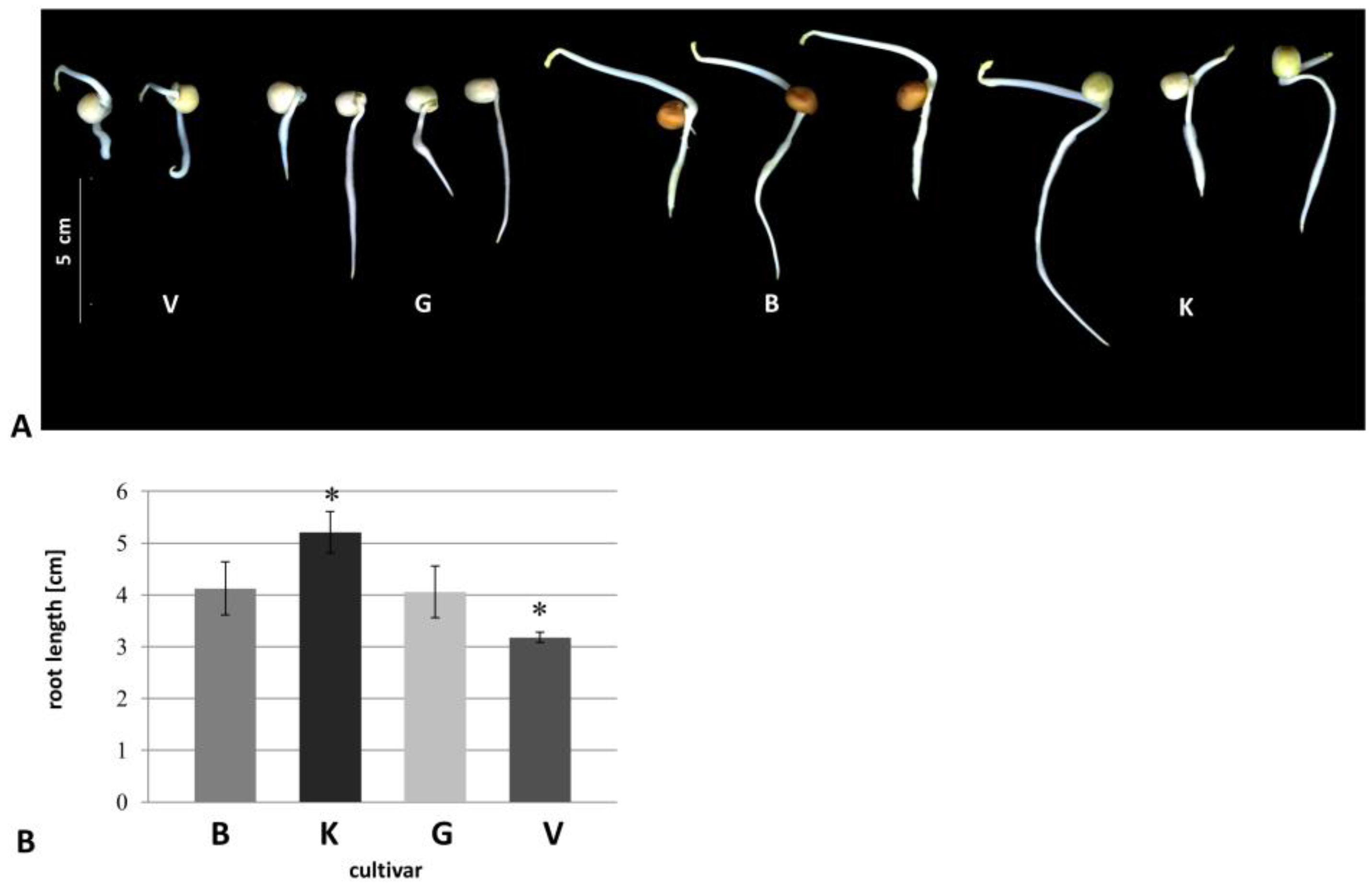

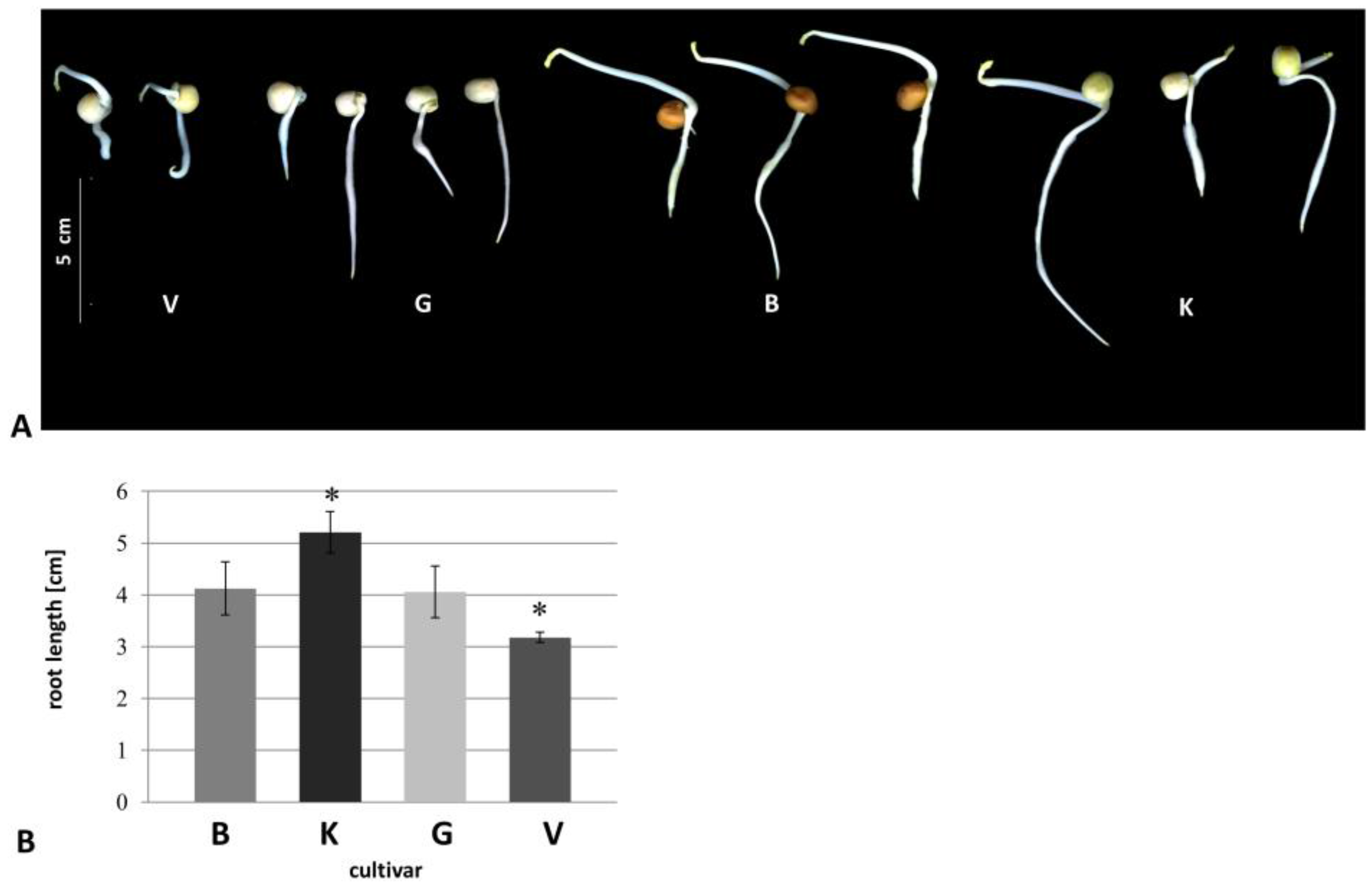{kind=link}
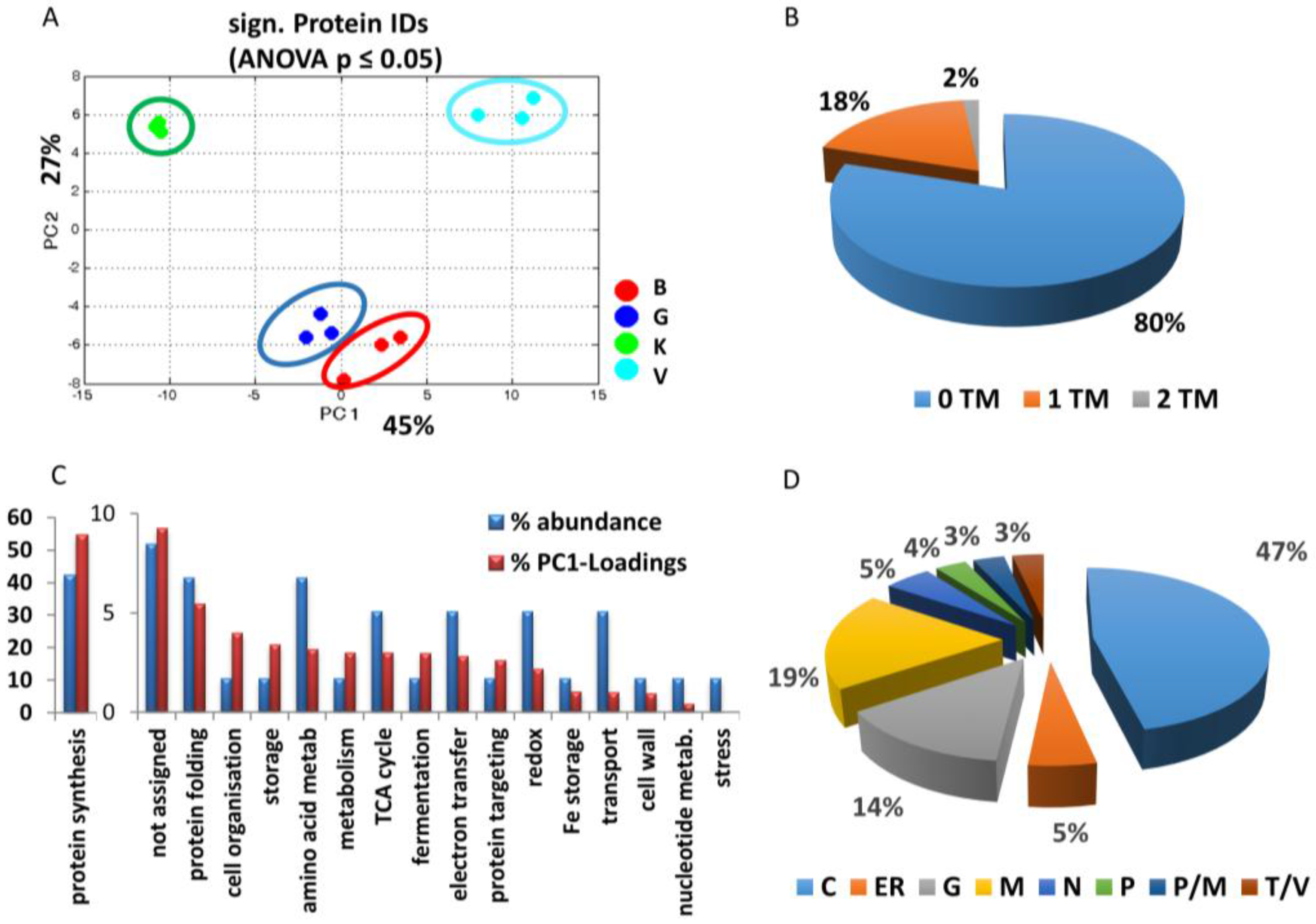{kind=link}
| Accession | Description | TM1 | TM2 | Function | SC | Ratio | PC1 loadings | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| loc | B | V | G | K | V/K | B/K | G/K | V/G | V/B | G/B | ||||||
| contig12248 | Annexin-like protein RJ4 | 0 | 0 | cell organisation | N * | 5.7 | 18.7 | 3.7 | 0.5 | 37.3 | 11.3 | 7.3 | 5.1 | 3.3 | 0.7 | 0.2490 |
| p.sativum_wa1_contig21697 | 60S ribosomal protein L4-1 | 0 | 0 | protein synthesis | C | 10.3 | 15.3 | 8.7 | 0.5 | 30.7 | 20.7 | 17.3 | 1.8 | 1.5 | 0.8 | 0.2379 |
| contig10741 | Convicilin | 0 | 0 | storage | G * | 1.0 | 11.7 | 0.5 | 0.5 | 23.3 | 2.0 | 1.0 | 23.3 | 11.1 | 0.5 | 0.2130 |
| p.sativum_wa1_contig28348 | 40S ribosomal protein S8 | 0 | 0 | protein synthesis | C | 3.2 | 8.0 | 1.7 | 0.5 | 16.0 | 6.3 | 3.3 | 4.8 | 2.5 | 0.5 | 0.2017 |
| p.sativum_wa1_contig16856 | 60S acidic ribosomal protein P2-2 | 0 | 0 | protein synthesis | C | 2.0 | 10.7 | 4.0 | 0.5 | 21.3 | 4.0 | 8.0 | 2.7 | 5.3 | 2.0 | 0.1961 |
| contig17941 | Endoplasmin homolog | 0 | 0 | protein folding | ER | 2.5 | 9.0 | 6.0 | 0.5 | 18.0 | 5.0 | 12.0 | 1.5 | 3.6 | 2.5 | 0.1904 |
| 9955324 pdb | Dihydrolipoamide dehydrogenase | 0 | 0 | metabolism | M* | 1.3 | 8.0 | 0.5 | 0.5 | 16.0 | 2.7 | 1.0 | 16.0 | 5.9 | 0.4 | 0.1878 |
| Pisum_sativum_v2_Contig4072 | ADH family 2 member B7 | 0 | 1 | fermentation | M | 12.3 | 10.3 | 5.7 | 1.0 | 10.3 | 12.3 | 5.7 | 1.8 | 0.8 | 0.5 | 0.1850 |
| contig02118 | 60S ribosomal protein L13-1 | 0 | 0 | protein synthesis | C | 1.7 | 6.7 | 0.5 | 0.5 | 13.3 | 3.3 | 1.0 | 13.3 | 4.0 | 0.3 | 0.1830 |
| p.sativum_wa1_contig25888 | 40S ribosomal protein S4-2 | 0 | 0 | protein synthesis | C | 3.0 | 6.0 | 2.2 | 0.5 | 12.0 | 6.0 | 4.3 | 2.8 | 2.0 | 0.7 | 0.1766 |
| Contig3080 | 40S ribosomal protein S17-4 | 0 | 0 | protein synthesis | C | 4.0 | 5.0 | 0.5 | 0.5 | 10.0 | 8.0 | 1.0 | 10.0 | 1.3 | 0.1 | 0.1763 |
| EX568861 | 40S ribosomal protein S17 | 0 | 0 | protein synthesis | C | 4.0 | 5.0 | 0.5 | 0.5 | 10.0 | 8.0 | 1.0 | 10.0 | 1.3 | 0.1 | 0.1763 |
| GH720005 | 60S ribosomal protein L9 | 0 | 0 | protein synthesis | C | 5.0 | 6.3 | 5.0 | 0.5 | 12.7 | 10.0 | 10.0 | 1.3 | 1.3 | 1.0 | 0.1755 |
| p.sativum_wa1_contig17528 | 60S ribosomal protein L13-1 | 0 | 0 | protein synthesis | C | 1.7 | 5.3 | 1.3 | 0.5 | 10.7 | 3.3 | 2.7 | 4.0 | 3.2 | 0.8 | 0.1642 |
| Pisum_sativum_v2_Contig5665 | 60S ribosomal protein L4 | 0 | 0 | protein synthesis | C | 2.5 | 5.3 | 1.3 | 0.5 | 10.7 | 5.0 | 2.7 | 4.0 | 2.1 | 0.5 | 0.1621 |
| p.sativum_wa1_contig24787 | 40S ribosomal protein S2-4 | 0 | 0 | protein synthesis | C | 1.5 | 5.3 | 0.5 | 0.5 | 10.7 | 3.0 | 1.0 | 10.7 | 3.6 | 0.3 | 0.1604 |
| contig13719 | 40S ribosomal protein SA | 0 | 0 | protein synthesis | C | 0.5 | 6.0 | 0.5 | 0.5 | 12.0 | 1.0 | 1.0 | 12.0 | 12.5 | 1.0 | 0.1585 |
| p.sativum_wa1_contig21882 | 60S ribosomal protein L4-1 | 0 | 0 | protein synthesis | C | 2.2 | 5.0 | 1.3 | 0.5 | 10.0 | 4.3 | 2.7 | 3.8 | 2.3 | 0.6 | 0.1570 |
| p.sativum_wa1_contig07673 | 40S ribosomal protein S15-4 | 0 | 0 | protein synthesis | C | 5.0 | 4.7 | 6.0 | 0.5 | 9.3 | 10.0 | 12.0 | 0.8 | 0.9 | 1.3 | 0.1541 |
| p.sativum_wa1_contig22133 | 40S ribosomal protein SA | 0 | 0 | protein synthesis | C | 0.5 | 5.3 | 0.5 | 0.5 | 10.7 | 1.0 | 1.0 | 10.7 | 11.1 | 1.0 | 0.1517 |
| ACU20293.1 | 40S ribosomal protein S30 | 0 | 0 | protein synthesis | C | 4.3 | 4.3 | 4.7 | 0.5 | 8.7 | 8.7 | 9.3 | 0.9 | 1.0 | 1.1 | 0.1486 |
| ACJ85955.1 | 60S ribosomal L12-like protein | 0 | 0 | protein synthesis | C | 1.3 | 4.3 | 1.5 | 0.5 | 8.7 | 2.7 | 3.0 | 2.9 | 3.2 | 1.1 | 0.1429 |
| BAB40231.1 | S-type apyrase | 0 | 1 | not assigned | N* | 8.0 | 3.3 | 8.0 | 0.5 | 6.7 | 16.0 | 16.0 | 0.4 | 0.4 | 1.0 | 0.1345 |
| p.sativum_wa1_contig03572 | 40S ribosomal protein S3a | 0 | 0 | protein synthesis | C | 0.5 | 3.7 | 0.5 | 0.5 | 7.3 | 1.0 | 1.0 | 7.3 | 7.1 | 1.0 | 0.1274 |
| contig09310 | Uncharacterized protein At5g10860 | 0 | 0 | not assigned | M | 1.7 | 2.7 | 0.5 | 0.5 | 5.3 | 3.3 | 1.0 | 5.3 | 1.6 | 0.3 | 0.1241 |
| Pisum_sativum_v2_Contig1288 | 40S ribosomal protein S3a | 0 | 0 | protein synthesis | C | 0.5 | 3.3 | 0.5 | 0.5 | 6.7 | 1.0 | 1.0 | 6.7 | 6.7 | 1.0 | 0.1209 |
| Pisum_sativum_v2_Contig4787 | Protein disulfide isomerase-like 1-4 | 0 | 1 | redox | ER | 4.7 | 6.0 | 7.7 | 1.7 | 3.6 | 2.8 | 4.6 | 0.8 | 1.3 | 1.7 | 0.1198 |
| Pisum_sativum_v2_Contig857 | 40S ribosomal protein S18 | 0 | 0 | protein synthesis | C | 0.5 | 3.0 | 0.5 | 0.5 | 6.0 | 1.0 | 1.0 | 6.0 | 5.9 | 1.0 | 0.1132 |
| p.sativum_wa1_contig04121 | LOX homol. domain-containing protein 1 | 0 | 0 | not assigned | G* | 2.3 | 2.7 | 3.7 | 0.5 | 5.3 | 4.7 | 7.3 | 0.7 | 1.1 | 1.7 | 0.1119 |
| Pisum_sativum_v2_Contig5583 | Succinyl-CoA ligase sub. b | 0 | 1 | TCA cycle | G* | 0.5 | 5.3 | 0.5 | 1.3 | 4.0 | 0.4 | 0.4 | 10.7 | 11.1 | 1.0 | 0.1109 |
| GH720468 | 40S ribosomal protein S25-2 | 0 | 0 | protein synthesis | C | 4.3 | 3.5 | 3.3 | 0.5 | 7.0 | 8.7 | 6.7 | 1.1 | 0.8 | 0.8 | 0.1089 |
| Contig386 | 40S ribosomal protein S14 | 0 | 0 | protein synthesis | C | 11.3 | 15.0 | 12.0 | 4.7 | 3.2 | 2.4 | 2.6 | 1.3 | 1.3 | 1.1 | 0.0823 |
| GH720846 | 60S ribosomal protein L22-2 | 0 | 0 | protein synthesis | C | 3.0 | 1.8 | 1.5 | 0.5 | 3.7 | 6.0 | 3.0 | 1.2 | 0.6 | 0.5 | 0.0781 |
| contig09315 | Cysteine synthase | 0 | 0 | amino acid metab | G | 16.7 | 15.7 | 6.7 | 6.3 | 2.5 | 2.6 | 1.1 | 2.4 | 0.9 | 0.4 | 0.0702 |
| AAB24082.1 | ferritin | 0 | 0 | Fe storage | P | 4.3 | 8.7 | 4.3 | 3.3 | 2.6 | 1.3 | 1.3 | 2.0 | 2.0 | 1.0 | 0.0643 |
| contig21497 | Cysteine synthase | 0 | 0 | amino acid metab. | P/M | 18.7 | 16.0 | 7.7 | 7.3 | 2.2 | 2.5 | 1.0 | 2.1 | 0.9 | 0.4 | 0.0628 |
| Pisum_sativum_v2_Contig4779 | UDP-glucuronic acid decarboxylase 1 | 1 | 1 | cell wall | G/N * | 0.5 | 1.8 | 3.7 | 0.5 | 3.7 | 1.0 | 7.3 | 0.5 | 3.7 | 10.0 | 0.0584 |
| contig20699 | NADH-Q oxidoreductase 40 kDa sub. | 0 | 0 | electron transfer | M | 3.0 | 3.3 | 0.5 | 2.5 | 1.3 | 1.2 | 0.2 | 6.7 | 1.1 | 0.2 | 0.0532 |
| p.sativum_wa1_contig18574 | Chaperonin CPN60-2 | 0 | 0 | protein folding | M | 17.7 | 18.7 | 7.3 | 10.3 | 1.8 | 1.7 | 0.7 | 2.5 | 1.1 | 0.4 | 0.0515 |
| contig22493 | Cysteine synthase | 0 | 0 | amino acid metab | G | 18.7 | 10.0 | 5.3 | 6.3 | 1.6 | 2.9 | 0.8 | 1.9 | 0.5 | 0.3 | 0.0472 |
| Pisum_sativum_v2_Contig6323 | component of 2-oxoglutarate dehydrogenase | 0 | 1 | TCA cycle | G* | 5.0 | 4.7 | 1.0 | 3.3 | 1.4 | 1.5 | 0.3 | 4.7 | 0.9 | 0.2 | 0.0455 |
| p.sativum_wa1_contig18562 | Chaperonin CPN60-2 | 0 | 0 | protein folding | M | 14.3 | 17.7 | 5.3 | 11.0 | 1.6 | 1.3 | 0.5 | 3.3 | 1.2 | 0.4 | 0.0439 |
| Pisum_sativum_v2_Contig1332 | Probable arginase | 1 | 1 | amino acid metab. | G | 4.3 | 8.0 | 11.3 | 5.3 | 1.5 | 0.8 | 2.1 | 0.7 | 1.9 | 2.5 | 0.0191 |
| 189095946 pdb | Mitochondrial Type Ii Peroxiredoxin | 0 | 0 | redox | M | 0.5 | 5.7 | 0.5 | 4.0 | 1.4 | 0.1 | 0.1 | 11.3 | 11.1 | 1.0 | 0.0130 |
| p.sativum_wa1_contig02960 | Elongation factor 1-alpha | 0 | 0 | protein synthesis | C | 1.3 | 4.3 | 5.3 | 3.3 | 1.3 | 0.4 | 1.6 | 0.8 | 3.2 | 3.3 | 0.0026 |
| P37900.585272 sp | HSP70 | 0 | 0 | stress | M | 0.5 | 6.0 | 0.5 | 5.3 | 1.1 | 0.1 | 0.1 | 12.0 | 12.5 | 1.0 | -0.0022 |
| Pisum_sativum_v2_Contig5744 | Probable protein disulfide-isomerase A6 | 0 | 1 | redox | ER | 12.7 | 25.7 | 18.3 | 25.3 | 1.0 | 0.5 | 0.7 | 1.4 | 2.0 | 1.4 | -0.0040 |
| Pisum_sativum_v2_Contig5499 | NADH-Q oxidoreductase subunit | 0 | 1 | electron transfer | G* | 5.0 | 7.7 | 2.5 | 9.3 | 0.8 | 0.5 | 0.3 | 3.1 | 1.5 | 0.5 | -0.0090 |
| G9JKP3 | Plastid OEP 16.2 | 0 | 2 | transport | P | 0.5 | 5.7 | 2.5 | 5.7 | 1.0 | 0.1 | 0.4 | 2.3 | 11.1 | 5.0 | -0.0138 |
| CBD35496.1 | Elongation factor 1-alpha | 0 | 0 | protein synthesis | C | 0.5 | 3.7 | 4.0 | 3.3 | 1.1 | 0.2 | 1.2 | 0.9 | 7.1 | 10.0 | -0.0179 |
| p.sativum_wa1_contig06534 | V-type proton ATPase 116 kDa sub.a 1 | 0 | 0 | transport | T/V | 3.0 | 0.5 | 0.5 | 1.3 | 0.4 | 2.3 | 0.4 | 1.0 | 0.2 | 0.2 | -0.0201 |
| p.sativum_wa1_contig17185 | Elongation factor 1-alpha | 0 | 0 | protein synthesis | C | 0.5 | 3.0 | 2.5 | 3.0 | 1.0 | 0.2 | 0.8 | 1.2 | 5.9 | 5.0 | -0.0217 |
| 56554368 pdb | Nucleoside diphosphate kinase | 0 | 1 | not assigned | M | 1.0 | 4.0 | 2.5 | 5.0 | 0.8 | 0.2 | 0.5 | 1.6 | 4.0 | 2.5 | -0.0246 |
| p.sativum_wa1_contig12891 | Adenylate kinase B | 0 | 0 | nucleotide metab. | C | 0.5 | 8.3 | 4.0 | 8.0 | 1.0 | 0.1 | 0.5 | 2.1 | 16.7 | 10.0 | -0.0259 |
| p.sativum_wa1_contig19033 | V-type proton ATPase subunit E | 0 | 0 | transport | T/V | 8.0 | 2.7 | 6.0 | 4.7 | 0.6 | 1.7 | 1.3 | 0.4 | 0.3 | 0.8 | -0.0281 |
| contig10734 | SDH flavoprotein subunit 1 | 0 | 0 | TCA cycle | M | 2.0 | 6.7 | 1.7 | 10.0 | 0.7 | 0.2 | 0.2 | 4.0 | 3.3 | 0.8 | -0.0313 |
| contig20456 | HSP 70 kDa | 0 | 0 | protein folding | M | 0.5 | 3.5 | 0.5 | 4.3 | 0.8 | 0.1 | 0.1 | 7.0 | 7.1 | 1.0 | -0.0558 |
| p.sativum_wa1_contig16648 | ferredoxin | 0 | 0 | electron transfer | P/M | 4.3 | 1.0 | 3.3 | 5.0 | 0.2 | 0.9 | 0.7 | 0.3 | 0.2 | 0.8 | -0.1138 |
| Pisum_sativum_v2_Contig2233 | TOM40 homolog 1 | 1 | 1 | protein targeting | M | 0.5 | 1.3 | 0.5 | 8.3 | 0.2 | 0.1 | 0.1 | 2.7 | 2.6 | 1.0 | -0.1632 |
| Q43877 | Duplicate protein - HMG-I/Y | 0 | 0 | not assigned | N | 0.5 | 0.5 | 6.0 | 5.3 | 0.1 | 0.1 | 1.1 | 0.1 | 1.0 | 10.0 | -0.1842 |
| Significant fold change | 36 | 31 | 20 | 31 | 29 | 23 | ||||||||||
| 41 | 46 | 36 | 42 | 37 | 29 | |||||||||||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meisrimler, C.-N.; Wienkoop, S.; Lüthje, S. Proteomic Profiling of the Microsomal Root Fraction: Discrimination of Pisum sativum L. Cultivars and Identification of Putative Root Growth Markers. Proteomes 2017, 5, 8. https://doi.org/10.3390/proteomes5010008
Meisrimler C-N, Wienkoop S, Lüthje S. Proteomic Profiling of the Microsomal Root Fraction: Discrimination of Pisum sativum L. Cultivars and Identification of Putative Root Growth Markers. Proteomes. 2017; 5(1):8. https://doi.org/10.3390/proteomes5010008
Chicago/Turabian StyleMeisrimler, Claudia-Nicole, Stefanie Wienkoop, and Sabine Lüthje. 2017. "Proteomic Profiling of the Microsomal Root Fraction: Discrimination of Pisum sativum L. Cultivars and Identification of Putative Root Growth Markers" Proteomes 5, no. 1: 8. https://doi.org/10.3390/proteomes5010008