Fatty Liver and Fatty Heart—Where do They Stand in the AMIS Syndrome?

Abstract
:
1. Introduction
2. Experimental
2.1. Animals and Groups (n = 12–18/group)

2.2. Surgical Preparation
2.3. Rapid Insulin Sensitivity Test (RIST)
2.4. Tissue and Blood Samples
2.5. Chemicals
2.6. Statistical Analysis
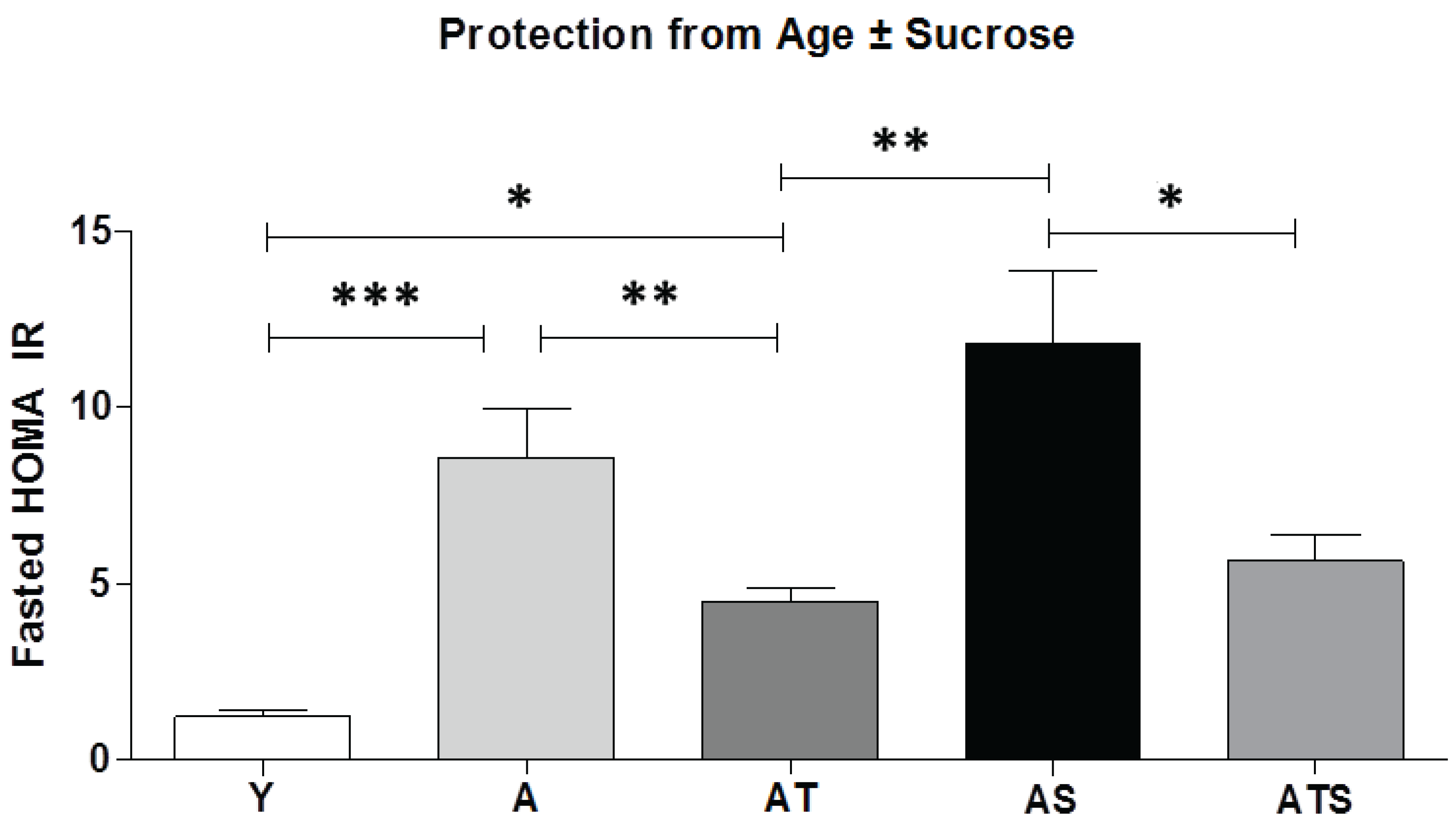
3. Results and Discussion
3.1. Insulin and HISS Action
3.2. Fasting and Postprandial Metabolic Parameters


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nine-Week-Old Rats | 52-Week-Old Rats | |||||
|---|---|---|---|---|---|---|
| Parameters | Young Controls (Y) | Aged (A) | Aged Sucrose (AS) | Aged Treated (AT) | Aged Treated Sucrose (ATS) | p < 0.05 |
| Body Weight g | 366 ± 112.8 (12) | 795 ± 24.1 * (12) | 853 ± 26.6 * (12) | 783 ± 21.4 * (12) | 802 ± 19.4 * (12) | * vsY |
| MAP mmHg | 114 ± 3.7(12) | 121 ± 3.7 (12) | 114 ± 4.3 (12) | 114 ± 5.2 (12) | 118 ± 5.0 (12) | Ns |
| Fasted Glucose mg/dl | 80.1 ± 3.0 (11) | 99.0 ± 2.6 * (12) | 102.8 ± 3.2 * (12) | 89.7 ± 1.7 (12) | 91.3 ± 2.4 *§ (11) | * vsY, § vsAS |
| Fed Glucose mg/dl | 105.4 ± 5.0 (11) | 122.5 ± 4.7 (12) | 147.3 ± 11.6 * (12) | 117.2 ± 5.0 (11) | 121.6 ± 5.0 § (11) | * vsY, § vsAS |
| Fasted Insulin μg/L | 0.307 ± 0.04 (11) | 1.65 ± 0.26 * (11) | 1.72 ± 0.28 * (11) | 0.90 ± 0.07 (11) | 1.17 ± 0.16 * (11) | * vsY, § vsAS |
| Fed Insulin μg/L | 2.63 ± 0.42 (11) | 9.79 ± 2.34 § (11) | 17.22 ± 3.13 * (11) | 7.12 ± 0.91 (11) | 7.26 ± 0.85 § (11) | * vsY, § vsAS |
| Whole Body FM g | 31.9 ± 2.1 (14) | 263.6 ± 13.7 *§ (12) | 331.6 ± 17.3 * (13) | 221.5 ± 17.0 * (14) | 249.0 ± 13.1 *§ (13) | * vsY, § vsAS |
| Whole Body FM % | 8.7 ± 0.4 (14) | 33.4 ± 1.2 *€ (12) | 38.2 ± 1.0 * (13) | 28.4 ± 1.6 * (14) | 30.8 ± 1.4 *§ (13) | * vsY, § vsAS € vsAT |
| Total Visceral FM g | 11.6 ± 0.8 (14) | 83.4 ± 3.9 *§ (12) | 105.8 ± 6.1 * (13) | 69.3 ± 5.6 * (14) | 80.8 ± 4.9 *§ (13) | * vsY, § vsAS |
| Total Visceral FM % | 3.18 ± 0.17 (14) | 10.6 ± 0.3 *€ (12) | 12.2 ± 0.38 * (13) | 8.9 ± 0.57 * (14) | 10.0 ± 0.54 *§ (13) | * vsY, § vsAS € vsAT |
| Plasma Triglycerides mmol/L | 0.57 ± 0.11 (14) | 1.76 ± 0.24 *§ (11) | 3.24 ± 0.40 * (12) | 1.81 ± 0.19 * (12) | 1.90 ± 0.25 *§ (12) | * vsY, § vsAS |
| Total Chol/HDL Chol | 1.26 ± 0.03 (14) | 1.69 ± 0.12 * (12) | 1.80 ± 0.11 * (11) | 1.65 ± 0.09 * (12) | 1.62 ± 0.06 *§ (13) | * vsY, § vsAS |
| AST | 212.3 ± 13.1 (14) | 316.7 ± 48.9 (12) | 309.0 ± 31.0 (14) | 293.4 ± 26.7 (11) | 295.4 ± 23.8 (14) | Ns |
| ALT | 69.5 ± 4.7 (14) | 117.3 ± 13.8* (12) | 121.6 ± 14.0 * (14) | 117.5 ± 14.1 * (11) | 107.3 ± 10.7 (14) | * vsY |

3.3. Hepatic and Cardiac Triglycerides (TG), and Body Fat



3.4. AMIS, Hepatic and Plasma Triglycerides and NEFA
4. Current Status of the AMIS Syndrome Hypothesis
5. Conclusions
Acknowledgments
Author Contributions
Conflict of Interest
References
- Erickson, S.K. Nonalcoholic fatty liver disease. J. Lipid Res. 2009, 50, S412–S416. [Google Scholar]
- Browning, J.D.; Szczepaniak, L.S.; Dobbins, R.; Nuremberg, P.; Horton, J.D.; Cohen, J.C.; Grundy, S.M.; Hobbs, H.H. Prevalence of hepatic steatosis in an urban population in the United States: Impact of ethnicity. Hepatology 2004, 40, 1387–1395. [Google Scholar]
- Farrell, G.C.; Larter, C.Z. Nonalcoholic fatty liver disease: From steatosis to cirrhosis. Hepatology 2006, 43, S99–S112. [Google Scholar]
- Catta-Preta, M.; Mendonca, L.S.; Fraulob-Aquino, J.; Aguila, M.B.; Mandarim-de-Lacerda, C.A. A critical analysis of three quantitative methods of assessment of hepatic steatosis in liver biopsies. Virchows Arch. 2011, 459, 477–485. [Google Scholar] [PubMed]
- Christman, A.L.; Lazo, M.; Clark, J.M.; Selvin, E. Low glycated hemoglobin and liver disease in the U.S. population. Diabetes Care 2011, 34, 2548–2550. [Google Scholar] [PubMed]
- Magkos, F. Putative factors that may modulate the effect of exercise on liver fat: Insights from animal studies. J. Nutr. MeTable 2012. [Google Scholar] [CrossRef]
- Lattuada, G.; Ragogna, F.; Perseghin, G. Why does NAFLD predict type 2 diabetes? Curr. Diab. Rep. 2011, 11, 167–172. [Google Scholar] [PubMed]
- Lautt, W.W.; Wang, H.H. Obesity as an early symptom of the AMIS syndrome. J. Clin. Med. 2014, 3, 1178–1198. [Google Scholar] [PubMed]
- Sadri, P.; Reid, M.A.; Afonso, R.A.; Schafer, J.; Legare, D.J.; Macedo, P.M.; Lautt, W.W. Meal-induced insulin sensitization in conscious and anaesthetized rat models comparing liquid mixed meal with glucose and sucrose. Br. J. Nutr. 2006, 95, 288–295. [Google Scholar] [PubMed]
- Chang, C.L.; Lin, Y.; Bartolome, A.P.; Chen, Y.C.; Chiu, S.C.; Yang, W.C. Herbal therapies for type 2 diabetes mellitus: Chemistry, biology, and potential application of selected plants and compounds. Evid. Based Complement. Alternat. Med. 2013. [Google Scholar] [CrossRef]
- Lautt, W.W. The HISS story overview: A novel hepatic neurohumoral regulation of peripheral insulin sensitivity in health and diabetes. Can. J. Physiol. Pharmacol. 1999, 77, 553–562. [Google Scholar] [PubMed]
- Fernandes, A.B.; Patarrao, R.S.; Videira, P.A.; Macedo, M.P. Understanding postprandial glucose clearance by peripheral organs: The role of the hepatic parasympathetic system. J. Neuroendocrinol. 2011, 23, 1288–1295. [Google Scholar] [PubMed]
- Lautt, W.W.; Macedo, M.P.; Sadri, P.; Takayama, S.; Duarte Ramos, F.; Legare, D.J. Hepatic parasympathetic (HISS) control of insulin sensitivity determined by feeding and fasting. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G29–G36. [Google Scholar] [PubMed]
- Patarrao, R.S.; Lautt, W.W.; Afonso, R.A.; Ribeiro, R.T.; Guarino, M.P.; Fernandes, A.B.; Boavida, J.M.; Macedo, M.P. Meal-induced insulin sensitization and its parasympathetic regulation in humans. Can. J. Physiol. Pharmacol. 2008, 86, 880–888. [Google Scholar] [PubMed]
- Lautt, W.W.; Ming, Z.; Legare, D.J. Attenuation of age- and sucrose-induced insulin resistance and syndrome X by a synergistic antioxidant cocktail: The AMIS syndrome and HISS hypothesis. Can. J. Physiol. Pharmacol. 2010, 88, 313–323. [Google Scholar] [PubMed]
- Wang, H.H.; Chowdhury, K.K.; Lautt, W.W. A synergistic, balanced antioxidant cocktail, protects aging rats from insulin resistance and absence of meal-induced insulin sensitization (AMIS) syndrome. Molecules 2015, 20, 669–682. [Google Scholar] [PubMed]
- Ming, Z.; Lautt, W.W. HISS, not insulin, causes vasodilation in response to administered insulin. J. Appl. Physiol. 2011, 110, 60–68. [Google Scholar] [PubMed]
- Lautt, W.W.; Ming, Z.; Macedo, M.P.; Legare, D.J. HISS-dependent insulin resistance (HDIR) in aged rats is associated with adiposity, progresses to syndrome X, and is attenuated by a unique antioxidant cocktail. Exp. Gerontol. 2008, 43, 790–800. [Google Scholar] [PubMed]
- Ming, Z.; Legare, D.J.; Lautt, W.W. Absence of meal-induced insulin sensitization (AMIS) in aging rats is associated with cardiac dysfunction that is protected by antioxidants. J. Appl. Physiol. 2011, 111, 704–714. [Google Scholar] [PubMed]
- Ming, Z.; Legare, D.J.; Lautt, W.W. Obesity, syndrome X, and diabetes: The role of HISS-dependent insulin resistance altered by sucrose, an antioxidant cocktail, and age. Can. J. Physiol. Pharmacol. 2009, 87, 873–882. [Google Scholar] [PubMed]
- Ribeiro, R.T.; Lautt, W.W.; Legare, D.J.; Macedo, M.P. Insulin resistance induced by sucrose feeding in rats is due to an impairment of the hepatic parasympathetic nerves. Diabetologia 2005, 48, 976–983. [Google Scholar] [PubMed]
- Afonso, R.A.; Lautt, W.W.; Schafer, J.; Legare, D.J.; Oliveira, A.G.; Macedo, M.P. High-fat diet results in postprandial insulin resistance that involves parasympathetic dysfunction. Br. J. Nutr. 2010, 104, 1450–1459. [Google Scholar] [PubMed]
- Chowdhury, K.K.; Legare, D.J.; Lautt, W.W. Insulin sensitization by voluntary exercise in aging rats is mediated through hepatic insulin sensitizing substance (HISS). Exp. Gerontol. 2011, 46, 73–80. [Google Scholar] [PubMed]
- Chowdhury, K.K.; Legare, D.J.; Lautt, W.W. Exercise enhancement of hepatic insulin-sensitising substance-mediated glucose uptake in diet-induced prediabetic rats. Br. J. Nutr. 2013, 109, 844–852. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, K.K.; Legare, D.J.; Lautt, W.W. Interaction of antioxidants and exercise on insulin sensitivity in healthy and prediabetic rats. Can. J. Physiol. Pharmacol. 2013, 91, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, K.K.; Legare, D.J.; Lautt, W.W. Lifestyle impact on meal-induced insulin sensitization in health and prediabetes: A focus on diet, antioxidants, and exercise interventions. Can. J. Physiol. Pharmacol. 2013, 91, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Dirkx, E.; van Eys, G.J.; Schwenk, R.W.; Steinbusch, L.K.; Hoebers, N.; Coumans, W.A.; Peters, T.; Janssen, B.J.; Brans, B.; Vogg, A.T.; et al. Protein kinase-D1 overexpression prevents lipid-induced cardiac insulin resistance. J. Mol. Cell. Cardiol. 2014, 76, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Kok, B.P.; Brindley, D.N. Myocardial fatty acid metabolism and lipotoxicity in the setting of insulin resistance. Heart Fail. Clin. 2012, 8, 643–661. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Muoio, D.M.; Shiota, M.; Fujimoto, Y.; Cline, G.W.; Shulman, G.I.; Koves, T.R.; Stevens, R.; Millington, D.; Newgard, C.B. Hepatic expression of malonyl-CoA decarboxylase reverses muscle, liver and whole-animal insulin resistance. Nat. Med. 2004, 10, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.H.; Stein, D.T.; Barzilai, N.; Cui, M.H.; Tonelli, J.; Kishore, P.; Hawkins, M. Increased intrahepatic triglyceride is associated with peripheral insulin resistance: In vivo MR imaging and spectroscopy studies. Am. J. Physiol. Endocrinol. MeTable 2007, 293, E1663–E1669. [Google Scholar] [CrossRef] [PubMed]
- Perseghin, G. The role of non-alcoholic fatty liver disease in cardiovascular disease. Dig. Dis. 2010, 28, 210–213. [Google Scholar] [CrossRef] [PubMed]
- Marchesini, G.; Brizi, M.; Morselli-Labate, A.M.; Bianchi, G.; Bugianesi, E.; McCullough, A.J.; Forlani, G.; Melchionda, N. Association of nonalcoholic fatty liver disease with insulin resistance. Am. J. Med. 1999, 107, 450–455. [Google Scholar] [CrossRef]
- Ribeiro, R.T.; Afonso, R.A.; Guarino, M.P.; Macedo, M.P. Loss of postprandial insulin sensitization during aging. J. Gerontol. A Biol. Sci. Med. Sci. 2008, 63, 560–565. [Google Scholar] [CrossRef] [PubMed]
- Lautt, W.W.; Wang, X.; Sadri, P.; Legare, D.J.; Macedo, M.P. Rapid insulin sensitivity test (RIST). Can. J. Physiol. Pharmacol. 1998, 76, 1080–1086. [Google Scholar] [CrossRef] [PubMed]
- Lautt, W.W. Practice and principles of pharmacodynamic determination of HISS-dependent and HISS-independent insulin action: Methods to quantitate mechanisms of insulin resistance. Med. Res. Rev. 2003, 23, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Snyder, F.; Stephens, N. A simplified spectrophotometric determination of ester groups in lipids. Biochim. Biophys. Acta 1959, 34, 244–245. [Google Scholar]
- Tardi, P.G.; Mukherjee, J.J.; Choy, P.C. The quantitation of long-chain acyl-CoA in mammalian tissue. Lipids 1992, 27, 65–67. [Google Scholar] [CrossRef]
- Poanta, L.I.; Albu, A.; Fodor, D. Association between fatty liver disease and carotid atherosclerosis in patients with uncomplicated type 2 diabetes mellitus. Med. Ultrason. 2011, 13, 215–219. [Google Scholar] [PubMed]
- Guarino, M.P.; Afonso, R.A.; Raimundo, N.; Raposo, J.F.; Macedo, M.P. Hepatic glutathione and nitric oxide are critical for hepatic insulin-sensitizing substance action. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 284, G588–G594. [Google Scholar] [CrossRef] [PubMed]
- Lautt, W.W.; Schafer, J.; Macedo, M.P.; Legare, D.J. Bethanechol and N-acetylcysteine mimic feeding signals and reverse insulin resistance in fasted and sucrose-induced diabetic rats. Can. J. Physiol. Pharmacol. 2011, 89, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Lautt, W.W. A new paradigm for diabetes and obesity: The hepatic insulin sensitizing substance (HISS) hypothesis. J. Pharmacol. Sci. 2004, 95, 9–17. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lautt, W.W.; Ming, Z.; Legare, D.J.; Chowdhury, K.K.; Hatch, G.M.; Wang, H.H. Fatty Liver and Fatty Heart—Where do They Stand in the AMIS Syndrome? Healthcare 2015, 3, 666-682. https://doi.org/10.3390/healthcare3030666
Lautt WW, Ming Z, Legare DJ, Chowdhury KK, Hatch GM, Wang HH. Fatty Liver and Fatty Heart—Where do They Stand in the AMIS Syndrome? Healthcare. 2015; 3(3):666-682. https://doi.org/10.3390/healthcare3030666
Chicago/Turabian StyleLautt, W. Wayne, Zhi Ming, Dallas J. Legare, Kawshik K. Chowdhury, Grant M. Hatch, and Hui Helen Wang. 2015. "Fatty Liver and Fatty Heart—Where do They Stand in the AMIS Syndrome?" Healthcare 3, no. 3: 666-682. https://doi.org/10.3390/healthcare3030666