
Selectivity and Efficiency of Conductive Molecularly Imprinted Polymer (c-MIP) Based on 5-Phenyl-Dipyrromethane and 5-Phenol-Dipyrromethane for Quorum Sensing Precursors Detection


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Instrumentations and Procedures
2.3. Synthesis of 5-Phenyl-Dipyrromethane (5-ph-DP)
2.4. Synthesis and Characterization of 5-Phenol-Dipyrromethane (5-pOH-DP)
2.5. Synthesis and Characterisation of N-Acetyl Homoserine Lactone
2.6. Electrochemical Investigations of 5-ph-DP, 5-pOH-DP, and Homoserine Lactone Derivatives
2.7. Electromicrogravimeric (EQCM) Preparation of cNIP and cMIP
2.8. Gravimetric (QCM) Evaluation of cMIP Sensor Performances
3. Results and Discussion
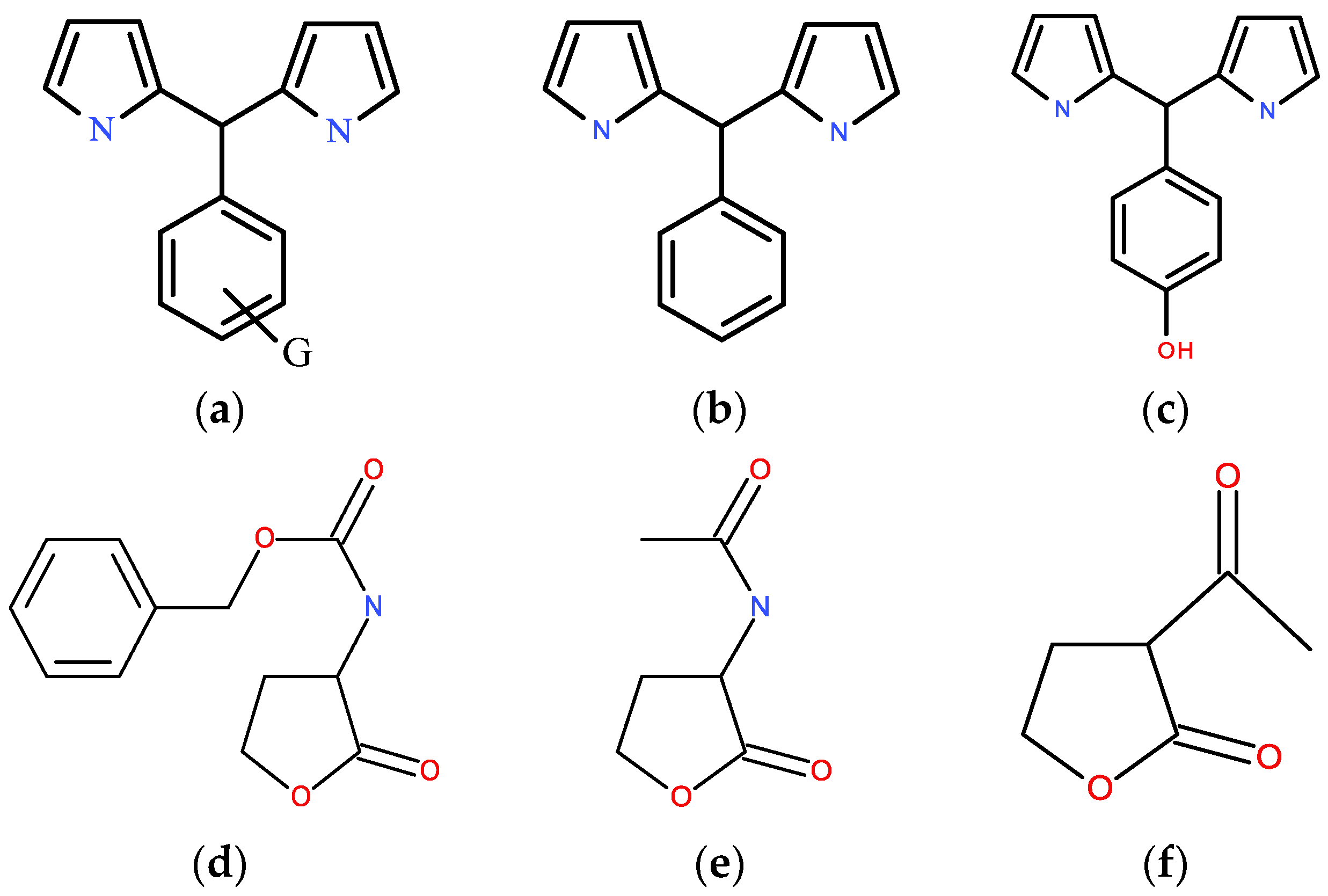
3.1. NMR Data
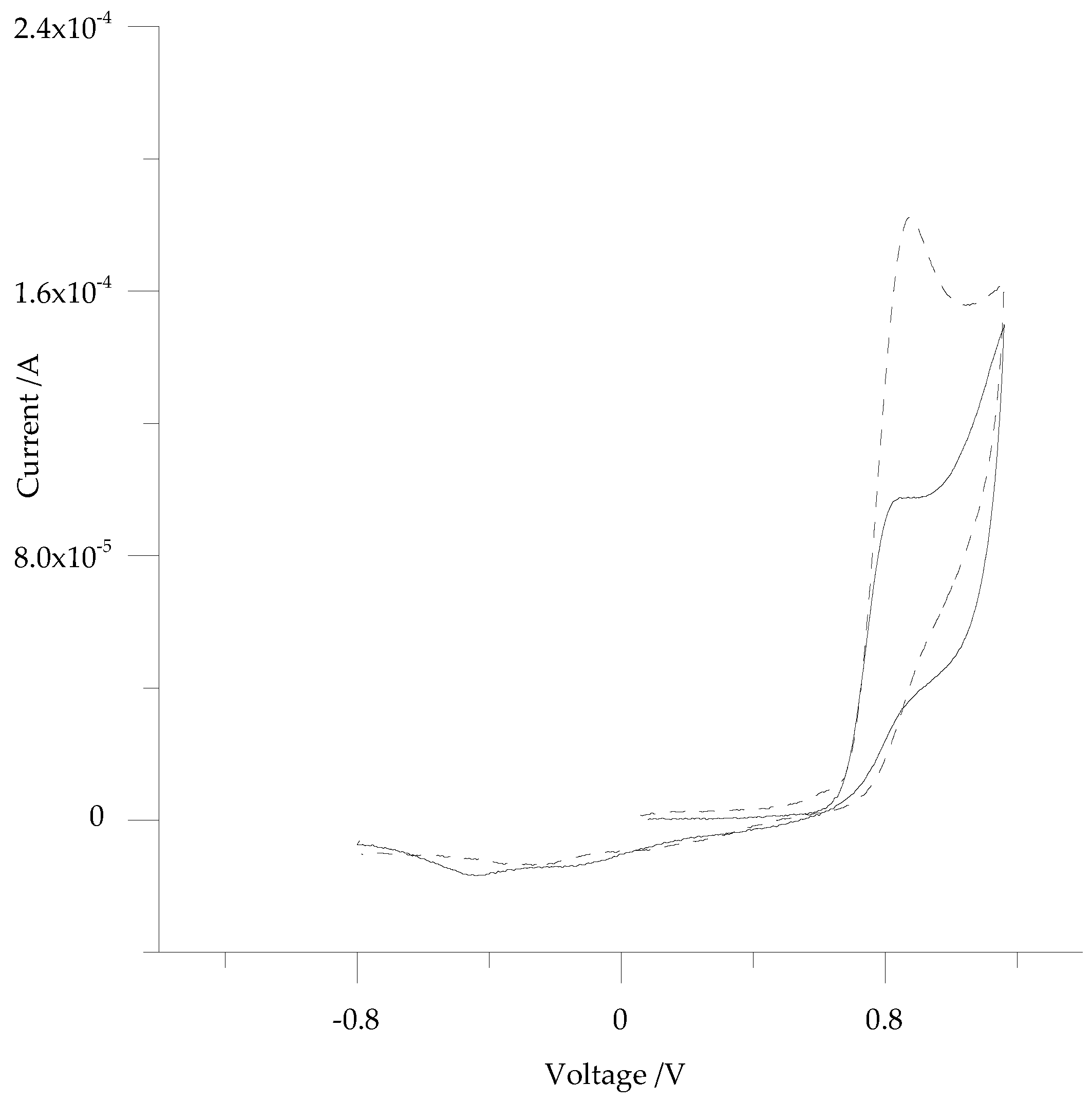
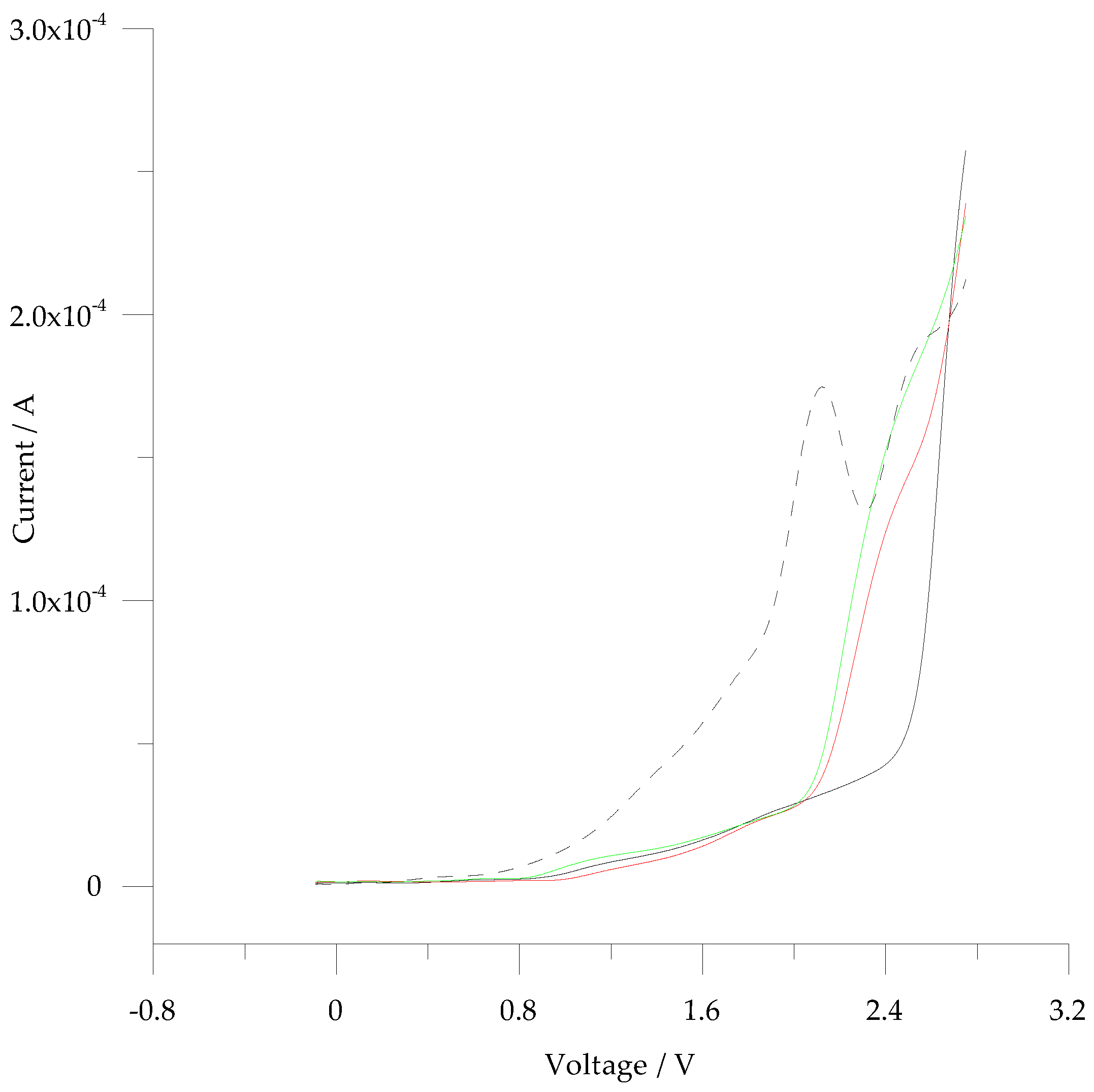
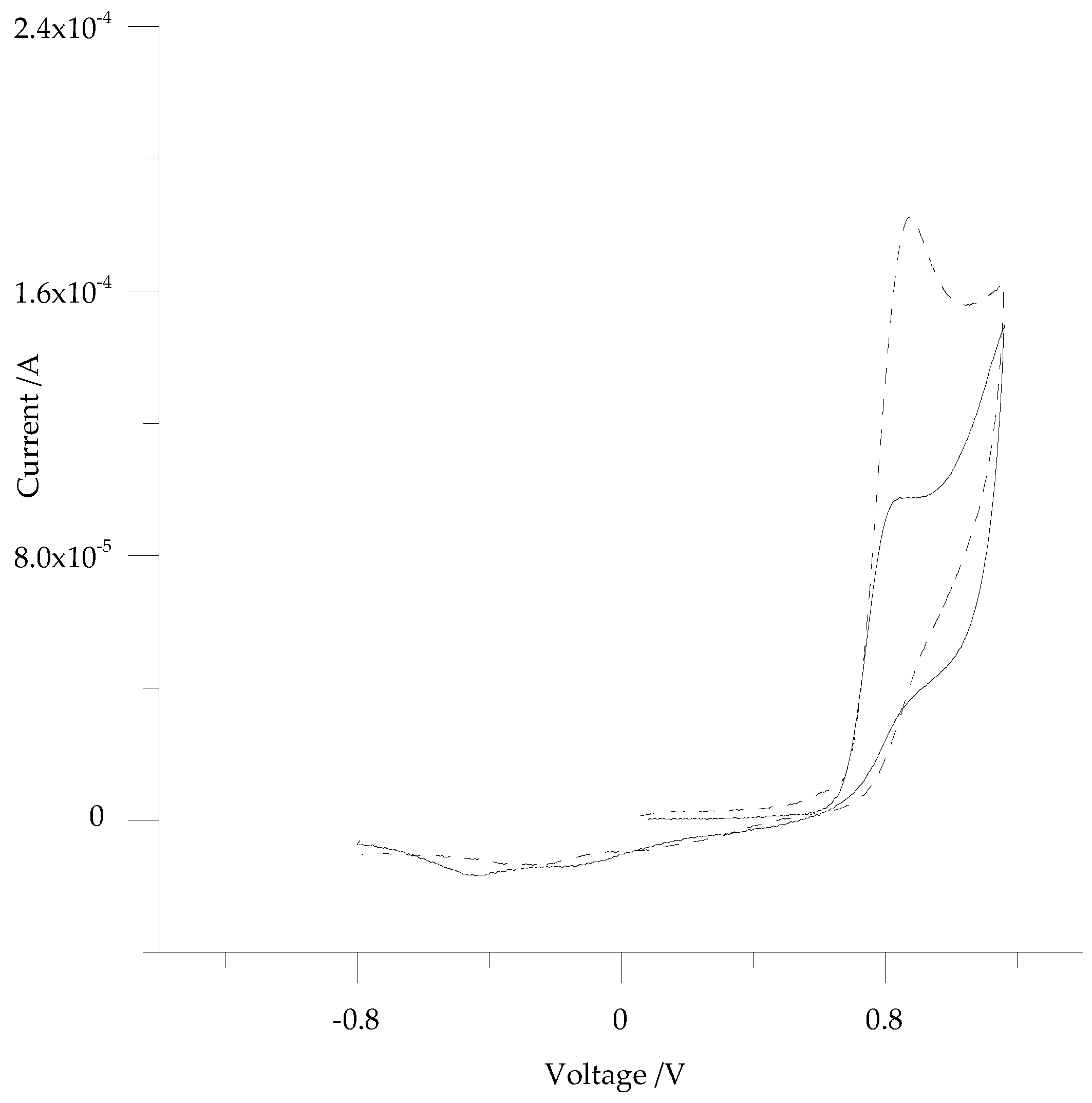
3.2. Electrochemistry of 5-ph-DP and 5-pOH-DP and Homoserine Lactone Derivatives in Solution
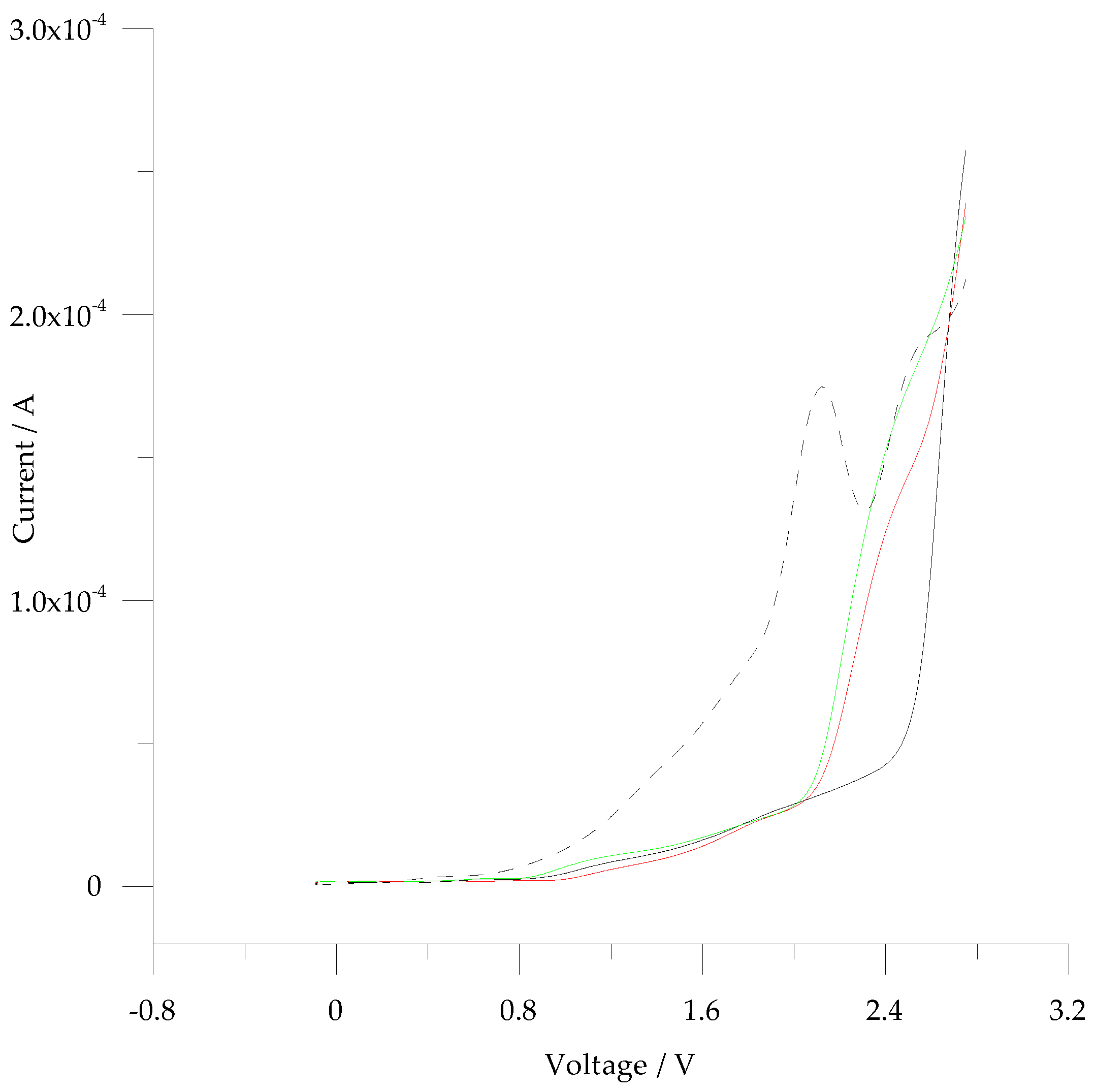
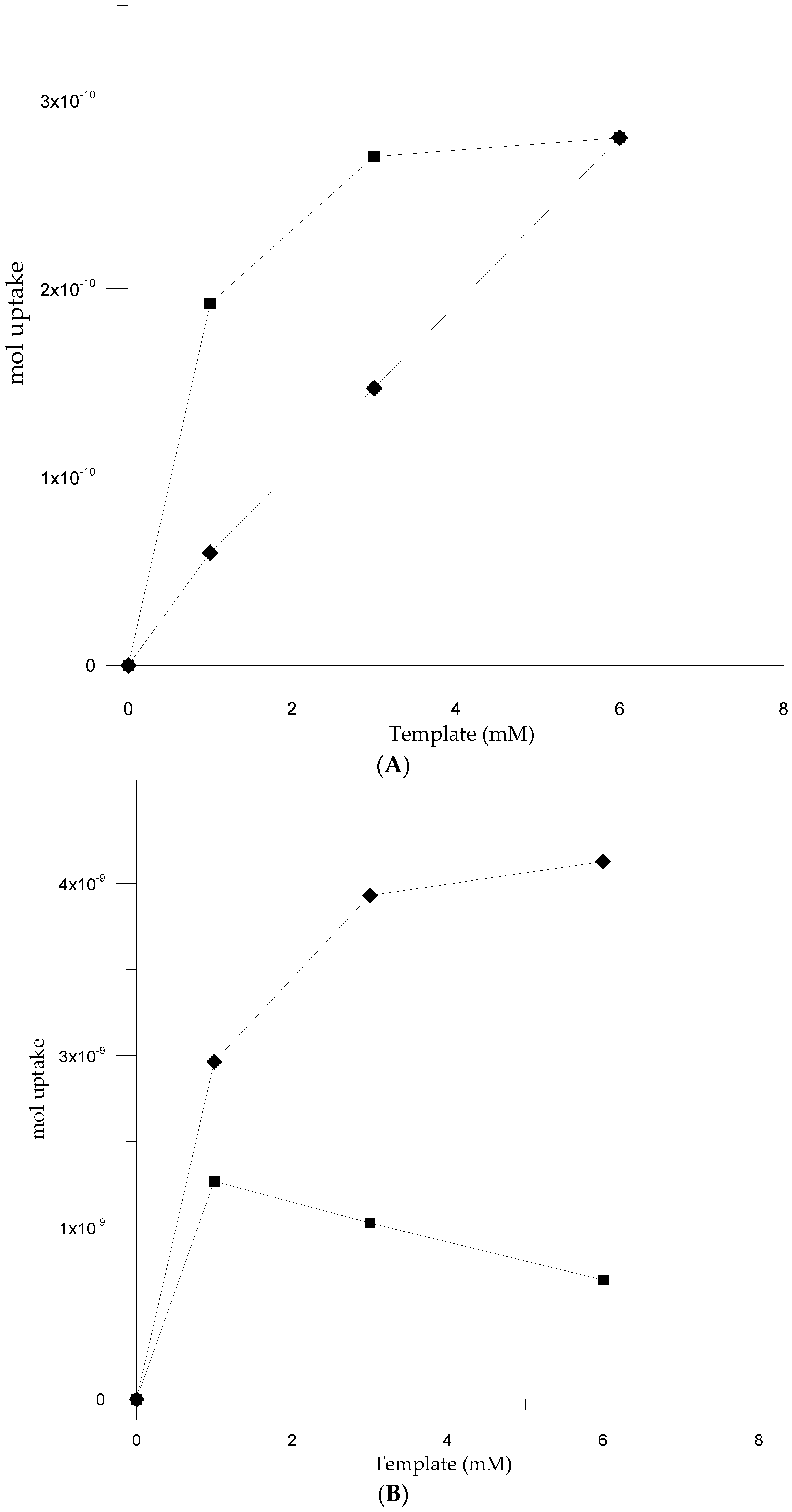
3.3. Electromicrogravimetric (EQCM) Preparation of cNIP and cMIP
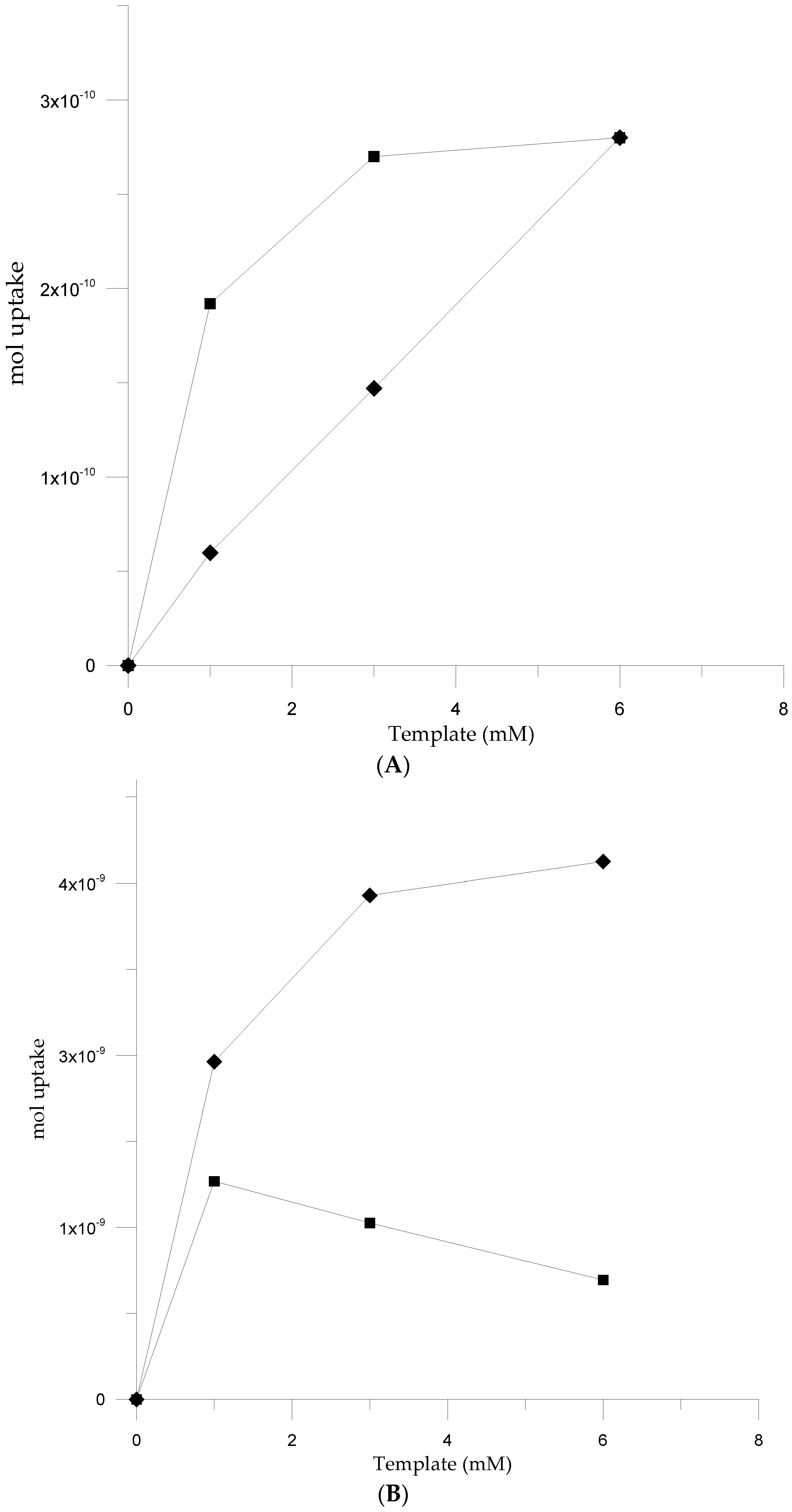
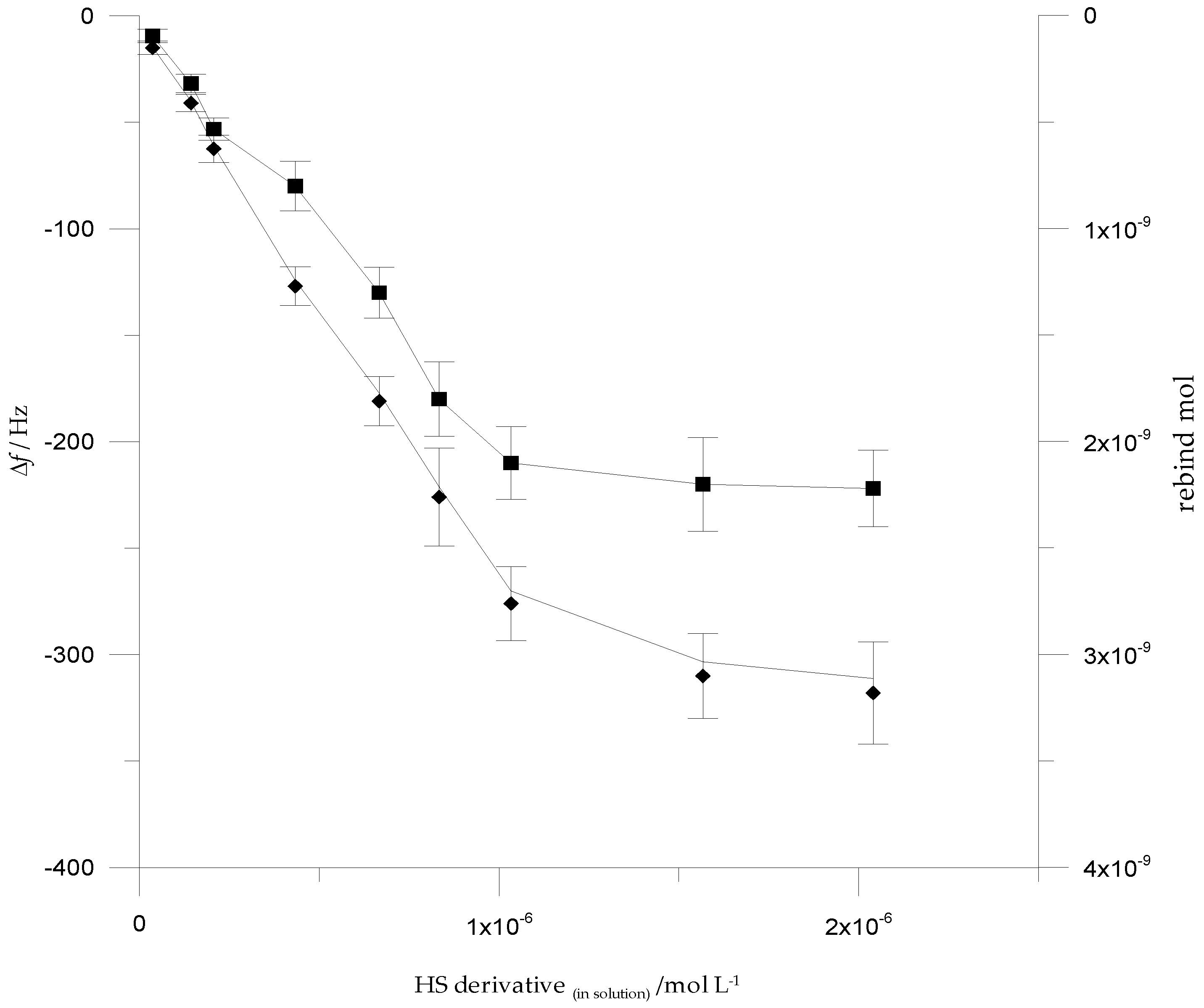
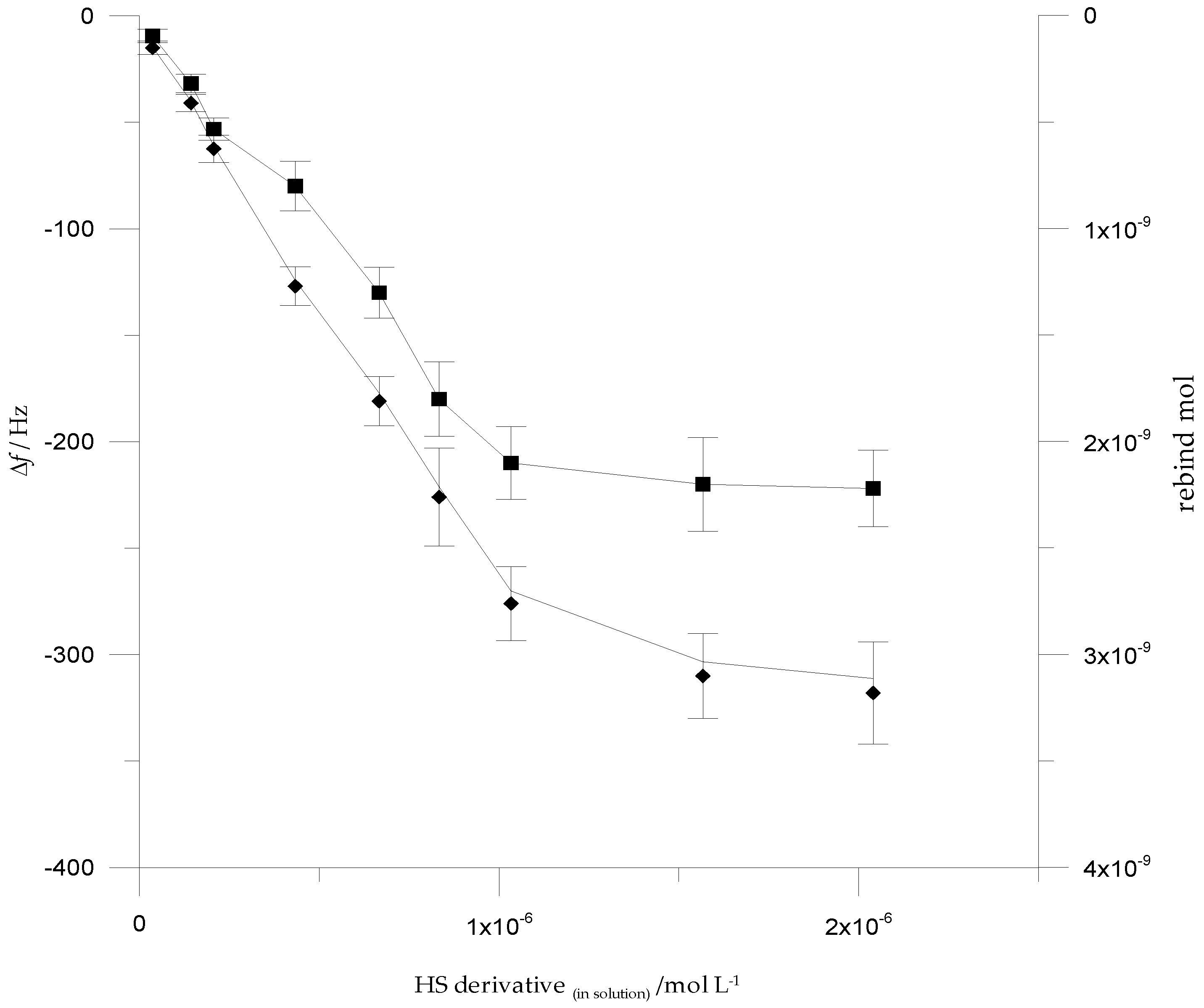
3.4. Analytical Performances of QCM Sensor cMIPs Modified
3.5. Selectivity Assessment
4. Conclusions
Supplementary Materials
Author Contributions
Conflicts of Interest
References
- Lattach, Y.; Archirel, P.; Remita, S. Influence of the Chemical Functionalities of a Molecularly Imprinted Conducting Polymer on Its Sensing Properties: Electrochemical Measurements and Semiempirical DFT Calculations. J. Phys. Chem. B 2012, 116, 1467–1481. [Google Scholar] [CrossRef] [PubMed]
- Özcan, L.; Şahyn, Y. Determination of paracetamol based on electropolymerized-molecularly imprinted polypyrrole modified pencil graphite electrode. Sens. Actuators B Chem. 2007, 127, 362–369. [Google Scholar] [CrossRef]
- EL-Sharifa, H.F.; Aizawab, H.; Reddy, S.M. Spectroscopic and quartz crystal microbalance (QCM) characterisation of protein-based MIPs. Sens. Actuators B Chem. 2015, 206, 239–245. [Google Scholar] [CrossRef]
- Guardia, L.; Badia, R.; Diaz-Garcia, M.E. Assessment of molecularly imprinted sol-gel materials for selective room temperature phosphorescence recognition of nafcillin. J. Chromatogr. B 2004, 804, 247–254. [Google Scholar]
- Ye, L.; Haupt, K. Molecularly imprinted polymers as antibody and receptor mimics for assays, sensors and drug discovery. Anal. Bioanal. Chem. 2004, 378, 1887–1897. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wang, X.; Lu, W.; Wu, X.; Li, J. Molecular imprinting: Perspectives and applications. Chem. Soc. Rev. 2016, 45, 2137–2211. [Google Scholar] [CrossRef] [PubMed]
- Garcia, R.; Freitas, A.M. Application of Molecularly Imprinted Polymers for the Analysis of Pesticide Residues in Food—A Highly Selective and Innovative Approach. Am. J. Anal. Chem. 2011, 2, 16–25. [Google Scholar] [CrossRef]
- Poma, A.; Brahmbhatt, H.; Pendergraff, H.M.; Watts, J.K.; Turner, N.W. Generation of Novel Hybrid Aptamer–Molecularly Imprinted Polymeric Nanoparticles. Adv. Mater. 2015, 27, 750–758. [Google Scholar] [CrossRef] [PubMed]
- Poma, A.; Piletsky, S.A.; Whitcombe, M.J. Plastic Antibodiesin Designing Receptors for the Next Generation of Biosensors, 2nd ed.; Piletsky, S.A., Whitcombe, M.J., Eds.; Springer: New York, NY, USA, 2012; pp. 105–129. [Google Scholar]
- Canfarotta, F.; Poma, A.; Guerreiro, A.; Piletsky, S. Solid-phase synthesis of molecularly imprinted nanoparticles. Nat. Protoc. 2016, 11, 443–455. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Zhao, W.; Yao, S.; Xu, J.; Wang, W.; Chen, Z. Molecularly Imprinted Polypyrrole Prepared by Electrodeposition for the Selective Recognition of Tryptophan Enantiomers. J. Appl. Polym. Sci. 2010, 115, 1952–1957. [Google Scholar] [CrossRef]
- Syritski, V.; Reut, J.; Menaker, A.; Gyurcsányi, R.E.; Öpik, A. Electrosynthesized molecularly imprinted polypyrrole films for enantioselective recognition of l-aspartic acid. Electrochim. Acta 2008, 53, 2729–2736. [Google Scholar] [CrossRef]
- Pietrzyk, A.; Suriyanarayanan, S.; Kutner, W.; Chitta, R.; D’Souza, F. Selective histamine piezoelectric chemosensor using a recognition film of the molecularly imprinted polymer of bis(bithiophene) derivatives. Anal. Chem. 2009, 81, 2633–2643. [Google Scholar] [CrossRef] [PubMed]
- Malitesta, C.; Mazzotta, E.; Picca, R.A.; Poma, A.; Chianella, I.; Piletsky, S.A. MIP sensors—The electrochemical approach. Anal. Bioanal. Chem. 2012, 402, 1827–1846. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.S.; Pietrzyk-Le, A.; D’Souza, F.; Kutner, W. Electrochemically synthesized polymers in molecular imprinting for chemical sensing. Anal. Bioanal. Chem. 2012, 402, 3177–3204. [Google Scholar] [CrossRef] [PubMed]
- Whitcombe, M.J.; Chianella, I.; Larcombe, L.; Piletsky, S.A.; Noble, J.; Porter, R.; Horgan, A. The rational development of molecularly imprinted polymer-based sensors for protein detection. Chem. Soc. Rev. 2011, 40, 1547–1571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartlett, P.N.; Chung, L.Y.; Moore, P. Conducting polymer films. Attachment of pyrrole groups to aza-macrocycles and attempted electrochemical polymerisation of the resulting monomers. Electrochim. Acta 1990, 35, 1051–1055. [Google Scholar] [CrossRef]
- Curran, D.; Grimshaw, J.; Perera, S.D. Poly(pyrrole) as a support for electrocatalytic materials. Chem. Soc. Rev. 1991, 20, 391–404. [Google Scholar] [CrossRef]
- Janata, J. Principles of Chemical Sensors, 2nd ed.; Springer: New York, NY, USA, 2008; pp. 13–50. [Google Scholar]
- Sellergen, B.; Hall, A.J. Synthetic Chemistry in Molecular Imprinting. In Molecular Imprinting: Principles and Applications of Micro- and Nanostructure Polymers, 1st ed.; Ye, L., Ed.; Pan Stanford Publishing, CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2013; pp. 25–65. [Google Scholar]
- Susmel, S.; Comuzzi, C. 5-phenyl-dipyrromethane and 5-(4-pyridyl)-dipyrromethane as modular building blocks for bio-inspired conductive molecularly imprinted polymer (cMIP). An Electrochemical and Piezoelectric investigation. RSC Adv. 2015, 5, 78379–78388. [Google Scholar] [CrossRef]
- Lintz, M.J.; Oinuma, K.I.; Wysoczynski, C.L.; Greenberg, E.P.; Churchill, M.E.A. Crystal structure of QscR, a Pseudomonas aeruginosa quorum sensing signal receptor. Proc. Natl. Acad. Sci. USA 2011, 108, 15763–15768. [Google Scholar] [CrossRef] [PubMed]
- Dias, D.M.; Ciulli, A. NMR approaches in structure-based lead discovery: Recent developments and new frontiers for targeting multi-protein complexes. Prog. Biophys. Mol. Biol. 2014, 116, 101–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Geet, A.L. Calibration of methanol nuclear magnetic resonance thermometer at low temperature. Anal. Chem. 1970, 42, 679–680. [Google Scholar] [CrossRef]
- Sauerbrey, G. Verwendung von Schwingquarzen zur Wägung dünner Schichten und zur Mikrowägung. Z. Phys. 1959, 155, 206–222. (In German) [Google Scholar] [CrossRef]
- Ka, J.W.; Lee, C.H. Optimizing the synthesis of 5,10-disubstituted tripyrromethanes. Tetrahedron Lett. 2000, 41, 4609–4613. [Google Scholar] [CrossRef]
- Briñas, R.P.; Brückner, C. Triarylcorroles by Oxidative Coupling of Triaryltetrapyrranes. Synlett 2001, 3, 442–444. [Google Scholar]
- Littler, B.L.; Ciringh, Y.; Lindsey, J.S. Investigation of Conditions Giving Minimal Scrambling in the Synthesis of trans-Porphyrins from Dipyrromethanes and Aldehydes. J. Org. Chem. 1999, 64, 2864–2872. [Google Scholar] [CrossRef] [PubMed]
- Ak, M.; Gancheva, V.; Terlemezyan, L.; Tanyeli, C.; Toppare, L. Synthesis of a dipyrromethane functionalized monomerand optoelectrochromic properties of its polymer. Eur. Polym. J. 2008, 44, 2567–2573. [Google Scholar] [CrossRef]
- Ohnishi, M.; Urry, D.W. Temperature dependence of amide proton chemical shifts: The secondary structures of gramicidin S and valinomycin. Biochem. Biophys. Res. Commun. 1969, 36, 194–202. [Google Scholar] [CrossRef]
- Kopple, K.D.; Ohnishi, M.; Go, A. Conformations of cyclic peptides. III. Cyclopentaglycyltyrosyl and related compounds. J. Am. Chem. Soc. 1969, 91, 4264–4272. [Google Scholar] [CrossRef]
- Merutka, G.; Dyson, H.J.; Wright, P.E. Random coil’ 1H chemical shifts obtained as a function of temperature and trifluoroethanol concentration for the peptide series GGXGG. J. Biomol. NMR 1995, 5, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Kontogianni, V.G.; Charisiadis, P.; Primikyri, A.; Pappas, C.G.; Exarchou, V.; Tzakosa, A.G.; Gerothanassis, I.P. Hydrogen bonding probes of phenol-OH groups. Org. Biomol. Chem. 2013, 11, 1013–1025. [Google Scholar] [CrossRef] [PubMed]
- Zotti, G.; Schiavon, G.; Zecchin, S.; Sannicolo, F.; Brenna, E. Anion Assisted Anodic Coupling of 2,2′-Bipyrrole. Role of Tosylate Anion in the Electrochemical Synthesis of Polypyrrole. Chem. Mater. 1995, 7, 1464–1468. [Google Scholar] [CrossRef]
- Zheng, W.; Razal, J.M.; Spinks, G.M.; Truong, V.T.; Whitten, P.G.; Wallace, G.G. The Role of Unbound Oligomers in the Nucleation and Growth of Electrodeposited Polypyrrole and Method for Preparing High Strength, High Conductivity Films. Langmuir 2012, 28, 10891–10897. [Google Scholar] [CrossRef] [PubMed]
- Martina, S.; Enklemann, V.; Schluter, A.D.; Wegner, G.; Zotti, G.; Zerbi, G. Synthesis and electrochemical and spectroscopical studies of 2.5-pyrrole oligomers and well-defined short-chain poly(2.5-pyrrole). Synth. Met. 1993, 55, 1096–1101. [Google Scholar] [CrossRef]
- Miles, M.J.; Smith, W.T.; Shapiro, J.S. Morphological investigation by atomic force microscopy and light microscopy of electropolymerised polypyrrole films. Polymer 2000, 41, 3349–3356. [Google Scholar] [CrossRef]
- Suarez, M.F.; Compton, R.G. In situ atomic force microscopy study of polypyrrole synthesis and the volume changes induced by oxidation and reduction of the polymer. J. Electroanal. Chem. 1999, 462, 211–221. [Google Scholar] [CrossRef]
- Higghins, S. Conjugated polymers incorporating pendant functional groups-synthesis and characterization. Chem. Soc. Rev. 1997, 26, 247–257. [Google Scholar] [CrossRef]
- Khairunnisa, A.; Liao, W.; Yau, S. Adsorption and Electrochemical Polymerization of Pyrrole on Au(100) Electrodes as Examined by In Situ Scanning Tunneling Microscopy. J. Phys. Chem. C 2016, 120, 26425–26434. [Google Scholar] [CrossRef]
- Ratautaitea, V.; Plausinaitisa, D.; Baleviciutea, I.; Mikoliunaitea, L.; Ramanaviciene, A.; Ramanavicius, A. Characterization of caffeine-imprinted polypyrrole by a quartz crystal microbalance and electrochemical impedance spectroscopy. Sens. Actuators B 2015, 212, 63–71. [Google Scholar] [CrossRef]
- Sadki, S.; Schottland, P.; Brodie, N.; Sabouraud, G. The mechanisms of pyrrole electropolymerization. Chem. Soc. Rev. 2000, 29, 283–293. [Google Scholar]
- Reed, C.E.; Kanazawa, K.K.; Kaufmann, J.H. Physical description of a viscoelastically loaded AT-cut quartz resonator. J. Appl. Phys. 1990, 68, 1993–2001. [Google Scholar] [CrossRef]
- Susmel, S.; Toniolo, R.; Pizzariello, A.; Dossi, N.; Bontempelli, G. A piezoelectric immunosensor based on antibody entrapment within a non-totally rigid polymeric film. Sens. Actuators B 2005, 111, 331–338. [Google Scholar] [CrossRef]
- Nakluaa, W.; Suedeea, R.; Lieberzeit, P.A. Dopaminergic receptor–ligand binding assays based on molecularly imprinted polymers on quartz crystal microbalance sensors. Biosens. Bioelectron. 2016, 81, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Schweiger, B.; Kim, Y.J.; Ulbricht, M. Electropolymerized Molecularly Imprinted Polypyrrole Film for Sensing of Clofibric Acid. Sensors 2015, 15, 4870–4889. [Google Scholar] [CrossRef] [PubMed]
- Oral, E.; Peppas, N.A. Dynamic studies of molecular imprinting polymerizations. Polymer 2004, 45, 6163–6173. [Google Scholar] [CrossRef]
- Kan, X.; Xing, Z.; Zhu, A.; Zhao, Z.; Xu, G.; Li, C.; Zhou, H. Molecularly imprinted polymers based electrochemical sensor for bovine hemoglobin recognition. Sens. Actuators B 2012, 168, 395–401. [Google Scholar] [CrossRef]
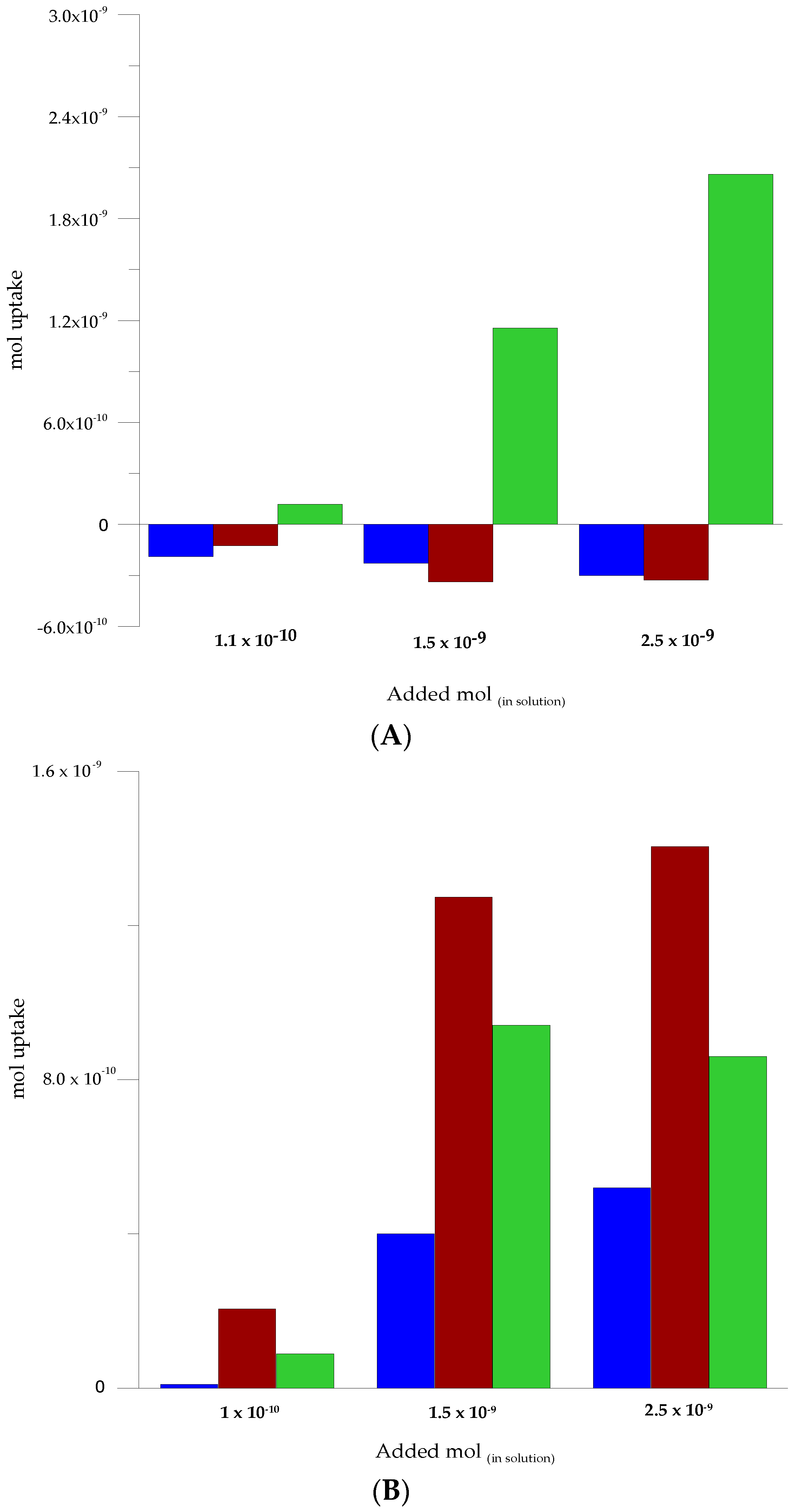

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
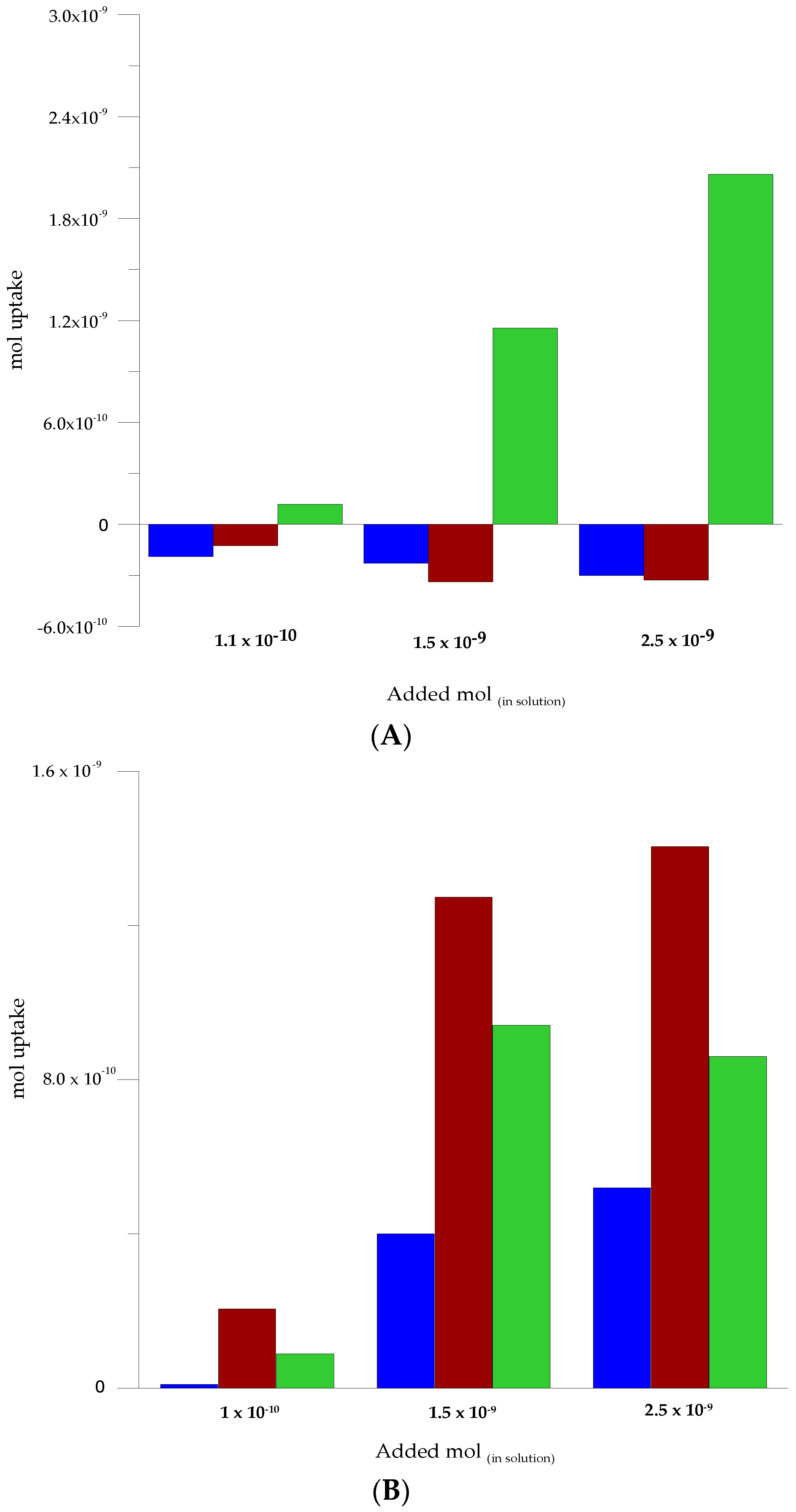{kind=link}
| NMR Sample Solution | Δδ (ppm) | |||
|---|---|---|---|---|
| NHCbz-HS | NHAcetyl-HS | NHpy | OHphenol | |
| 5-pOH-DP/Cbz-HS | −0.01 | 0.01 | 0.03 | |
| 5-pOH-DP/Acetyl-HS | 0.09 | 0.03 | 0.07 | |
| 5-ph-DP/Cbz-HS | −0.01 | −0.03 | ||
| 5-ph-DP/Acetyl-HS | 0.03 | 0.01 | ||
| NMR Sample Solution | Δδ/K (ppb/K) | |||
|---|---|---|---|---|
| NHpy | OHphenol | NHAcetyl-HS | NHCbz-HS | |
| Acetyl-HS | −3.1 (±0.1) | |||
| Cbz-HS | −2.8 (±0.2) | |||
| 5-pOH-DP | −3.0 (±0.1) | −6.4 (±0.2) | ||
| 5-ph-DP | −2.6 (±0.1) | |||
| 5-pOH-DP/Acetyl-HS | −2.7 (±0.2) | −5.8 (±0.1) | −1.6 (±0.2) | |
| 5-ph-DP/Acetyl-HS | −2.6 (±0.3) | −2.8 (±0.1) | ||
| 5-pOH-DP/Cbz-HS | −2.9 (±0.2) | −6.2 (±0.1) | −2.9 (±0.3) | |
| 5-ph-DP/Cbz-HS | −2.9 (±0.2) | −2.8 (±0.1) | ||
| Monomer * | HS-Derivative | Epa/mV | ja/A·cm−2 |
|---|---|---|---|
| 5-ph-DP | / | 873 | 8.42 × 10−4 (cv % 4.5; n = 5) |
| 5-ph-DP | Cbz-HS (1 mM) | 876 | 7.86 × 10−4 (cv % 3.5; n = 3) |
| Cbz-HS (3 mM) | 874 | 6.84 × 10−4 (cv % 5.3; n = 3) | |
| Cbz-HS (6 mM) | 874 | 6.67 × 10−6 (cv % 5.1; n = 3) | |
| 5-ph-DP | Acetyl-HS (1 mM) | 879 | 9.23 × 10−4 (cv % 5.2; n = 3) |
| Acetyl-HS (3 mM) | 876 | 6.48 × 10−4 (cv % 5.2; n = 3) | |
| Acetyl-HS (6 mM) | 876 | 6.22 × 10−4 (cv % 8.5; n = 3) | |
| 5-pOH-DP | / | 810 | 4.36 × 10−4 (cv % 5.5; n = 5) |
| 5-pOH-DP | Cbz-HS (1 mM) | 850 | 5.15 × 10−4 (cv % 2.5; n = 3) |
| Cbz-HS (3 mM) | 870 | 6.33 × 10−4 (cv % 4.5; n = 3) | |
| Cbz-HS (6 mM) | 850 | 5.51 × 10−6 (cv % 3.9; n = 3) | |
| 5-pOH-DP | Acetyl-HS (1 mM) | 889 | 9.39 × 10−4 (cv % 3.2; n = 3) |
| Aceyl-HS (3 mM) | 855 | 6.38 × 10−4 (cv % 5.5; n = 3) | |
| Acetyl-HS (6 mM) | 855 | 6.63 × 10−4 (cv % 7.5; n = 3) |
| cNIP § | Sweep | Δf/Hz | cMIP § | Sweep | Δf/Hz | Net Δf/Hz* | Δm (ng) |
|---|---|---|---|---|---|---|---|
| poly-5-ph-DP | 1 | −303.88 | poly-5-ph-DP+Acetyl-HS | 1 | −257.25 | 46.63 | −65.28 |
| 2 | −327.36 | 23.28 | −32.59 | ||||
| 2 | −350.64 | 3 | −346.80 | 23.28 | −32.59 | ||
| 4 | −346.8 | 30.96 | −43.34 | ||||
| 3 | −370.08 | 5 | −338.88 | −11.52 | 16.13 | ||
| 6 | −307.68 | −46.56 | 65.18 | ||||
| 4 | −377.76 | poly-5-ph-DP+Cbz-HS | 1 | −257.3 | 46.6 | −65.24 | |
| 2 | −322.64 | 28 | −39.20 | ||||
| 5 | −327.36 | 3 | −342.96 | 27.1 | −37.97 | ||
| 4 | −343.0 | 34.8 | −48.72 | ||||
| 6 | −261.12 | 5 | −364.4 | −37.04 | 51.86 | ||
| 6 | −307.68 | −46.56 | 65.18 | ||||
| poly-5-pOH-DP | 1 | −364.5 | poly-5-pOH-DP+Acetyl-HS | 1 | −486.80 | −112.1 | 171.74 |
| 2 | −512.4 | −112.4 | 157.36 | ||||
| 2 | −400.0 | 3 | −532.8 | −117.12 | 163.97 | ||
| 4 | −520.0 | −109.44 | 153.22 | ||||
| 3 | −415.7 | 5 | −528.8 | −108 | 151.2 | ||
| 6 | −344.0 | 71.36 | −99.96 | ||||
| 4 | −410.6 | poly-5-pOH-DP+Cbz-HS | 1 | −408.8 | −44.3 | 62.02 | |
| 2 | −454.8 | −54.8 | 76.72 | ||||
| 5 | −420.8 | 3 | −460.8 | −45.12 | 63.16 | ||
| 4 | −467.6 | −57.04 | 79.86 | ||||
| 6 | −415.4 | 5 | −474 | −53.2 | 74.48 | ||
| 6 | −468 | −52.64 | 73.65 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Susmel, S.; Comuzzi, C. Selectivity and Efficiency of Conductive Molecularly Imprinted Polymer (c-MIP) Based on 5-Phenyl-Dipyrromethane and 5-Phenol-Dipyrromethane for Quorum Sensing Precursors Detection. Chemosensors 2017, 5, 5. https://doi.org/10.3390/chemosensors5010005
Susmel S, Comuzzi C. Selectivity and Efficiency of Conductive Molecularly Imprinted Polymer (c-MIP) Based on 5-Phenyl-Dipyrromethane and 5-Phenol-Dipyrromethane for Quorum Sensing Precursors Detection. Chemosensors. 2017; 5(1):5. https://doi.org/10.3390/chemosensors5010005
Chicago/Turabian StyleSusmel, Sabina, and Clara Comuzzi. 2017. "Selectivity and Efficiency of Conductive Molecularly Imprinted Polymer (c-MIP) Based on 5-Phenyl-Dipyrromethane and 5-Phenol-Dipyrromethane for Quorum Sensing Precursors Detection" Chemosensors 5, no. 1: 5. https://doi.org/10.3390/chemosensors5010005