1. Introduction
Basal cell nevus syndrome (BCNS), also known as Gorlin–Goltz syndrome, is characterized by the development of multiple nevoid basal cell carcinomas at an early age and is often accompanied by clinical features such as odontogenic keratocysts, palmar pits, congenital skeletal abnormalities, and an increased risk for the development of medulloblastomas [
1]. BCNS is inherited in an autosomal dominant fashion and is, in a vast majority of cases, caused by a heterozygous loss of function variants in the tumor suppressor gene Patched 1 (
PTCH1), which is part of the Sonic Hedgehog (SHH) signalling pathway. More rarely, BCNS has been reported to be caused by a loss of function mutation in
PTCH2 and
SUFU [
2,
3], although the involvement of
PTCH2 has been disputed [
4]. The SHH pathway drives cell proliferation and differentiation via the transcription factor Gli1 [
5] and has a prominent role during embryonic development. Consequentially, BCNS patients often suffer from congenital growth abnormalities affecting the skull (e.g., macrocephaly), spine, and ribs. The development of BCCs, which is one of the major clinical criteria for the diagnosis of BCNS [
1], is dependent on a somatic second-hit pathogenic variant in the other
PTCH1 allele, which is typically introduced via UV light exposure, causing multiple BCCs to appear at a young age. Additional somatic variants may be introduced in the SHH pathway, for example, in
Smoothened (
SMO), which may drive BCC development [
6].
Although a clinical diagnosis of BCNS can be established based on the major and minor symptom criteria, a genetic diagnosis is important, not only to confirm the clinical diagnosis, but also to understand the disease segregation pattern and recurrence risk. The establishment of a genetic diagnosis for BCNS relies on the sequencing of the
PTCH1 gene in order to detect single-nucleotide variants (SNVs) and small indels but also requires accurate
PTCH1 copy-number analysis. Both large deletions and duplications in the
PTCH1 tumor suppressor gene have been reported to affect protein conformation and function, which results in PTCH1 inactivation [
7,
8]. Different methods exist for CNV analysis, including the conventional multiplex-ligation-probe (MLPA)-based methodology, targeted NGS-based approaches using, for example, smMIPs with unique molecular identifiers (UMIs), which help to detect CNVs in a more quantitative manner, and ddPCR, which allows absolute copy-number quantification [
9,
10,
11,
12,
13]. For NGS-based CNV detection, a large reference dataset of control samples is needed for normalization, which may only be available after having analyzed enough samples. The choice of a specific CNV analysis methodology in a diagnostic setting is highly important, as the sensitivity and resolution eventually define the diagnostic yield of the genetic analysis [
14]. This especially applies to CNV analysis in patients with a postzygotic mosaicism, who can be asymptomatic, and where the variant allele frequency in blood DNA can be very low due to spatiotemporal postzygotic occurrences in the embryonic tissues. Such CNVs might easily be missed when using conventional Sanger-sequencing-based approaches or MLPA due to a relatively low sensitivity [
15]. Therefore, if available, the testing of different tissue types besides blood is an important diagnostic approach in suspected mosaic cases.
In this case study, we compared the test outcomes for PTCH1 CNV detection using the more conventional multiplex ligation-probe (MLPA) methodology and digital droplet PCR (ddPCR) based on DNA isolated from the peripheral blood lymphocytes of a female BCNS patient (index patient), where MLPA detected a heterozygous CNV duplication. A smMIP-NGS approach for CNV detection was not possible since a large reference dataset was unavailable. The patient’s father, only presenting with multiple BCCs, was suspected to have a low-grade mosaic form of BCNS, and was therefore tested for CNVs based on multiple tissues, including blood, hair, and saliva. PTCH1 CNV analysis in these two cases allowed us to compare the accuracy of the more conventional multiplex ligation-probe (MLPA) methodology with digital droplet PCR (ddPCR) for the detection of CNVs.
2. Materials and Methods
2.1. Patients and DNA Samples
At the Department of Clinical Genetics (MUMC+, Maastricht, The Netherlands), a request for genetic analysis was obtained for a female patient (index) from the Oulu University Hospital in Finland, who was clinically diagnosed with BCNS. Also, her father was analysed after the molecular diagnosis of BCNS was established. The 38-year-old patient met four major clinical criteria for BCNS, including multiple BCCs, palmoplantar pits, an odontogenic keratocyst in her jaw, and calcification of the falx cerebri [
1]. She also met some minor criteria, including macrocephaly, kyphoscoliosis, an ovarian fibroma, and ocular abnormalities [
1]. There were no family members with anamnestic symptoms of BCNS, but the patient’s father (68 years of age) had developed multiple BCCs at older age. To exclude BCNS, his blood was also analyzed after molecular diagnosis of BCNS was established in his daughter. The index patient’s mother, brother, and two children have no symptoms suggestive of BCNS.
Peripheral blood samples were collected from the index patient and her father, and genomic DNA was extracted using automated QIAsymphony device and Qiagen Qiasymphony DSP DNA Midi kit (Qiagen, Hilden, Germany) in Nordlab genetics laboratory, Oulu, Finland and sent to the MUMC+ for further genetic analysis. DNA isolation from the tissues (DNeasy Blood & Tissue Kit, Qiagen, Hilden, Germany) was performed in MUMC+ and used for genetic analysis.
Both patients provided written informed consent for genetic analysis, approved by the ethics committee of the Northern Ostrobothnia Hospital District (EETTMK: 45/2015 and amendment 2020).
2.2. PCR and Sanger Sequencing
PTCH1 exons and flanking intronic regions were PCR-amplified using KAPA2G FastHotStart DNA polymerase combined with HotStart Buffer A (Merck, Darmstadt, Germany) using M13-labeled primers with a reaction concentration of 0.33 pmol/μL and 20 ng DNA input (isolated from blood lymphocytes).
PTCH1 primer sequences are specified in
Supplementary Table S1, and PCR amplification was performed according to the manufacturer’s protocol. PCR products were purified using exo-sap treatment (Isogen life science, de Meern, The Netherlands). Sanger sequencing was performed using the BigDye Terminator Cycle Sequencing Kit (V1.1) (Thermofisher Scientific, Waltham, MA, USA) and the ABI 3730 automatic sequencer (Applied Biosystems, Foster City, CA, USA). Sequencing results were analyzed using the Mutation Surveyor software version 5.1 (Softgenetics, State College, PA, USA).
PTCH1 Sanger sequencing was performed only for the index patient.
2.3. MLPA
Multiplex Ligation-dependent Probe Amplification (MLPA) (MRC Holland, Amsterdam, The Netherlands) was performed using a DNA input of 200 ng, as measured by nanodrop, and diluted in MQ to a total volume of 5 μL. MLPA analysis was performed on DNA isolated from the (control) tissues. Also, a negative control condition was included. The sample hybridization mixture was prepared according to the manufacturer’s protocol, using SALSA MLPA Probe Mix P067-B2
PTCH1 (MRC Holland, Amsterdam, The Netherlands). The MLPA kit contains 33 MLPA probes for 23 out of 25 exons of the
PTCH1 gene (no probes are included for exons 1a and 8, NCBI RefSeq: NM_000264.5), where amplification products sizes range from 142 to 454 nt. The probe mix contains ten reference probes located at different autosomal chromosome locations and nine quality control fragments with amplicons between 64 and 105 nt. The complete probe sequences in the P067
PTCH1 mix can be found at the manufacturer’s website (
https://www.mrcholland.com/ (accessed on 20 January 2024)). The MLPA probe hybridization mixture, MLPA ligation mixture, and PCR mixture were used according to the manufacturer’s protocol, as were the corresponding thermal cycle programs; 1 μL PCR product of each reaction (FAM-labeled) was used for capillary electrophoresis using the ABI 3730 automatic sequencer. Fragment sizes and signal intensities were quantified using the software tool GeneMarker V2.4.0 (Softgenetics, State College, PA, USA), and normalized to the reference probes (expected to have a normal copy number). A relative copy-number ratio was calculated with respect to the wild-type control DNA condition. All quality control fragments were checked to ensure they met the minimal quality criteria, ensuring sufficient DNA input (Q-fragments) and proper DNA denaturation (D-fragments) and avoiding sample mix up (X and Y fragments, no DNA control). A relative copy-number peak ratio of 0.75–1.25 was considered equivocal, whereas a ratio of >1.25 was interpreted as a copy-number gain, and <0.75 as a copy-number loss.
2.4. ddPCR
ddPCR-based CNV analysis of PTCH1 was performed using the commercial BioRad PrimePCR copy-number assay for PTCH1 (BioRad, Hercules, CA, USA), consisting of a duplex PCR with FAM-labeled PTCH1 probe (channel 1) and HEX-labeled EIF2C1 probe (channel 2). The 68nt PTCH1 amplicon crosses an exon–intron junction and is detected by the FAM-labeled PTCH1 probe. PTCH1 copy-number quantification is corrected for the EIF2C1 reference assay, which is based on a 69 nt amplicon detected using a HEX-labeled EIF2C1 probe. Amplicon context sequences are indicated in .
Table 1.
ddPCR probe context sequences as indicated by the manufacturer (BioRad) and exonic–intronic location.
As germline CNVs are most likely to involve less than 10 copies per cell, ddPCR was performed using ~20 ng DNA input of each tissue. ddPCR PTCH1 CNV analysis was applied to the DNA samples, including a negative control. DNA fragmentation using the HaeIII restriction enzyme (New England Biolabs, Ipswich, MA, USA) was performed during the ddPCR assay in order to improve template accessibility, as recommended by the manufacturer (BioRad, Hercules, CA, USA). The reaction mixture was generated according to the manufacturer’s manual for ddPCR CNV analysis; 20 μL of the reaction mixture, containing the DNA, was added to the DG8 cartridge, as well as 70 μL of droplet generation oil for droplet generation in the QX200 Digital Droplet Generator. Thermal cycling was performed, followed by reading of the droplets using the QX200 Droplet Reader, all according to the manufacturer’s protocol (Biorad, Hercules, CA, USA). Data acquisition and analysis were performed using the BioRad QuantaSoft software v1.7.4, according to the manufacturer’s instructions. The PTCH1 copy-number ratio was determined by calculating the ratio of the PTCH1 target molecule concentration to the EIF2C1 reference molecule concentration, multiplied by the number of copies of the reference molecule in the genome, which is 2. An average copy-number ratio was calculated based on five technical duplicates.
4. Discussion
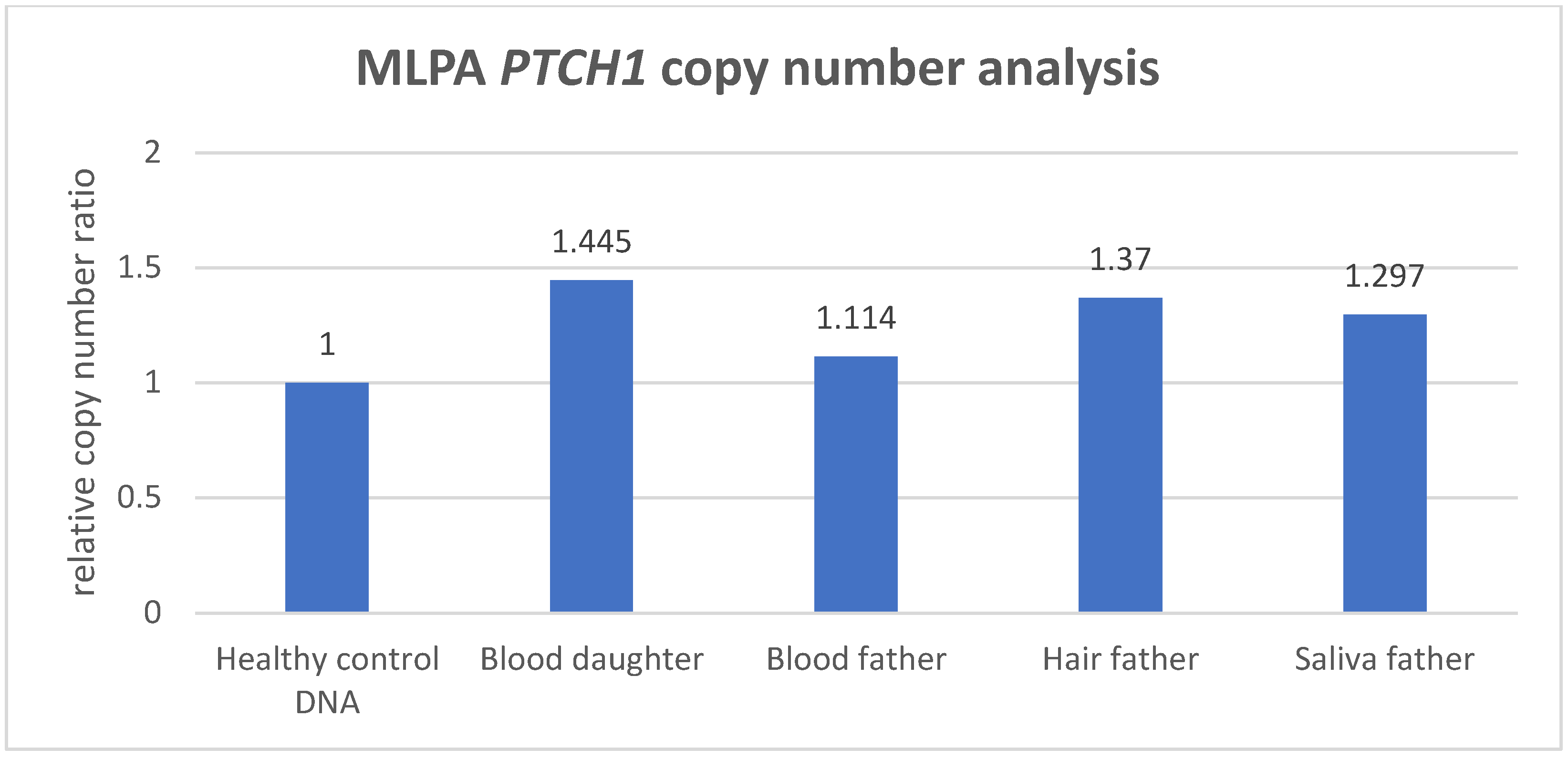
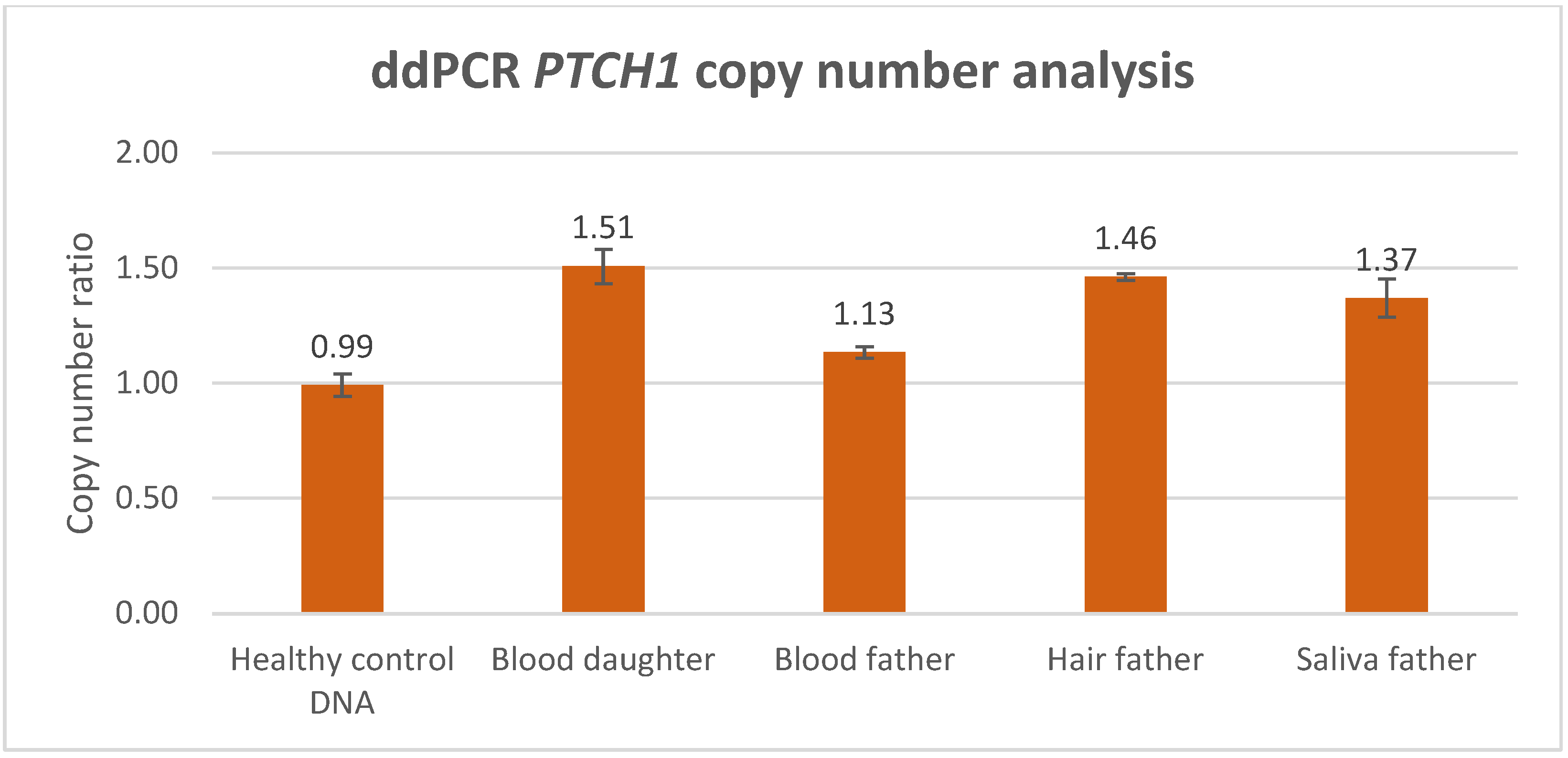
As illustrated in this case study, when performing targeted PTCH1 genetic analysis in BCNS, non-quantitative detection methods such as Sanger sequencing might reveal indications for an allelic imbalance. Still, the testing for PTCH1 copy-number variants in BCNS requires a sensitive and quantitative technique in order to establish reliable detection and an accurate diagnosis. A routinely used quantitative CNV detection method is MLPA, which is performed to exclude gross genomic deletion or duplication variants in DNA diagnostics for the PTCH1 gene. In this study, we compared CNV analysis outcomes generated with MLPA and ddPCR methodology, based on a BCNS patient who was likely to harbor a germline PTCH1 variant, as well as the mosaic father who presented with multiple BCCs only. Both ddPCR and MLPA analyses detected a significantly increased PTCH1 copy number in the peripheral lymphocyte DNA of the BCNS patient (index) with an average duplication factor of 1.49 and an average relative copy-number ratio of 1.445, respectively (the average of the MLPA probes in the gained region). Although both outcomes are clearly indicative of a PTCH1 copy-number increase, only the ddPCR outcome accurately reflected the presence of a germline CNV, in this case involving a PTCH1 gene gain consisting of three copies. This also implies that MLPA detects the PTCH1 gene gain in only 89% of the patient’s blood lymphocytes, which is approximately 11% lower than theoretically would be expected in a germline gene gain (copy-number ratio of 1.5), suggesting a less-accurate copy-number quantification. Moreover, the distribution of the MLPA probes revealed that PTCH1 exons 3 to 12 were aberrant (with an increased copy-number factor (>1.25)), indicating a partial gene gain.
In contrast to the MLPA method, the ddPCR assay consists of only a single probe localized within the exon-10–intron-10 region of PTCH1 and could therefore be considered less reliable, assessing only one genetic data point. However, in the peripheral blood lymphocytes of the index’s father, ddPCR analysis also detected an increased PTCH1 copy number. The average duplication factor (1.13) indicated that the gene gain was present in ~26% of the blood lymphocytes, indicating that the PTCH1 gene gain was inherited via the father, who was likely to harbor the gene defect as a low grade, postzygotic (gonadal) mosaicism. Interestingly, MLPA analysis was not able to detect an increased PTCH1 copy number in the peripheral blood lymphocytes of the mosaic father based on the standard, validated cutoff threshold (>1.25) for copy-number gain (dept. of Clinical Genetics, MUMC+), where none of the 23 probes exceeded this threshold. Despite being considered as not significantly increased, the average relative copy-number ratio of 1.114 within the exon 3–12 region would suggest that the gain is present in ~23% of the father’s blood lymphocytes, with the latter showing a quantification difference of ~3% between ddPCR and MLPA.
The testing for CNVs in multiple tissues of a suspected mosaic patient is desirable, as it reduces the risk of missing the genetic defect due to an allele frequency that is below the technique’s detection sensitivity. The importance of such an approach is strengthened with regard to the mosaic patient, in whom the CNV was significantly enriched in DNA isolated from hair and saliva, and where both techniques were able to detect and confirm the PTCH1 copy-number variant. Whereas ddPCR analysis showed that the PTCH1 gain was present in hair cells (approximately 92%) and in saliva cells (74%), MLPA detected the gain in 74% of the hair cells and 59% of the saliva cells, indicating that a rather large variation exists between the ddPCR and MLPA measurement outcomes. Although CNV analysis in the gonadal cells of a mosaic patient can be of significant value for the estimation of the disease recurrence risk, no such analysis was performed in the spermatocytes of the mosaic father due to his declining this request and the irrelevance because of his non-reproductive age. This may, however, be relevant in other identified mosaic patients of reproductive age who do not yet have affected offspring.
Our data show that, although both techniques were able to detect the germline CNV in the index patient’s blood lymphocytes, MLPA can miss CNVs in the peripheral blood of mosaic patients, especially in low-grade postzygotic mosaicism. Thw=e limitation of MLPA in the establishment of a genetic diagnosis in mosaic patients was among others illustrated by a case study on clinically unaffected parents of patients with tuberous sclerosis complex and neurofibromatosis type 1, where it was shown that MLPA was also less sensitive than FISH or PCR for detecting large rearrangements [
16,
17]. In diagnostic settings, but also in most of the reported MLPA studies, a theoretical or arbitrary ratio range is commonly used, which is uniform for all probes in the mix, and defines the cut-off value with respect to the normal range (e.g., 0.75–1.25, 0.8–1.2, or 0.95–1.05) [
16,
18,
19,
20]. As illustrated in our case study, such a “safe” arbitrary ratio range could easily result in missing a CNV. Therefore, it has been proposed that we establish a reference cut-off range for each individual probe, and provide unequivocal scoring criteria for the more accurate identification of CNVs using MLPA [
19,
21,
22]. Still, our data show that substantial differences exist with respect to the measured copy-number ratios in the different tissues between ddPCR and MLPA. Previously we showed the strength of ddPCR compared to RFLP in the detection of
PTCH1 mutations in low-grade mosaic BCNS [
23]. When performing CNV analysis on DNA isolated from tissue with a high wild-type background, as is the case in mosaicism, absolute quantification methods such as ddPCR are even more valuable, where a high detection accuracy is accomplished that is not achieved when using relative CNV detection methods such as MLPA, QPCR, or array-based technologies [
11,
24,
25]. Another example of the high sensitivity of ddPCR analysis was shown by detecting CNVs at an allele frequency of <1% in somatic skin mosaicisms [
26].
A drawback to using ddPCR analysis for CNV detection in BCNS patients is the limited number of commercial CNV probes available for
PTCH1 [
11]. In this study, a mutational analysis of the index patient’s blood revealed indications for an allelic imbalance covering the intron 10 region prior to MLPA and ddPCR analysis. Yet, with no such prior knowledge, the testing of BCNS patients in a routine diagnostic setting would require the development of
PTCH1 multiplex ddPCR CNV assays covering larger parts of
PTCH1, thereby allowing more accurate mapping of the aberrant region within the gene [
27]. The definition of a large reference dataset to normalize the smMIP-NGS data could overcome this limitation in CNV detection, and, in fact, cover the complete gene. One might consider cumulatively collecting diagnostic samples to feed this large reference set and eventually implementing this CNV analysis via NGS. However, this is time-consuming and therefore the design of custom-made probes for ddPCR may be faster and more sensitive in cases where a rare CNV variant is detected.
Although we concur that a study with only 2 cases and a small number of tissue samples is limiting, it has shown that accurate CNV analysis in BCNS patients is crucial to establish a reliable genetic diagnosis, which involves familial segregation testing. Furthermore, estimating the disease recurrence risk for offspring in mosaic patients to offer reliable genetic counseling in a prenatal diagnosis (PND) or preimplantation genetic test (PGT) context is pivotal. This especially applies to CNV testing in low-grade post-zygotic mosaicism patients that might not fulfill the diagnostic criteria for BCNS, and in which the genetic defect can be present in peripheral blood with a very low allele frequency. Particularly in these patients, ddPCR offers a more accurate method of copy-number quantification compared to conventional techniques such as MLPA.


 ,
, 



{kind=link}
{kind=link}
{kind=link}