Feasibility of Applying Helper-Dependent Adenoviral Vectors for Cancer Immunotherapy

Abstract
:
1. Introduction
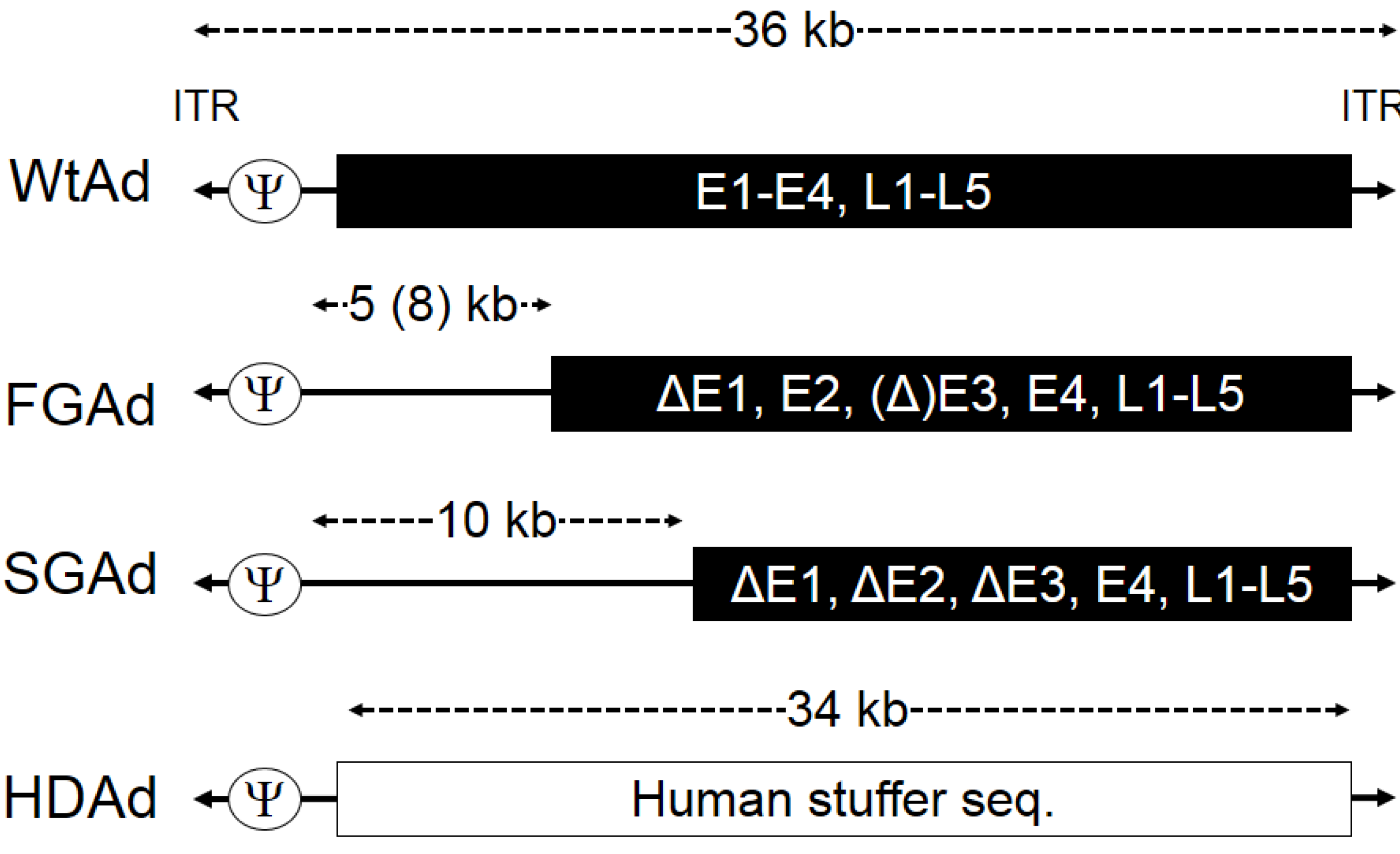
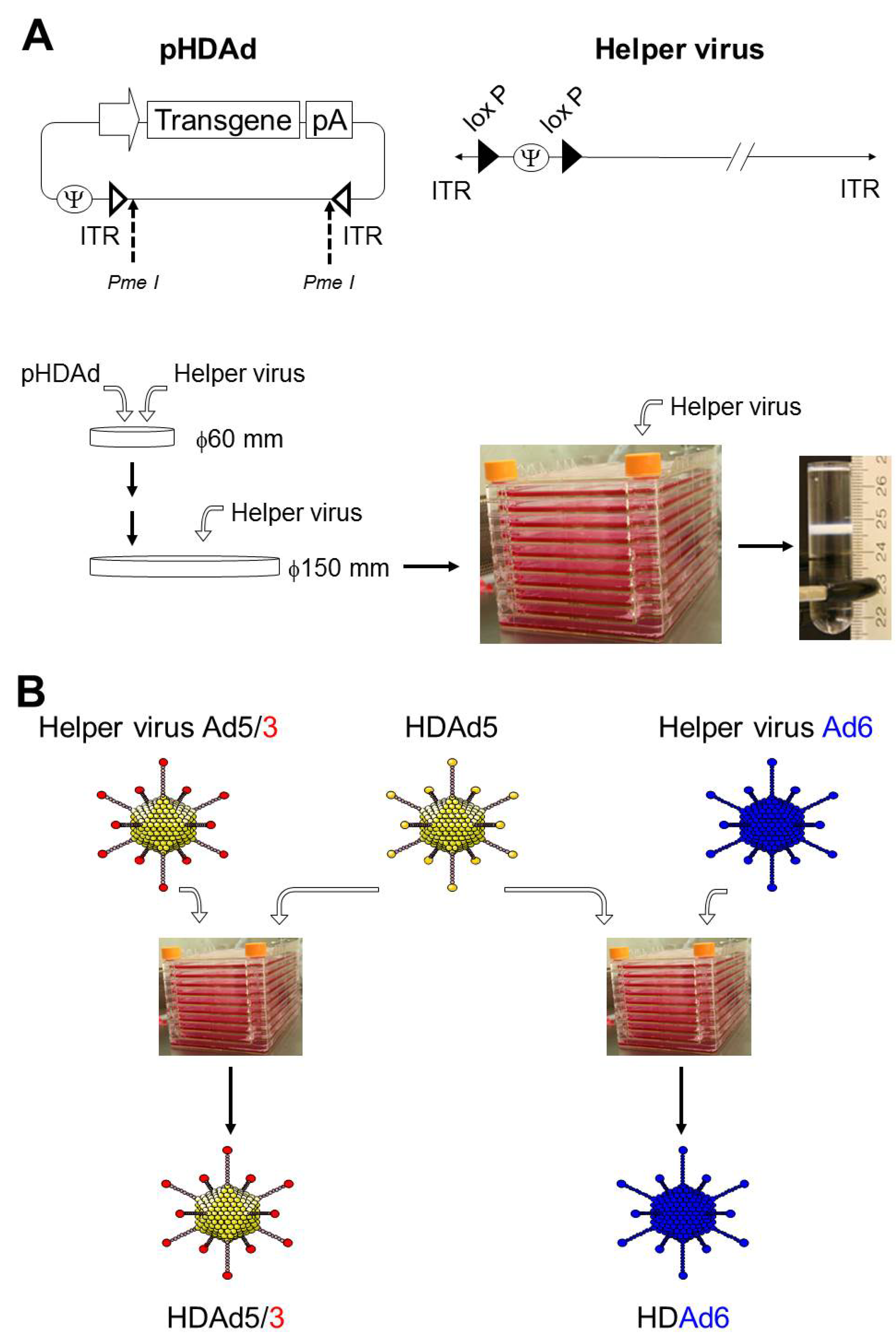
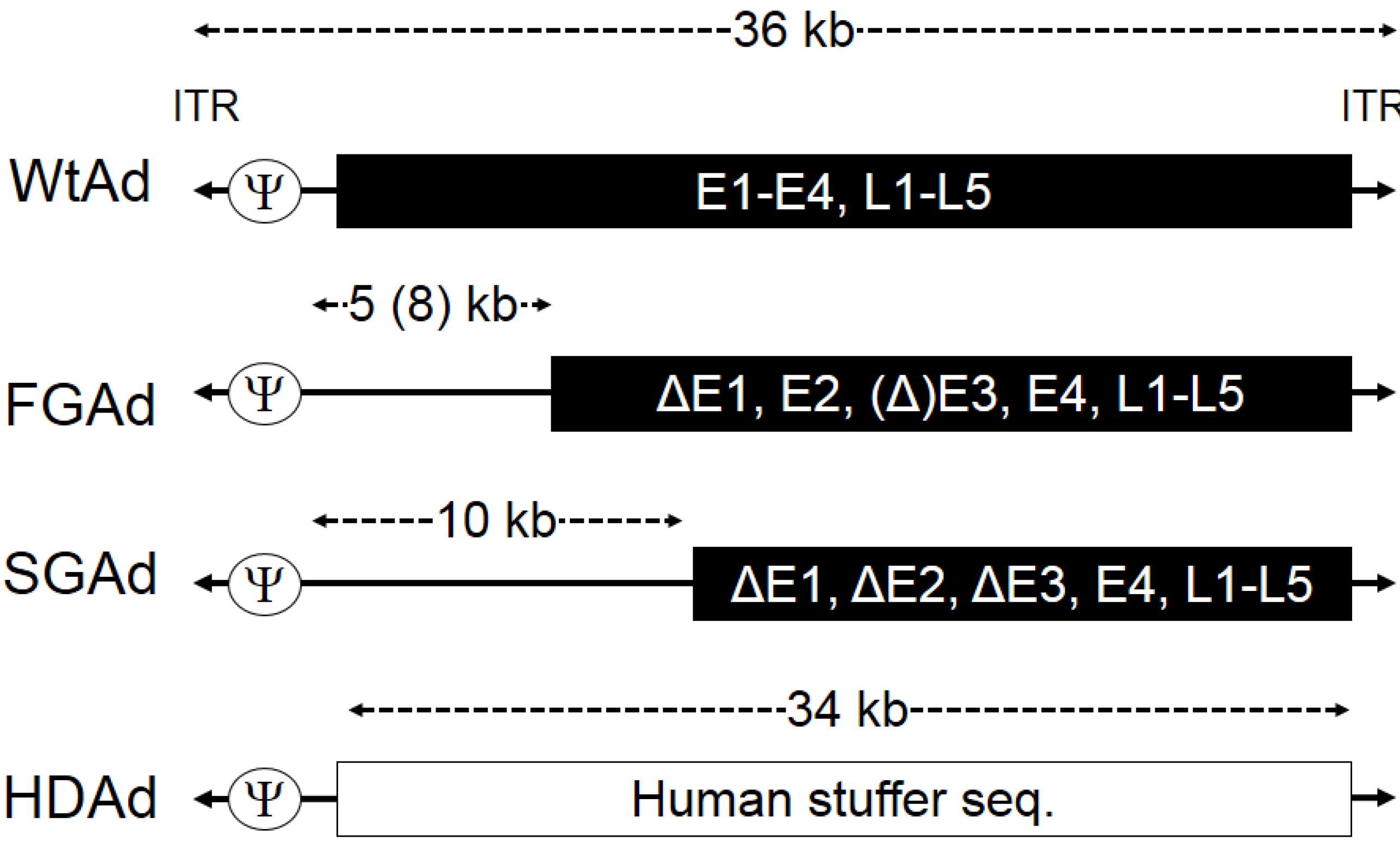
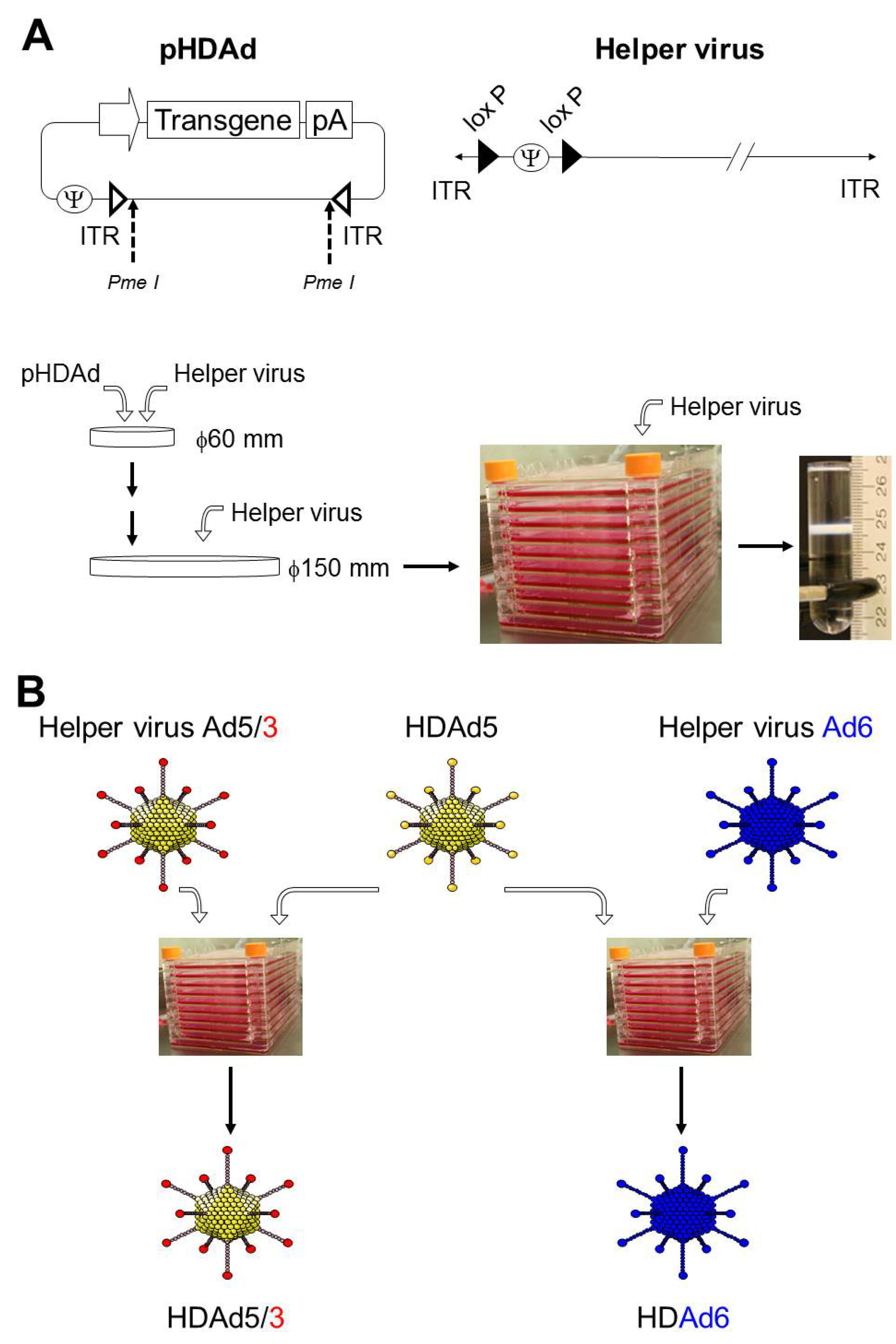
2. Production of HDAd
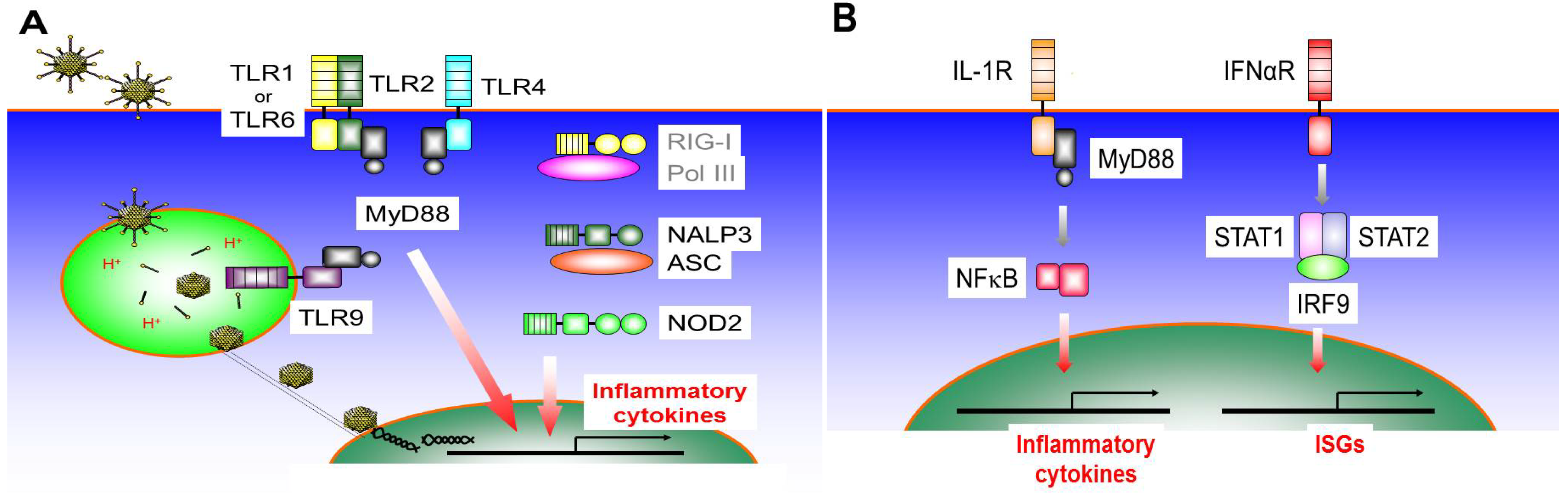
3. The Innate Inflammatory Response to Ad-Based Vectors may Contribute to Cancer Immunotherapy
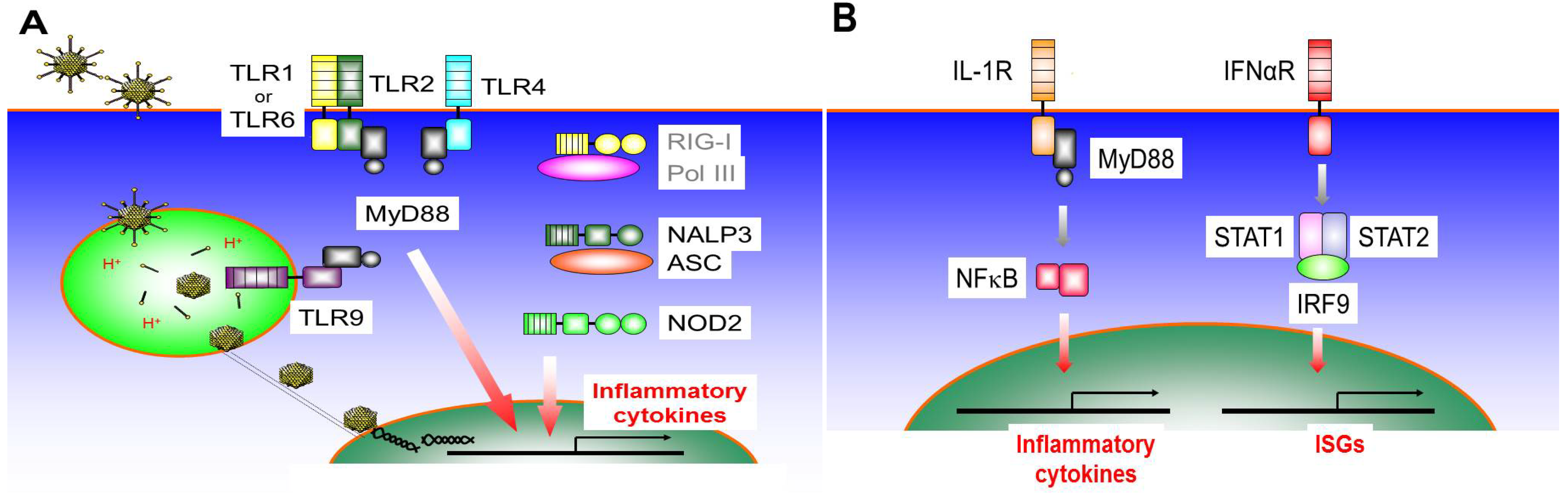
3.1. Toll-Like Receptor 9 (TLR9)
3.2. Toll-Like Receptor 4 (TLR4)
3.3. Toll-Like Receptor 2 (TLR2)
3.4. Nod-Like Receptors (NLRs)
3.5. RIG-I Like Receptors (RLRs)
3.6. IL-1R Signaling Pathway
3.7. IFNαR Signaling Pathway
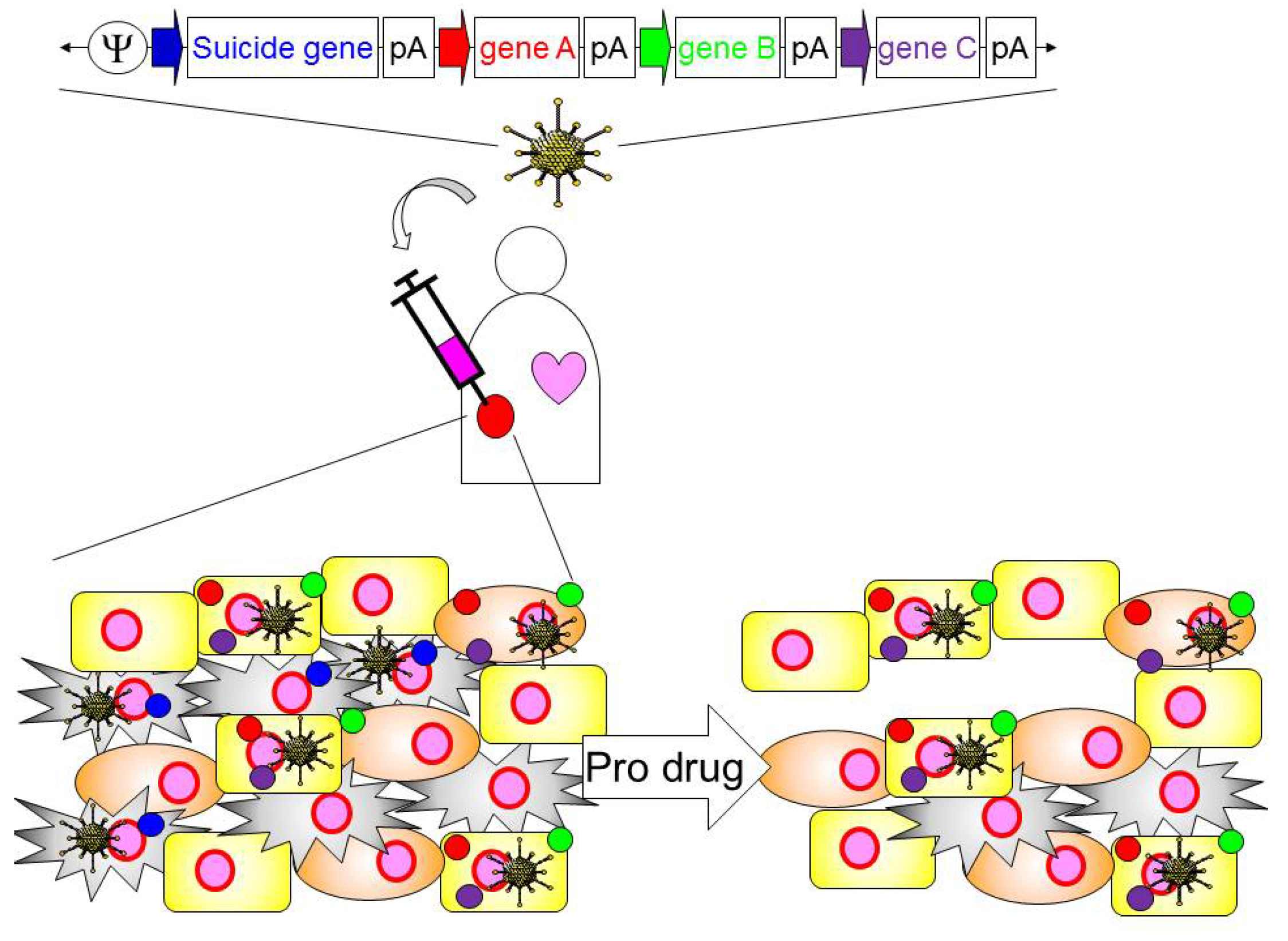
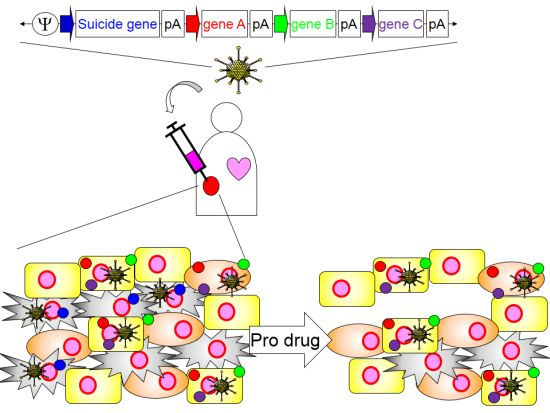
4. Application of HDAd for Cancer Immunotherapy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Function | Gene | Cancer type | Clinical trial Code |
|---|---|---|---|
| Cytokine | IFNβ | Pleural Mesothelioma, Colorectal Carcinoma | NCT00299962, NCT00107861 |
| IFNα2b | Mesothelioma | NCT01212367 | |
| IFNγ | B-Cell Lymphoma | NCT00394693 | |
| IL-12 | Breast Cancer, Colorectal Cancer, Prostate Cancer, Melanoma, Neoplasms | NCT00849459, NCT00072098, NCT00406939, NCT01397708, NCT00110526 | |
| IL-2 | Neuroblastoma | NCT00048386 | |
| MDA-7 (IL-24) | Malignant Melanoma | NCT00116363 | |
| TNFα | Esophageal Cancer, Pancreatic Cancer | NCT00051480, NCT00051467 | |
| GM-CSF | Malignant Solid Tumor | NCT01598129 | |
| FLt3L | Malignant Glioma | NCT01811992 | |
| Tumor suppressor | p53 | Squamous Carcinoma, Lip and Oral Cavity Cancer, Head and Neck Carcinoma, Brain Tumors, Liver Cancer, Ovarian Cancer, Lung Cancer, Bladder Cancer, Breast Cancer | NCT00041613, NCT00064103, NCT00004041, NCT00003147, NCT00003880, NCT00003649, NCT00003167 |
| REIC/Dkk-3 | Prostate cancer | NCT01197209 | |
| RTVP-1 | Prostatic Neoplasms | NCT00403221 | |
| Suicide molecule | TK | Malignant Glioma, Brain Tumors, Hepatocellular Carcinoma, Ovarian Cancer, Melanoma, Pancreatic Cancer | NCT01811992, NCT00002824, NCT00844623, NCT00638612, NCT00005057 |
| Costimulatory molecule | CD40L | Malignant Melanoma, Bladder Cancer, Breast Cancer, Neoplasms, Leukemia, Lymphoma | NCT01455259, NCT00706615, NCT00504322, NCT00942409 |
| Anti-angiogenic molecule | Endostatin | Head and Neck Squamous Carcinoma, Advanced solid tumors | NCT00634595, NCT00262327 |
| Antigen | PSA | Prostate cancer | NCT00583752 |
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Cerullo, V.; Koski, A.; Vaha-Koskela, M.; Hemminki, A. Chapter eight—Oncolytic adenoviruses for cancer immunotherapy: Data from mice, hamsters, and humans. Adv. Cancer Res. 2012, 115, 265–318. [Google Scholar] [CrossRef]
- Palmer, D.; Ng, P. Improved system for Helper-dependent adenoviral vector production. Mol. Ther. 2003, 8, 846–852. [Google Scholar] [CrossRef]
- Muruve, D.A.; Cotter, M.J.; Zaiss, A.K.; White, L.R.; Liu, Q.; Chan, T.; Clark, S.A.; Ross, P.J.; Meulenbroek, R.A.; Maelandsmo, G.M.; et al. Helper-dependent adenovirus vectors elicit intact innate but attenuated adaptive host immune responses in vivo. J. Virol. 2004, 78, 5966–5972. [Google Scholar] [CrossRef]
- Brunetti-Pierri, N.; Ng, P. Helper-dependent adenoviral vectors for liver-directed gene therapy. Hum. Mol. Genet. 2011, 20, R7–R13. [Google Scholar] [CrossRef]
- Cerullo, V.; Seiler, M.P.; Mane, V.; Cela, R.; Clarke, C.; Kaufman, R.J.; Pipe, S.W.; Lee, B. Correction of murine hemophilia A and immunological differences of factor VIII variants delivered by helper-dependent adenoviral vectors. Mol. Ther. 2007, 15, 2080–2087. [Google Scholar] [CrossRef]
- Guse, K.; Suzuki, M.; Sule, G.; Bertin, T.K.; Tyynismaa, H.; Ahola-Erkkila, S.; Palmer, D.; Suomalainen, A.; Ng, P.; Cerullo, V.; et al. Capsid-modified adenoviral vectors for improved muscle-directed gene therapy. Hum. Gene Ther. 2012, 23, 1065–1070. [Google Scholar] [CrossRef]
- Rauschhuber, C.; Noske, N.; Ehrhardt, A. New insights into stability of recombinant adenovirus vector genomes in mammalian cells. Eur. J. Cell Biol. 2012, 91, 2–9. [Google Scholar] [CrossRef]
- McCormack, W.M., Jr.; Seiler, M.P.; Bertin, T.K.; Ubhayakar, K.; Palmer, D.J.; Ng, P.; Nichols, T.C.; Lee, B. Helper-dependent adenoviral gene therapy mediates long-term correction of the clotting defect in the canine hemophilia A model. J. Thromb. Haemost. 2006, 4, 1218–1225. [Google Scholar] [CrossRef]
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer immunotherapy comes of age. Nature 2011, 480, 480–489. [Google Scholar] [CrossRef]
- Suzuki, M.; Cela, R.; Clarke, C.; Bertin, T.K.; Mourino, S.; Lee, B. Large-scale production of high-quality helper-dependent adenoviral vectors using adherent cells in cell factories. Hum. Gene Ther. 2010, 21, 120–126. [Google Scholar] [CrossRef]
- Parks, R.J.; Graham, F.L. A helper-dependent system for adenovirus vector production helps define a lower limit for efficient DNA packaging. J. Virol. 1997, 71, 3293–3298. [Google Scholar]
- Palmer, D.J.; Ng, P. Physical and infectious titers of helper-dependent adenoviral vectors: A method of direct comparison to the adenovirus reference material. Mol. Ther. 2004, 10, 792–798. [Google Scholar] [CrossRef]
- Weaver, E.A.; Hillestad, M.L.; Khare, R.; Palmer, D.; Ng, P.; Barry, M.A. Characterization of species C human adenovirus serotype 6 (Ad6). Virology 2011, 412, 19–27. [Google Scholar] [CrossRef]
- Zuckerman, J.B.; Robinson, C.B.; McCoy, K.S.; Shell, R.; Sferra, T.J.; Chirmule, N.; Magosin, S.A.; Propert, K.J.; Brown-Parr, E.C.; Hughes, J.V.; et al. A phase I study of adenovirus-mediated transfer of the human cystic fibrosis transmembrane conductance regulator gene to a lung segment of individuals with cystic fibrosis. Hum. Gene Ther. 1999, 10, 2973–2985. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef]
- Haas, T.; Metzger, J.; Schmitz, F.; Heit, A.; Muller, T.; Latz, E.; Wagner, H. The DNA sugar backbone 2' deoxyribose determines toll-like receptor 9 activation. Immunity 2008, 28, 315–323. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef]
- Medina-Kauwe, L.K. Endocytosis of adenovirus and adenovirus capsid proteins. Adv. Drug Deliv. Rev. 2003, 55, 1485–1496. [Google Scholar] [CrossRef]
- Cerullo, V.; Seiler, M.P.; Mane, V.; Brunetti-Pierri, N.; Clarke, C.; Bertin, T.K.; Rodgers, J.R.; Lee, B. Toll-like receptor 9 triggers an innate immune response to helper-dependent adenoviral vectors. Mol. Ther. 2007, 15, 378–385. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Kawabata, K.; Koizumi, N.; Sakurai, F.; Nakashima, K.; Sakurai, H.; Sasaki, T.; Okada, N.; Yamanishi, K.; Mizuguchi, H. Role of MyD88 and TLR9 in the innate immune response elicited by serotype 5 adenoviral vectors. Hum. Gene Ther. 2007, 18, 753–762. [Google Scholar] [CrossRef]
- Suzuki, M.; Cerullo, V.; Bertin, T.K.; Cela, R.; Clarke, C.; Guenther, M.; Brunetti-Pierri, N.; Lee, B. MyD88-dependent silencing of transgene expression during the innate and adaptive immune response to helper-dependent adenovirus. Hum. Gene Ther. 2010, 21, 325–336. [Google Scholar] [CrossRef]
- Krieg, A.M. Toll-like receptor 9 (TLR9) agonists in the treatment of cancer. Oncogene 2008, 27, 161–167. [Google Scholar] [CrossRef]
- Speiser, D.E.; Lienard, D.; Rufer, N.; Rubio-Godoy, V.; Rimoldi, D.; Lejeune, F.; Krieg, A.M.; Cerottini, J.C.; Romero, P. Rapid and strong human CD8+ T cell responses to vaccination with peptide, IFA, and CpG oligodeoxynucleotide 7909. J. Clin. Invest. 2005, 115, 739–746. [Google Scholar] [CrossRef]
- Cerullo, V.; Diaconu, I.; Romano, V.; Hirvinen, M.; Ugolini, M.; Escutenaire, S.; Holm, S.L.; Kipar, A.; Kanerva, A.; Hemminki, A. An oncolytic adenovirus enhanced for toll-like receptor 9 stimulation increases antitumor immune responses and tumor clearance. Mol. Ther. 2012, 20, 2076–2086. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Waddington, S.N.; McVey, J.H.; Bhella, D.; Parker, A.L.; Barker, K.; Atoda, H.; Pink, R.; Buckley, S.M.; Greig, J.A.; Denby, L.; et al. Adenovirus serotype 5 hexon mediates liver gene transfer. Cell 2008, 132, 397–409. [Google Scholar] [CrossRef]
- Kalyuzhniy, O.; di Paolo, N.C.; Silvestry, M.; Hofherr, S.E.; Barry, M.A.; Stewart, P.L.; Shayakhmetov, D.M. Adenovirus serotype 5 hexon is critical for virus infection of hepatocytes in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 5483–5488. [Google Scholar] [CrossRef]
- Doronin, K.; Flatt, J.W.; di Paolo, N.C.; Khare, R.; Kalyuzhniy, O.; Acchione, M.; Sumida, J.P.; Ohto, U.; Shimizu, T.; Akashi-Takamura, S.; et al. Coagulation factor X activates innate immunity to human species C adenovirus. Science 2012, 338, 795–798. [Google Scholar] [CrossRef]
- Adams, S. Toll-like receptor agonists in cancer therapy. Immunotherapy 2009, 1, 949–964. [Google Scholar] [CrossRef]
- Lapteva, N.; Aldrich, M.; Rollins, L.; Ren, W.; Goltsova, T.; Chen, S.Y.; Huang, X.F. Attraction and activation of dendritic cells at the site of tumor elicits potent antitumor immunity. Mol. Ther. 2009, 17, 1626–1636. [Google Scholar] [CrossRef]
- Mills, K.H. TLR-dependent T cell activation in autoimmunity. Nat. Rev. Immunol. 2011, 11, 807–822. [Google Scholar]
- Appledorn, D.M.; Patial, S.; McBride, A.; Godbehere, S.; van Rooijen, N.; Parameswaran, N.; Amalfitano, A. Adenovirus vector-induced innate inflammatory mediators, MAPK signaling, as well as adaptive immune responses are dependent upon both TLR2 and TLR9 in vivo. J. Immunol. 2008, 181, 2134–2144. [Google Scholar]
- Appledorn, D.M.; McBride, A.; Seregin, S.; Scott, J.M.; Schuldt, N.; Kiang, A.; Godbehere, S.; Amalfitano, A. Complex interactions with several arms of the complement system dictate innate and humoral immunity to adenoviral vectors. Gene Ther. 2008, 15, 1606–1617. [Google Scholar] [CrossRef]
- Lamm, D.L.; Blumenstein, B.A.; Crawford, E.D.; Montie, J.E.; Scardino, P.; Grossman, H.B.; Stanisic, T.H.; Smith, J.A., Jr.; Sullivan, J.; Sarosdy, M.F.; et al. A randomized trial of intravesical doxorubicin and immunotherapy with bacille Calmette-Guerin for transitional-cell carcinoma of the bladder. N. Engl. J. Med. 1991, 325, 1205–1209. [Google Scholar] [CrossRef]
- Curtin, J.F.; Liu, N.; Candolfi, M.; Xiong, W.; Assi, H.; Yagiz, K.; Edwards, M.R.; Michelsen, K.S.; Kroeger, K.M.; Liu, C.; et al. HMGB1 mediates endogenous TLR2 activation and brain tumor regression. PLoS Med. 2009, 6, e10. [Google Scholar] [CrossRef]
- Assi, H.; Candolfi, M.; Baker, G.; Mineharu, Y.; Lowenstein, P.R.; Castro, M.G. Gene therapy for brain tumors: Basic developments and clinical implementation. Neurosci. Lett. 2012, 527, 71–77. [Google Scholar] [CrossRef]
- Elinav, E.; Strowig, T.; Henao-Mejia, J.; Flavell, R.A. Regulation of the antimicrobial response by NLR proteins. Immunity 2011, 34, 665–679. [Google Scholar] [CrossRef]
- Fritz, J.H.; Ferrero, R.L.; Philpott, D.J.; Girardin, S.E. Nod-like proteins in immunity, inflammation and disease. Nat. Immunol. 2006, 7, 1250–1257. [Google Scholar] [CrossRef]
- Muruve, D.A.; Petrilli, V.; Zaiss, A.K.; White, L.R.; Clark, S.A.; Ross, P.J.; Parks, R.J.; Tschopp, J. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature 2008, 452, 103–107. [Google Scholar] [CrossRef]
- Suzuki, M.; Cela, R.; Bertin, T.K.; Sule, G.; Cerullo, V.; Rodgers, J.R.; Lee, B. NOD2 signaling contributes to the innate immune response against helper-dependent adenovirus vectors independently of MyD88 in vivo. Hum. Gene Ther. 2011, 22, 1071–1082. [Google Scholar] [CrossRef]
- Kobayashi, K.S.; Chamaillard, M.; Ogura, Y.; Henegariu, O.; Inohara, N.; Nunez, G.; Flavell, R.A. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science 2005, 307, 731–734. [Google Scholar] [CrossRef]
- Couturier-Maillard, A.; Secher, T.; Rehman, A.; Normand, S.; de Arcangelis, A.; Haesler, R.; Huot, L.; Grandjean, T.; Bressenot, A.; Delanoye-Crespin, A.; et al. NOD2-mediated dysbiosis predisposes mice to transmissible colitis and colorectal cancer. J. Clin. Invest. 2013, 123, 700–711. [Google Scholar]
- Loo, Y.M.; Gale, M., Jr. Immune signaling by RIG-I-like receptors. Immunity 2011, 34, 680–692. [Google Scholar] [CrossRef]
- Chiu, Y.H.; Macmillan, J.B.; Chen, Z.J. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 2009, 138, 576–591. [Google Scholar] [CrossRef]
- Minamitani, T.; Iwakiri, D.; Takada, K. Adenovirus virus-associated RNAs induce type I interferon expression through a RIG-I-mediated pathway. J. Virol. 2011, 85, 4035–4040. [Google Scholar] [CrossRef]
- Qu, J.; Hou, Z.; Han, Q.; Jiang, W.; Zhang, C.; Tian, Z.; Zhang, J. Intracellular poly(I:C) initiated gastric adenocarcinoma cell apoptosis and subsequently ameliorated NK cell functions. J. Interferon Cytokine Res. 2013, 34, 52–59. [Google Scholar]
- Sims, J.E.; Smith, D.E. The IL-1 family: Regulators of immunity. Nat. Rev. Immunol. 2010, 10, 89–102. [Google Scholar]
- Di Paolo, N.C.; Miao, E.A.; Iwakura, Y.; Murali-Krishna, K.; Aderem, A.; Flavell, R.A.; Papayannopoulou, T.; Shayakhmetov, D.M. Virus binding to a plasma membrane receptor triggers interleukin-1 alpha-mediated proinflammatory macrophage response in vivo. Immunity 2009, 31, 110–121. [Google Scholar] [CrossRef]
- Apte, R.N.; Voronov, E. Is interleukin-1 a good or bad ‘guy’ in tumor immunobiology and immunotherapy? Immunol. Rev. 2008, 222, 222–241. [Google Scholar] [CrossRef]
- Deans, D.A.; Wigmore, S.J.; Gilmour, H.; Paterson-Brown, S.; Ross, J.A.; Fearon, K.C. Elevated tumour interleukin-1beta is associated with systemic inflammation: A marker of reduced survival in gastro-oesophageal cancer. Br. J. Cancer 2006, 95, 1568–1575. [Google Scholar] [CrossRef]
- Esandi, M.C.; van Someren, G.D.; Bout, A.; Mulder, A.H.; van Bekkum, D.W.; Valerio, D.; Noteboom, J.L. IL-1/IL-3 gene therapy of non-small cell lung cancer (NSCLC) in rats using ‘cracked’ adenoproducer cells. Gene Ther. 1998, 5, 778–788. [Google Scholar]
- Lavi, G.; Voronov, E.; Dinarello, C.A.; Apte, R.N.; Cohen, S. Sustained delivery of IL-1 Ra from biodegradable microspheres reduces the number of murine B16 melanoma lung metastases. J. Control. Release 2007, 123, 123–130. [Google Scholar] [CrossRef]
- Allgayer, H.; Nicolaus, S.; Schreiber, S. Decreased interleukin-1 receptor antagonist response following moderate exercise in patients with colorectal carcinoma after primary treatment. Cancer Detect. Prev. 2004, 28, 208–213. [Google Scholar] [CrossRef]
- Sadler, A.J.; Williams, B.R. Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 2008, 8, 559–568. [Google Scholar] [CrossRef]
- Suzuki, M.; Bertin, T.K.; Rogers, G.L.; Cela, R.G.; Zolotukhin, I.; Palmer, D.J.; Ng, P.; Herzog, R.W.; Lee, B. Differential type I interferon-dependent transgene silencing of helper-dependent adenoviral vs. adeno-associated viral vectors in vivo. Mol. Ther. 2013, 21, 796–805. [Google Scholar] [CrossRef]
- Zhu, J.; Huang, X.; Yang, Y. Innate immune response to adenoviral vectors is mediated by both Toll-like receptor-dependent and -independent pathways. J. Virol. 2007, 81, 3170–3180. [Google Scholar] [CrossRef]
- Suzuki, M.; Chiocca, E.A.; Saeki, Y. Early STAT1 activation after systemic delivery of HSV amplicon vectors suppresses transcription of the vector-encoded transgene. Mol. Ther. 2007, 15, 2017–2026. [Google Scholar] [CrossRef]
- Ullman, A.J.; Reich, N.C.; Hearing, P. Adenovirus E4 ORF3 protein inhibits the interferon-mediated antiviral response. J. Virol. 2007, 81, 4744–4752. [Google Scholar] [CrossRef]
- Chiocca, E.A.; Smith, K.M.; McKinney, B.; Palmer, C.A.; Rosenfeld, S.; Lillehei, K.; Hamilton, A.; DeMasters, B.K.; Judy, K.; Kirn, D. A phase I trial of Ad.hIFN-beta gene therapy for glioma. Mol. Ther. 2008, 16, 618–626. [Google Scholar] [CrossRef]
- Hallden, G.; Hill, R.; Wang, Y.; Anand, A.; Liu, T.C.; Lemoine, N.R.; Francis, J.; Hawkins, L.; Kirn, D. Novel immunocompetent murine tumor models for the assessment of replication-competent oncolytic adenovirus efficacy. Mol. Ther. 2003, 8, 412–424. [Google Scholar] [CrossRef]
- Tuve, S.; Liu, Y.; Tragoolpua, K.; Jacobs, J.D.; Yumul, R.C.; Li, Z.Y.; Strauss, R.; Hellstrom, K.E.; Disis, M.L.; Roffler, S.; et al. In situ adenovirus vaccination engages T effector cells against cancer. Vaccine 2009, 27, 4225–4239. [Google Scholar] [CrossRef]
- Kanerva, A.; Nokisalmi, P.; Diaconu, I.; Koski, A.; Cerullo, V.; Liikanen, I.; Tahtinen, S.; Oksanen, M.; Heiskanen, R.; Pesonen, S.; et al. Antiviral and antitumor T-cell immunity in patients treated with GM-CSF-coding oncolytic adenovirus. Clin. Cancer Res. 2013, 19, 2734–2744. [Google Scholar]
- Koski, A.; Kangasniemi, L.; Escutenaire, S.; Pesonen, S.; Cerullo, V.; Diaconu, I.; Nokisalmi, P.; Raki, M.; Rajecki, M.; Guse, K.; et al. Treatment of cancer patients with a serotype 5/3 chimeric oncolytic adenovirus expressing GMCSF. Mol. Ther. 2010, 18, 1874–1884. [Google Scholar] [CrossRef]
- Edukulla, R.; Woller, N.; Mundt, B.; Knocke, S.; Gurlevik, E.; Saborowski, M.; Malek, N.; Manns, M.P.; Wirth, T.; Kuhnel, F.; et al. Antitumoral immune response by recruitment and expansion of dendritic cells in tumors infected with telomerase-dependent oncolytic viruses. Cancer Res. 2009, 69, 1448–1458. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef]
- Bett, A.J.; Prevec, L.; Graham, F.L. Packaging capacity and stability of human adenovirus type 5 vectors. J. Virol. 1993, 67, 5911–5921. [Google Scholar]
- Russell, S.J.; Peng, K.W.; Bell, J.C. Oncolytic virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef]
- Miest, T.S.; Cattaneo, R. New viruses for cancer therapy: Meeting clinical needs. Nat. Rev. Microbiol. 2014, 12, 23–34. [Google Scholar] [CrossRef]
- Di Stasi, A.; Tey, S.K.; Dotti, G.; Fujita, Y.; Kennedy-Nasser, A.; Martinez, C.; Straathof, K.; Liu, E.; Durett, A.G.; Grilley, B.; et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N. Engl. J. Med. 2011, 365, 1673–1683. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Available online: http://clinicaltrials.gov/ (accessed on 1 August 2013).
- Xiong, W.; Candolfi, M.; Kroeger, K.M.; Puntel, M.; Mondkar, S.; Larocque, D.; Liu, C.; Curtin, J.F.; Palmer, D.; Ng, P.; et al. Immunization against the transgene but not the TetON switch reduces expression from gutless adenoviral vectors in the brain. Mol. Ther. 2008, 16, 343–351. [Google Scholar] [CrossRef]
- Curtin, J.F.; Candolfi, M.; Puntel, M.; Xiong, W.; Muhammad, A.K.; Kroeger, K.; Mondkar, S.; Liu, C.; Bondale, N.; Lowenstein, P.R.; et al. Regulated expression of adenoviral vectors-based gene therapies: Therapeutic expression of toxins and immune-modulators. Methods Mol. Biol. 2008, 434, 239–266. [Google Scholar]
- King, G.D.; Kroeger, K.M.; Bresee, C.J.; Candolfi, M.; Liu, C.; Manalo, C.M.; Muhammad, A.K.; Pechnick, R.N.; Lowenstein, P.R.; Castro, M.G. Flt3L in combination with HSV1-TK-mediated gene therapy reverses brain tumor-induced behavioral deficits. Mol. Ther. 2008, 16, 682–690. [Google Scholar] [CrossRef]
- Conforti, A.; Cipriani, B.; Peruzzi, D.; Dharmapuri, S.; Kandimalla, E.R.; Agrawal, S.; Mori, F.; Ciliberto, G.; La Monica, N.; Aurisicchio, L. A TLR9 agonist enhances therapeutic effects of telomerase genetic vaccine. Vaccine 2010, 28, 3522–3530. [Google Scholar] [CrossRef]
- Lotze, M.T.; Tracey, K.J. High-mobility group box 1 protein (HMGB1): Nuclear weapon in the immune arsenal. Nat. Rev. Immunol. 2005, 5, 331–342. [Google Scholar] [CrossRef]
- Dranoff, G.; Jaffee, E.; Lazenby, A.; Golumbek, P.; Levitsky, H.; Brose, K.; Jackson, V.; Hamada, H.; Pardoll, D.; Mulligan, R.C. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc. Natl. Acad. Sci. USA 1993, 90, 3539–3543. [Google Scholar] [CrossRef]
- Kohanbash, G.; McKaveney, K.; Sakaki, M.; Ueda, R.; Mintz, A.H.; Amankulor, N.; Fujita, M.; Ohlfest, J.R.; Okada, H. GM-CSF Promotes the immunosuppressive activity of glioma-infiltrating myeloid cells through interleukin-4 receptor-alpha. Cancer Res. 2013, 73, 6413–6423. [Google Scholar] [CrossRef]
- Van Kooten, C.; Banchereau, J. CD40-CD40 ligand. J. Leukoc. Biol. 2000, 67, 2–17. [Google Scholar]
- Pesonen, S.; Diaconu, I.; Kangasniemi, L.; Ranki, T.; Kanerva, A.; Pesonen, S.K.; Gerdemann, U.; Leen, A.M.; Kairemo, K.; Oksanen, M.; et al. Oncolytic immunotherapy of advanced solid tumors with a CD40L-expressing replicating adenovirus: Assessment of safety and immunologic responses in patients. Cancer Res. 2012, 72, 1621–1631. [Google Scholar] [CrossRef]
- Del Vecchio, M.; Bajetta, E.; Canova, S.; Lotze, M.T.; Wesa, A.; Parmiani, G.; Anichini, A. Interleukin-12: Biological properties and clinical application. Clin. Cancer Res. 2007, 13, 4677–4685. [Google Scholar] [CrossRef]
- Sangro, B.; Mazzolini, G.; Ruiz, J.; Herraiz, M.; Quiroga, J.; Herrero, I.; Benito, A.; Larrache, J.; Pueyo, J.; Subtil, J.C.; et al. Phase I trial of intratumoral injection of an adenovirus encoding interleukin-12 for advanced digestive tumors. J. Clin. Oncol. 2004, 22, 1389–1397. [Google Scholar] [CrossRef]
- Mazzolini, G.; Prieto, J.; Melero, I. Gene therapy of cancer with interleukin-12. Curr. Pharm. Des. 2003, 9, 1981–1991. [Google Scholar] [CrossRef]
- Jakobisiak, M.; Golab, J.; Lasek, W. Interleukin 15 as a promising candidate for tumor immunotherapy. Cytokine Growth Factor Rev. 2011, 22, 99–108. [Google Scholar] [CrossRef]
- Drake, C.G. Prostate cancer as a model for tumour immunotherapy. Nat. Rev. Immunol. 2010, 10, 580–593. [Google Scholar] [CrossRef]
- Drake, C.G. Immunotherapy for prostate cancer: an emerging treatment modality. Urol. Clin. N. Am. 2010, 37, 121–129. [Google Scholar] [CrossRef]
- Bollard, C.M.; Rossig, C.; Calonge, M.J.; Huls, M.H.; Wagner, H.J.; Massague, J.; Brenner, M.K.; Heslop, H.E.; Rooney, C.M. Adapting a transforming growth factor beta-related tumor protection strategy to enhance antitumor immunity. Blood 2002, 99, 3179–3187. [Google Scholar] [CrossRef]
- Marchi, L.H.; Paschoalin, T.; Travassos, L.R.; Rodrigues, E.G. Gene therapy with interleukin-10 receptor and interleukin-12 induces a protective interferon-gamma-dependent response against B16F10-Nex2 melanoma. Cancer Gene Ther. 2011, 18, 110–122. [Google Scholar] [CrossRef]
- Dias, J.D.; Hemminki, O.; Diaconu, I.; Hirvinen, M.; Bonetti, A.; Guse, K.; Escutenaire, S.; Kanerva, A.; Pesonen, S.; Loskog, A.; et al. Targeted cancer immunotherapy with oncolytic adenovirus coding for a fully human monoclonal antibody specific for CTLA-4. Gene Ther. 2012, 19, 988–998. [Google Scholar] [CrossRef]
- Puntel, M.; Muhammad, A.K.; Candolfi, M.; Salem, A.; Yagiz, K.; Farrokhi, C.; Kroeger, K.M.; Xiong, W.; Curtin, J.F.; Liu, C.; et al. A novel bicistronic high-capacity gutless adenovirus vector that drives constitutive expression of herpes simplex virus type 1 thymidine kinase and tet-inducible expression of Flt3L for glioma therapeutics. J. Virol. 2010, 84, 6007–6017. [Google Scholar] [CrossRef]
- Puntel, M.; AKM, G.M.; Farrokhi, C.; Vanderveen, N.; Paran, C.; Appelhans, A.; Kroeger, K.M.; Salem, A.; Lacayo, L.; Pechnick, R.N.; et al. Safety profile, efficacy, and biodistribution of a bicistronic high-capacity adenovirus vector encoding a combined immunostimulation and cytotoxic gene therapy as a prelude to a phase I clinical trial for glioblastoma. Toxicol. Appl. Pharmacol. 2013, 268, 318–330. [Google Scholar] [CrossRef]
- Jiang, M.H.; Chen, L.; Li, L.F.; Wu, H.P.; Jiang, L.H.; Qian, Y.Z.; Fang, G.E.; Xue, X.C. A gutless adenoviral vector expressing full-length anti-Her2 antibody. Clin. Exp. Pharmacol. Physiol. 2009, 36, e26–e31. [Google Scholar] [CrossRef]
- King, G.D.; Muhammad, A.K.; Xiong, W.; Kroeger, K.M.; Puntel, M.; Larocque, D.; Palmer, D.; Ng, P.; Lowenstein, P.R.; Castro, M.G. High-capacity adenovirus vector-mediated anti-glioma gene therapy in the presence of systemic antiadenovirus immunity. J. Virol. 2008, 82, 4680–4684. [Google Scholar] [CrossRef]
- VanderVeen, N.; Paran, C.; Krasinkiewicz, J.; Zhao, L.; Palmer, D.; Hervey-Jumper, S.; Ng, P.; Lowenstein, P.R.; Castro, M.G. Effectiveness and preclinical safety profile of doxycycline to be used “off-label” to induce therapeutic transgene expression in a phase I clinical trial for glioma. Hum. Gene Ther. 2013, 24, 116–126. [Google Scholar]
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Farzad, L.M.; Suzuki, M. Feasibility of Applying Helper-Dependent Adenoviral Vectors for Cancer Immunotherapy. Biomedicines 2014, 2, 110-131. https://doi.org/10.3390/biomedicines2010110
Farzad LM, Suzuki M. Feasibility of Applying Helper-Dependent Adenoviral Vectors for Cancer Immunotherapy. Biomedicines. 2014; 2(1):110-131. https://doi.org/10.3390/biomedicines2010110
Chicago/Turabian StyleFarzad, Lisa M., and Masataka Suzuki. 2014. "Feasibility of Applying Helper-Dependent Adenoviral Vectors for Cancer Immunotherapy" Biomedicines 2, no. 1: 110-131. https://doi.org/10.3390/biomedicines2010110