Tumor Restrictions to Oncolytic Virus

1
Institute of Biotechnology, University of Helsinki, Helsinki 00790, Finland
2
A.I. Virtanen Institute for Molecular Sciences, University of Eastern Finland, Kuopio 70211, Finland
*
Authors to whom correspondence should be addressed.
Biomedicines 2014, 2(2), 163-194; https://doi.org/10.3390/biomedicines2020163
Submission received: 18 February 2014
/
Revised: 17 March 2014
/
Accepted: 28 March 2014
/
Published: 17 April 2014
(This article belongs to the Special Issue Gene Therapy Used in Cancer Treatment)
Abstract
:Oncolytic virotherapy has advanced since the days of its conception but therapeutic efficacy in the clinics does not seem to reach the same level as in animal models. One reason is premature oncolytic virus clearance in humans, which is a reasonable assumption considering the immune-stimulating nature of the oncolytic agents. However, several studies are beginning to reveal layers of restriction to oncolytic virotherapy that are present before an adaptive neutralizing immune response. Some of these barriers are present constitutively halting infection before it even begins, whereas others are raised by minute cues triggered by virus infection. Indeed, we and others have noticed that delivering viruses to tumors may not be the biggest obstacle to successful therapy, but instead the physical make-up of the tumor and its capacity to mount antiviral defenses seem to be the most important efficacy determinants. In this review, we summarize the constitutive and innate barriers to oncolytic virotherapy and discuss strategies to overcome them.
1. Introduction
In most solid tumors the tumor interior displays a high cellular density with multiple cell types, both malignant and normal (stromal) cells, interdigitated by strands of extracellular matrix (ECM) and other poorly defined tissue. Stromal cells typically consist of fibroblasts, macrophages, neutrophils and other immune cell types. Tumor-associated fibroblasts are a source of the bulk acellular tissue in tumors [1], which is largely made up of polysugars and glycoproteins, such as collagens, laminins, fibronectins, proteoglycans, hyaluronan and water, forming a dense matrix around and between the cancer cells [2]. This matrix provides structural support but also haptotactic and chemotactic guidance for cancer cells [3]. Moreover, the prevailing tumor interstitium is rich in soluble angiogenic factors, such as members of the vascular endothelial growth factor (VEGF) family [4], cellular growth factors, including endothelial growth factor (EGF), basic fibroblast growth factor (FGF), platelet-derived growth factor (PDGF) and pleiotrophic factors, particularly transforming growth factor (TGF) beta [5]. Additionally, both tumor- and stromal cells secrete cytokines and chemokines, such as interleukin (IL) 6, IL 10, tumor necrosis factor (TNF) alpha, stromal cell-derived factor (SDF) 1, macrophage migration inhibitory factor (MIF) 1, which on one hand recruit more stromal and progenitor cells into the tumors and on the other hand curb the activity of antigen-presenting cells and anti-tumor T cells [6,7,8]. In several independent cancer types, high serum levels of IL 6 and IL 10 are indicators of poor prognosis [9].
Aberrant tumor and stromal growth combined with an overproduction of matrix leads to high interstitial pressure in the tumor, preventing lateral diffusion of therapeutic compounds within the tumor. Tight cellular compactness combined with a high tumor proliferation rate also causes physical crowding and puts constraints on oxygenation, which due to aberrant angiogenesis and rupture of the haphazard vessels is mostly incomplete and results in chronic tissue hypoxia toward the tumor cores. As cancer cells exist in a crowding/hypoxic- and cytokine/growth factor-perpetuated chronic state of stress, they upregulate a number of key cellular molecules that function as a collective defense against a wide range of therapies, including oncolytic viruses. The properties of oncolytic viruses and their status in clinical development as well as various delivery options have been reviewed elsewhere [10,11,12,13]. In this review, we provide an overview of two of the main intratumoral barriers to oncolytic virus spread; the extracellular tumor make-up and the intracellular antiviral defense mechanisms.
2. Physical Barriers to Oncolytic Viruses
In this chapter we introduce the principal physical obstacles for successful oncolytic virotherapy and discuss ways to overcome them. As viruses lack autonomous motility and may potentially adsorb to any surfaces that display their specific receptors, they may only infect the physically delimited regions where they first entered and they may be prone to unfruitful sequestration by already dead cells. Nevertheless, oncolytic viruses vary in size and some are small enough to fit even through tight ECM networks, and efforts have been made through genetic engineering of the viruses to minimize their non-specific binding properties. Most interestingly, viruses may be engineered to express enzymes that directly break down the physical barriers to oncolytic viruses.
2.1. Interstitial Fluid Pressure
Tumor cellular structure and composition may in concert with abundant extracellular matrix (ECM) deposition pose a severe hurdle for systemic virus entry and propagation of infection within the tumor. The extracellular matrix is a complex dense network consisting of multiple proteins, glycoproteins and polysaccharides including collagens, laminins, fibronectins, proteoglycans and hyaluronic acid [14]. The interstitial fluid pressure (IFP) in the tumor surrounding blood and lymphatic vessels is mainly created by the high cell density that forms an increased physical pressure outwards and does not allow free diffusion of therapeutics [15,16,17]. A high IFP generally predicts an aggressive tumor phenotype as tumor cells tend to escape from tumor margins where the pressure drops, thus facilitating spread of metastatic cells [18]. On the other hand, for therapeutic virus, interstitial fluid concentration gradients may pose a hurdle as passive diffusion in viable regions, typically the rim, may occur mainly outward [19]. Given the limited arsenal of known chemicals alleviating tumor compactness and the poor penetration drugs in general into the tumor tissue, this is a considerable problem that may not be easy to tackle. Some strategies are discussed below.
2.2. Extracellular Matrix Deposits
Viruses are passive particles and rely either on radial cell-to-cell spread or on soluble diffusion across concentration gradients to reach their target cells and to propagate the infection. Tumor matrix is critical in this regard, as it can block both forms of spread. Passively diffusing viruses may physically not fit through the strands of the ECM. This was shown for oncolytic herpes virus, which has an outer diameter of over 100 nm, and whose spreading could be improved by matrix-degrading bacterial collagenase co-injection [20]. Bilbao and coworkers showed that adenovirus entry into experimental hepatocellular carcinoma nodules in the livers of immunocompetent rats following intravenous or intraportal injection was directly related to the thickness of the ECM capsule enveloping the nodules [21]. On the other hand, we found using Semliki Forest virus, which has an outer diameter of about 60 nm, that even upon intratumoral injection spread was blocked by ECM, which formed a physical barrier for virus infection (Figure 1).
The foremost option to reduce interstitial fluid pressure and to remove physical molecule barriers imposed by the ECM is to use matrix-degrading enzymes. These act by digesting collagen-, fibrin- and other types of fibrillar matrix deposits, creating more space between cell clusters, and glycosaminoglycan polymers, known to limit fluid movement in tumors. Simultaneously, such strategies may also expose more cell surface to viruses, increasing the likelihood of infection. In a study by Kuriyama, trypsin or collagenase/dispase was injected into subcutaneous U87 and U251 glioma xenografts in immunocompromised mice, followed by a reporter adenovirus [22]. Both types of ECM-degrading enzymes increased tumor transduction by the virus, but when doses got too high, transduction efficacy suffered, demonstrating a balance between ECM-degradation and oncolytic virus efficacy. In another study, treatment of PC3 tumors with vaccinia virus producing matrix metalloprotease (MMP) 9 resulted in reduction of collagen IV fibrils and increase in virus penetration into tumor, which yielded elevated virus titers [23]. Hong et al. targeted orthotopic neuroblastoma xenografts engineered to express MMP9 with oncolytic HSV, achieving increased virus distribution compared to control tumors [24]. Similar results were obtained when human soft tissue sarcoma HSTS26T overexpressing MMP1 or MMP8 were injected with oncolytic HSV [19]. MMP1/8-expressing tumors contained significantly less sulfated glycosaminoglycans compared to control tumors.
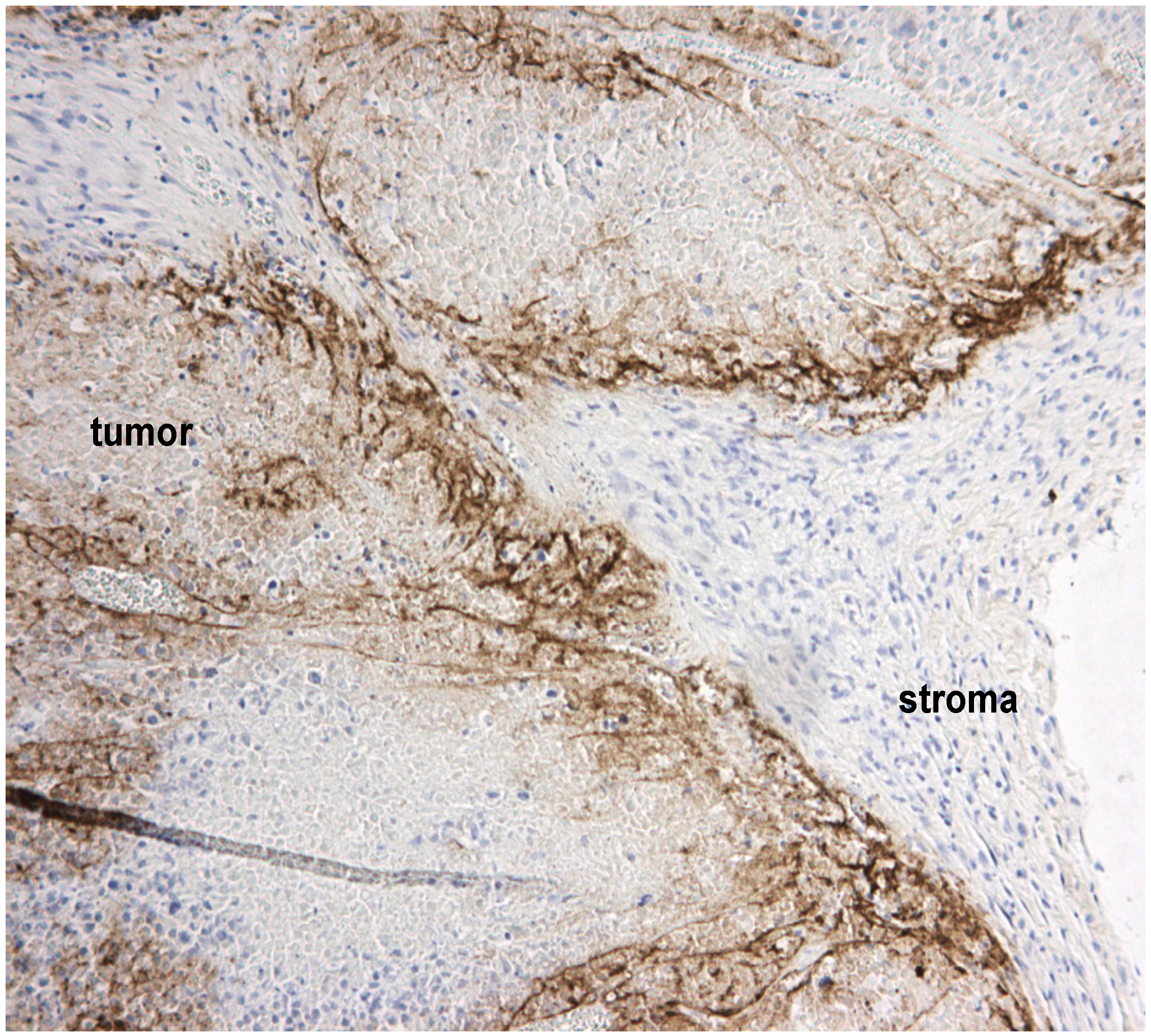
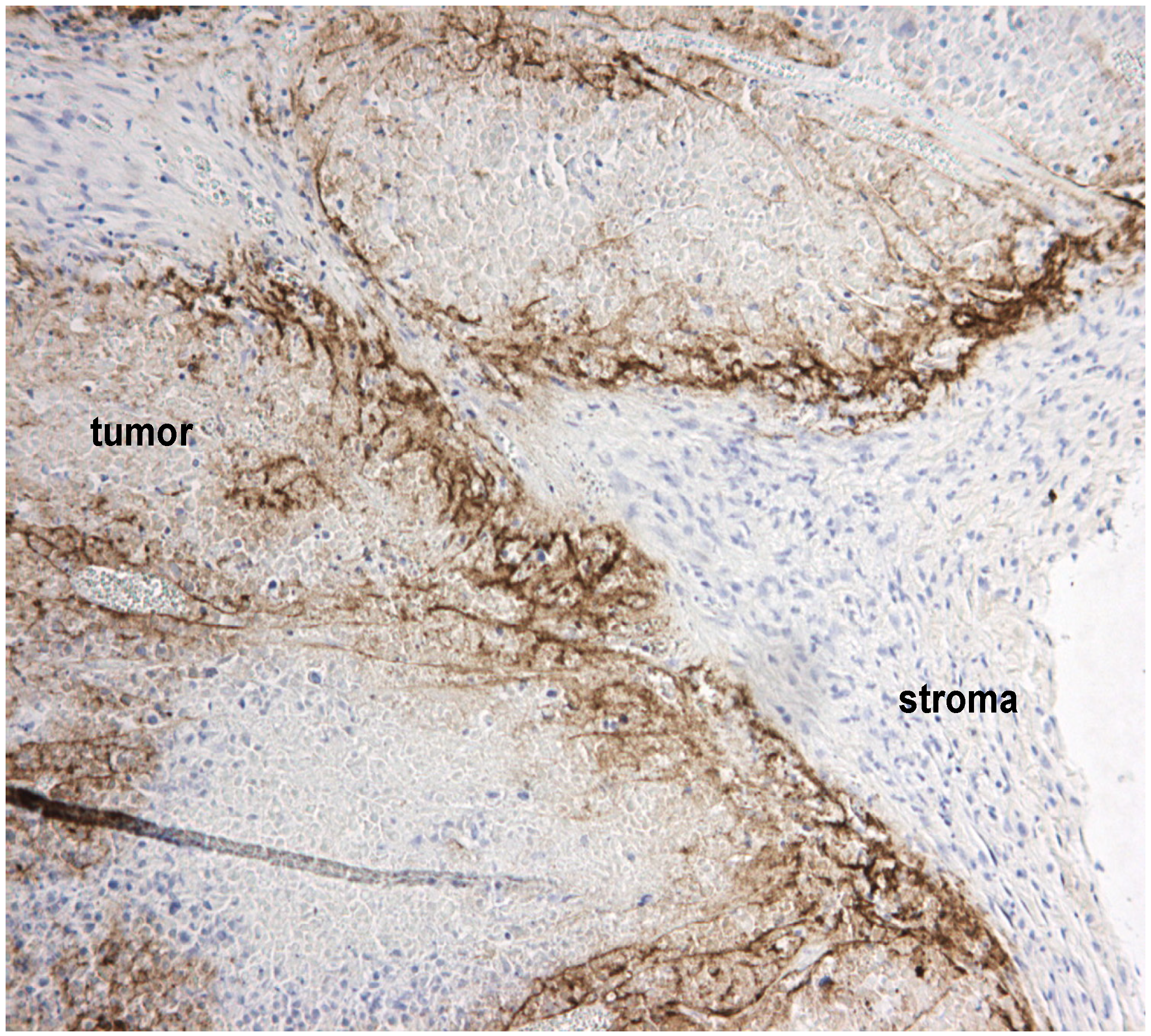
Figure 1.
Tumor stroma blocks virus spread within tumors. A representative section of human A2058 melanoma xenografts stained with polyclonal Semliki Forest virus (SFV) antibodies (in brown) shows that even following intratumoral injection, virus infection is delimited by non-permissive stromal cells and the extracellular matrix (collectively called stroma). We have studied these barriers in detail previously [25].
Figure 1.
Tumor stroma blocks virus spread within tumors. A representative section of human A2058 melanoma xenografts stained with polyclonal Semliki Forest virus (SFV) antibodies (in brown) shows that even following intratumoral injection, virus infection is delimited by non-permissive stromal cells and the extracellular matrix (collectively called stroma). We have studied these barriers in detail previously [25].
Other enzymes and protein effectors, either engineered into virus expression cassettes or provided exogenously, include hyaluronidase, decorin, various other MMPs, and notably relaxin—a peptide hormone normally expressed during particular phase of pregnancy that does not possess tissue-degrading activity itself, but instead induces a number of key collagen-degrading proteases seemingly in a tumor-specific manner [26]. Beyer showed that relaxin expressed by murine stem cells improved trastuzumab penetrance and therapy outcome in syngeneic tumor models [27]. In another study, chimeric adenovirus Ad5/35 expressing relaxin showed increased tumor transduction and virus dissemination [28].
Several other strategies to increase penetration of therapeutics in tumors have been developed [29]. While the stromal/ECM capsule of tumor nodules acts as a physical barrier to virus entry following intravenous injection, it was possible to improve tumor infection by administering vaso-active compounds (angiotensin II, histamine, nitroglycerine) before the virus [21]. This is interesting, as vasculature per se would not be expected to alter or influence the physical composition of the ECM, such as collagen strand thickness. Instead, virus access to the tumor was improved, most likely by increased access from the blood via tumor-stromal-adjoining vessels. Interestingly, in a study with oncolytic herpes virus, anti-VEGF monoclonal antibody Bevacizumab given before intravenous virus injection gave poorer anti-tumor efficacy than when given after the virus [30]. The study showed that while HSV is able to enter and infect tumors better through their leaky vasculature compared to Bevacizumab-normalized vasculature, vascular normalization by Bevacizumab still gave superior combination efficacy when the virus was already in the tumor. This effect was likely dependent on altering both physical and biological properties of the tumor, including interstitial pressure and oxygenation. In another study, anti-VEGF-A antibody injections in nude mice harboring U251 human glioma xenografts led to an increase in MMP2 expression and reduction in collagen fiber content, facilitating improved distribution of oncolytic adenovirus within the tumor tissue [31]. In general, targeting tumor vasculature by oncolytic viruses and including agents that affect tumor vasculature in combination regimens is gaining interest [32], and it will be interesting to see more specific studies on how vascular-acting agents may affect the physical barriers to oncolytic viruses.
Some viruses do not need an extracellular step for propagating infection, such as members of the herpes and poxvirus families, and may be able to spread from cell-to-cell. For example, HSV-1 is able to infect neighboring cells via lateral tight junctions in a manner dependent on its glycoproteins E and I [33]. Vaccinia virus, on the other hand, induces so called actin-tails, which are actin-filament-driven membranous protrusions of the plasma membrane harboring a single virus particle at the outer tip. These actin tails actively deliver and deposit virus particles onto or even into neighboring cells [34,35]. Because lateral spread without an extracellular step may circumvent neutralizing immunity as well as some of the physical obstacles the tumor microenvironment imposes, this ability has been engineered into viruses that normally do not possess it. A promising strategy is to engineer OVs to express membrane-fusogenic genes (MFGs), such as gibbon ape leukemia virus (GALV) glycoprotein, reptilian reovirus p14 protein (FAST) or the membrane glycoproteins H and F of measles virus [36,37]. Quite intriguingly, expression of several different MFGs by oncolytic adenovirus synergized with chemotherapy in anti-tumor efficacy both in vitro and in vivo [38], suggesting that membrane fusion facilitated lateral spread of also chemotherapeutics, which otherwise would not have occurred in a compact tumor. The potential limitation of cell-to-cell-dependent spread, however, may be that membrane-free physical barriers, such as the ECM, may still pose a barrier to spread. Also, it is unclear whether MFGs or other mechanisms of lateral spread may assist in reaching distant tumor nests.
2.3. Tight Junctions Block Virus Penetrance and Hide Virus Receptors
Tumors of epithelial origin mostly retain the firm cellular integrity seen in their original adhesive intercellular configuration. Several viruses use cellular receptors which are located in paracellular tight junctions, which may be problematic in tightly packed tumors [11]. Adenovirus C group viruses use coxsackie-adenovirus receptor (CAR) as their primary entry point, whereas adenovirus B1 group members and measles virus Edmonston use CD46 complement binding molecule as their cellular receptor [39]. Adenovirus B2 group (serotypes 3, 7, 11 and 14) entry occurs using desmoglein-2 (DSG-2) [40]. Of these, CAR and DSG-2 are preferentially located within tight junctions and are hidden from virus binding [41]. Also the complement receptor CD46 was ascribed a role in maintenance of epithelial cell integrity by interactions with the E-cadherin/catenin network [42]. These observations imply that before reaching the potential entry site on the tumor cell the virus must find its way into the junctional space. Moreover, it has become increasingly clear that for example glioma tumors traditionally targeted by Ad5 serotype adenoviruses often express only low levels of CAR and instead much higher levels of CD46 [43,44].
Beyond that tight junctions hide multiple virus receptors, they contain a network of adhesion molecules, including ZO-1, cadherins, claudins and occludin, which, if perturbed, is associated with more aggressive disease in many types of cancer [45,46]. Since tight junctions also contain important receptors that mediate tumor-promoting signaling and which have been targeted by monoclonal antibodies, notably Her2, it would be highly useful to develop strategies that temporarily loosen the tight junction contacts—even at cost of a transient increase in tumor metastatic risk. One of the most interesting approaches has been to exploit the natural propensity of adenovirus serotype 3, which as part of its natural life cycle creates dodecahedral particles (PtDd) consisting of viral capsid proteins, penton base and fiber that open tight junctions by binding to and dissolving desmoglein-2 dimers and reducing E-cadherin expression, to develop a specific tight-junction opening molecule, JO-1 [40,47,48]. Adenovirus type 3 uses PtDds to promote its own infection, opening the tight junctions ahead of infection to maximize access to desmoglein-2. Analogously, when administered to human tumor xenografts, JO-1 facilitated penetration of trastuzumab much deeper into the tumor tissue than when the monoclonal antibody was administered on its own [47]. Moreover, JO-1 synergized with several chemotherapeutics in solid tumor models [48]. Backed by our own findings with oncolytic SFV, showing that both extracellular matrix and tumor cell compactness restrict virus spread and oncolytic efficacy, we believe combination approaches that target both tight junctions and extracellular matrix will prove effective in future virotherapy development (Figure 2).
An alternative approach to increasing tumor penetration by oncolytic viruses is to alter tumor cell morphology and status. Notably, cell death and in particular the type of cell death induced by virus has been shown to affect virus distribution within a tumor mass; in a study by Nagano et al., administration of apoptosis-inducing paclitaxel before injecting oncolytic herpes simplex virus increased virus dissemination in the tumor, allowing its diffuse in “tunnels” created by shrinking/dying tumor cells [49].
Maintenance of physiological adherence is essential for proper ECM function and for retaining cellular integrity. Therefore, a potential caveat of using ECM degrading proteolytic enzymes or tight-junction openers is the risk of neoplastic cell detachment from the tumor ECM and increased risk of metastasis. While ECM-degradation or tight junction opening may operate innocuously, there is also a chance that loss of E-cadherins via proteases or via non-specific deregulation of tight junction integrity during ECM-modulating therapy could trigger pro-tumorigenic Wnt/β-catenin signaling, possibly driving epithelial-to-mesenchymal transition [50]. One study showed that ectopic relaxin expression stimulated MMP expression and enhanced breast cancer invasiveness [51], whereas another paper found short term relaxin exposure to increase breast cancer cell motility whereas long term expression reduced both motility and cancer invasiveness [52]. In the context of oncolytic viruses, Lavilla-Alonso tested several proteases, including hyaluronidase, relaxin, and macrophage metalloelastase, and showed they all could assist adenovirus entry into tumors, yet, the authors did not detect treatment-induced metastases or increase in tumor invasiveness [53]. Another strategy possibly balancing some of the putative risks of tight-junction disruption could be to use Wnt-dependent oncolytic viruses [54].
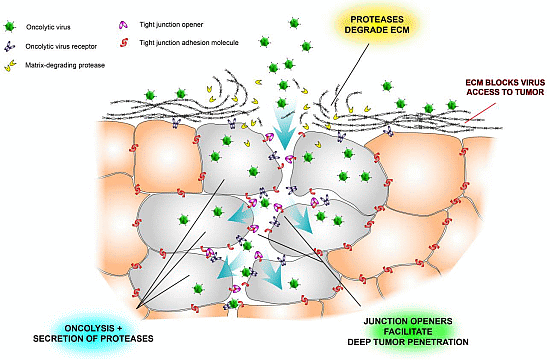
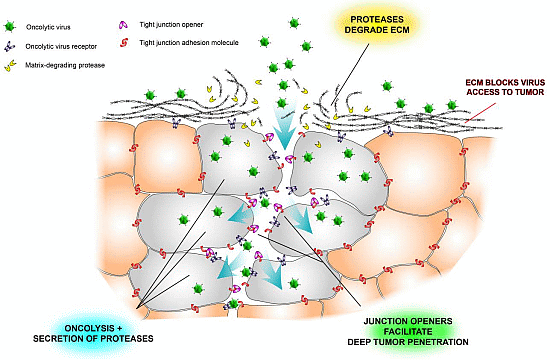
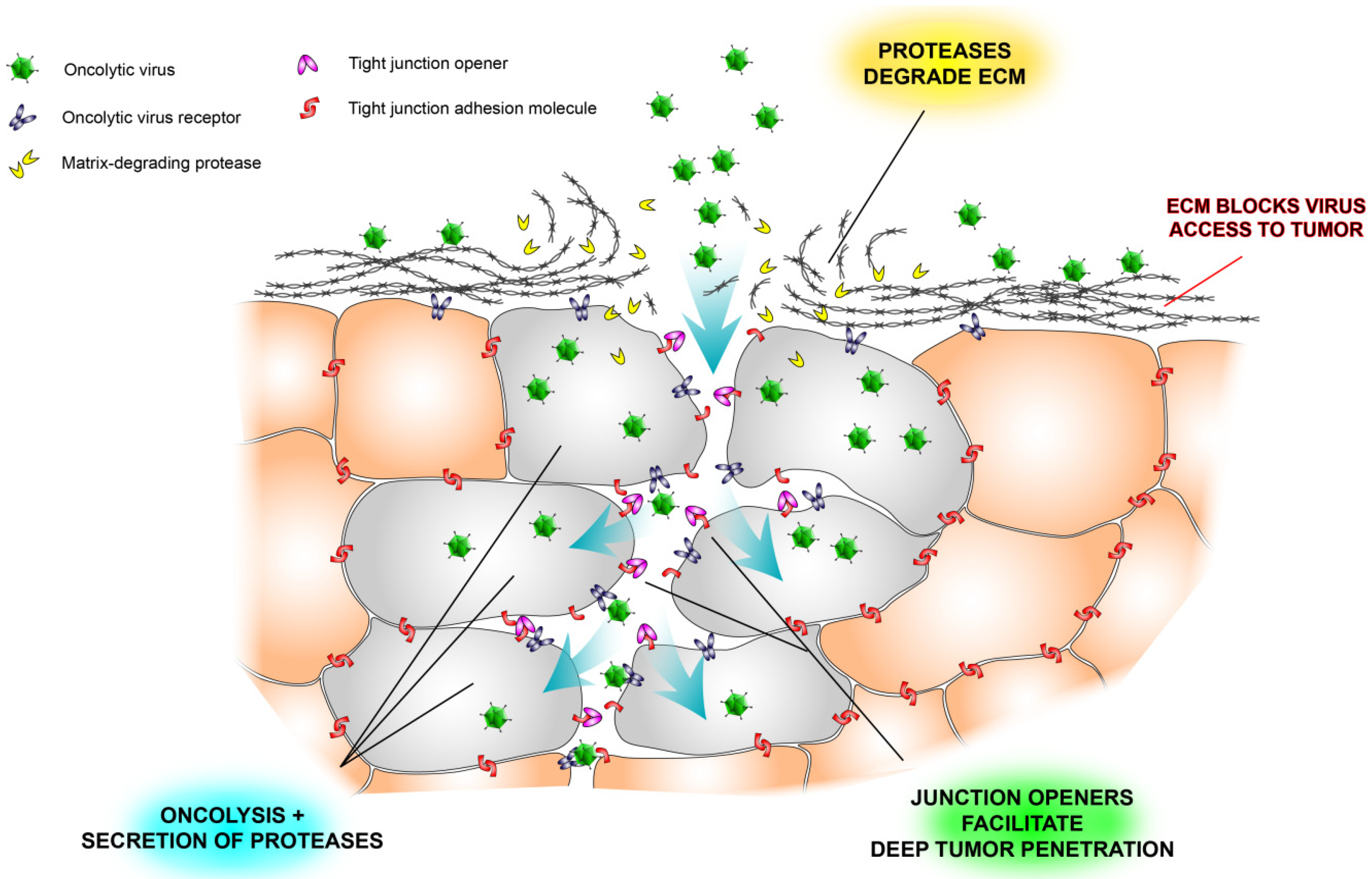
Figure 2.
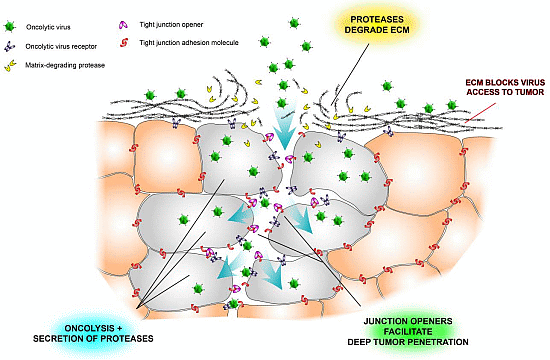
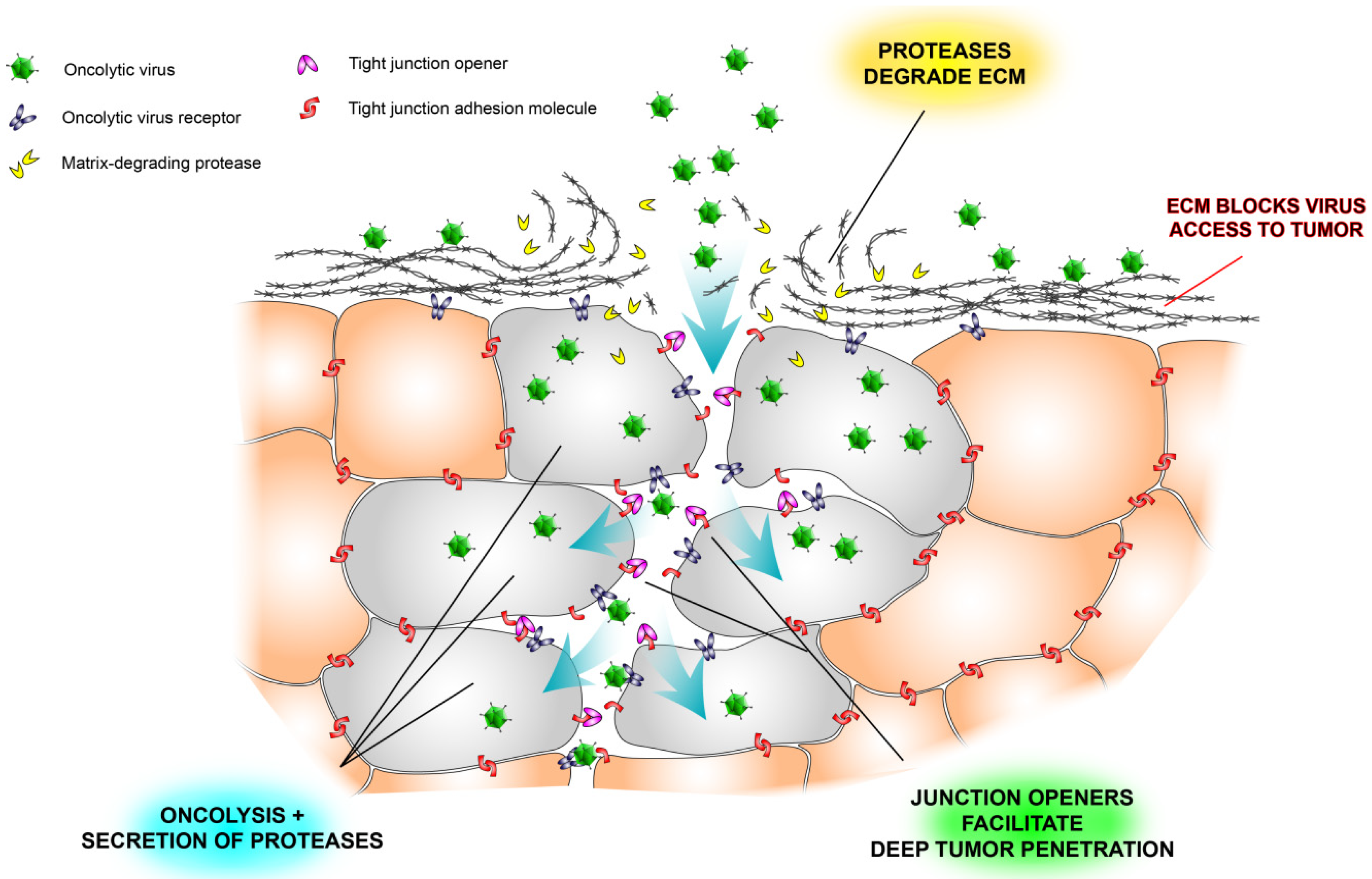
Schematic illustration of some physical barriers to oncolytic viruses and ways to overcome them. Tumor nests are often surrounded by extracellular matrix (ECM), which prevents viruses from reaching the tumor cells (see also Figure 1). Another problem is that tight junctions hide virus receptors and limit diffusion of viruses into the tumor tissue. Newly developed tight junction openers [40,47,48] may facilitate virus infection by exposing the hidden receptors, and virus-encoded proteases may degrade the stromal shield surrounding tumor nests [26].
Figure 2.
Schematic illustration of some physical barriers to oncolytic viruses and ways to overcome them. Tumor nests are often surrounded by extracellular matrix (ECM), which prevents viruses from reaching the tumor cells (see also Figure 1). Another problem is that tight junctions hide virus receptors and limit diffusion of viruses into the tumor tissue. Newly developed tight junction openers [40,47,48] may facilitate virus infection by exposing the hidden receptors, and virus-encoded proteases may degrade the stromal shield surrounding tumor nests [26].
2.4. Stromal Cells Hinder Viruses
Tumors are invariably fenestrated by host fibroblasts, myeloid cells and other non-transformed cells, which driven by cancer-induced cues can adopt various reprogrammed phenotypes to promote tumor angiogenesis and growth and to alter tumor responses to therapies [1,55]. As most of them normally play a role in immune homeostasis and pathogen sensing, they also respond to and influence oncolytic virus infection of tumors.
Fulci et al. reported that injection of oncolytic HSV-1 directly into glioblastoma tumor parenchyma triggered upregulation of CD68 and CD163 monocytic markers and rapid clearance of virus, likely executed by infiltrating phagocytic cells [56]. When clodronate liposomes were used to deplete macrophages in vivo, the authors observed a 5-fold increase in virus titers in the brain tumors concomitant with an 80% reduction in peripheral CD163+ macrophages in animal spleens, suggesting that CD163+ cells migrate into the tumors upon virus injection and limit overall oncolytic efficacy. While CD68+ cells were not reduced by peripheral macrophage depletion, arguing that these cells had been recruited to the tumors before the treatment, they could be eliminated in live glioma slices ex vivo, resulting in a 10-fold increase in virus replication. Macrophages may secrete antiviral type I interferon constitutively at very low (subnanomolar) levels [57] and recently Liu et al. showed that tumor-resident CD68+ macrophages induced a protective antiviral state in ovarian and breast tumors which led to resistance to oncolytic vesicular stomatitis virus (VSV) infection [58]. Type I IFN signaling (JAK1/2) inhibitor Ruxolitinib reversed the resistance, facilitating VSV replication in the macrophage-protected cancer cells. It is tempting to speculate that tumor-resident macrophages also rendered syngeneic GL261 and DBT mouse gliomas completely refractory to VA7 oncolytic alphavirus, as these cells were quite infectable in vitro and live glioma slices regained their sensitivity to virus within 24 h, indicating dilution of IFN-I [59,60].
Macrophages or the closely related myeloid suppressor cells are the predominant stromal cell type in many tumors and are actively recruited during cancer progression [61,62]. Heavy macrophage presence in the tumors indicates that such tumors may be resistant to oncolytic virotherapy, unless depleting preconditioning would be used. Moreover, as part of the early immune response to virus infection, monocytic cells are rapidly recruited to tumors following intratumoral virus injection. In one study, depletion of either CD11b+ cells or VEGF secreted by these cells in mice treated with oncolytic HSV-1 greatly increased oncolytic efficacy, tentatively linking pro-tumorigenic and antiviral angiogenic mechanisms [63].
Another key pro-tumorigenic signaling employed by many tumors is up-regulation of CXCR4, the receptor for chemokine CXCL12 (also known as SDF-1), and recruitment of stromal cells expressing CXCL12 which maintain the tumor and recruit additional cells to support tumor expansion and invasion, such as endothelial cell progenitors which contribute to neovascularization [64]. Indeed, high CXCR4 expression is a negative prognostic factor for many cancers. Oncolytic vaccinia virus engineered to express a soluble antagonist of CXCR4 showed improved intratumoral virus replication, increased vascular disruption and a markedly reduced infiltration of the tumors by putative immune-suppressive and tumor promoting stromal cells [65]. This study highlights how reducing pro-tumor stromal cell effects may alleviate antiviral resistance and yield increased overall therapeutic efficacy. On the other hand, if the paracrine antiviral effects of tumor-homing monocytic cells could be selectively abrogated these cells could favorably be used to ferry oncolytic viruses into tumors. Indeed, a significant delay in metastatic tumor potential was achieved when macrophages were harnessed to deliver a tumor-specific oncolytic adenovirus [66].
3. Tumor Cellular Defenses against Viruses
Improved oncolytic efficacy by means of increased tumor transduction is dependent on getting the viruses into the tumors and then on how many tumor cells are infected. Additionally, host immune status and the kinetics of the ensuing antiviral immune response are critical determinants of therapeutic efficacy with OVs. In order to improve virus infection of tumor cells and/or to limit infection of normal off-target cells, viruses may themselves be modified for increased receptor binding or uptake. Such strategies, summarized in greater detail elsewhere [67], include incorporation of receptor-binding motifs on the virion spikes, diverting infection to desired receptor-expressing (cancer) cells. Another strategy is to ferry the viruses in on the surface of or inside various secondary carrier entities, such as stem- or immune cells, which may enter the tumor from the circulation while paradoxically shielding the viruses from immune detection or antibody neutralization [68]. While these strategies ultimately increase the amount of virus that reaches the tumor bed, they are unlikely to alter tumor intracellular permissiveness to virus, which is still a prerequisite for oncolysis and likely primary determinant of virus-induced anti-tumor immune responses. In other words, tumor cells must allow viruses to replicate, otherwise there is no oncolysis and no therapy.
Central to cancer resistance to oncolytic viruses is the capacity to mount antiviral defense, and the quality of that defense. All human viruses have co-evolved with their hosts to achieve productive co-existence, and while innate antiviral defenses protect normal tissue from excessive virus replication, with the signaling and mechanisms described in great detail elsewhere [69,70], in tumors such defenses are an undesirable element that may restrict therapeutic efficacy. Propagating the defense are soluble cytokines, with type I (alpha and beta species) interferons playing an essential role against most viruses and type II (gamma) interferon providing a non-redundant auxiliary protective role in controlling pathogenesis of certain viruses. Mice knocked out for the type I IFN receptor (IFNAR) typically succumb within a day of multi-organ infection when challenged with viruses that may not even be pathogenic in normal adult hosts [71,72]. There have been no reported cases of genetic defects in the type I IFN receptor in humans, but three unrelated cases of complete signal transducer and activator of T cells (STAT) 1, the essential signaling mediator of type I IFNs, deficiency in humans have been recorded to date, all of which were lethal due to multi-organ virus infection [73,74]. While genetic defects in either type I or type II IFN genes are extremely rare in humans, genetic mutation of the IFN gamma receptor has been documented on some occasions, with the patients displaying high sensitivity to mycobacterial infections [75].
Some interferon-like proteins, such as limitin [76], and many unrelated and structurally diverse “danger”-associated endogenous molecules, including HMGB1 and heat-shock proteins, collectively called alarmins [77], likely signal via the type I IFN receptor or induce its expression, and therefore, in the coming chapters we consider tumor defense against viruses as an equation of the degree of type I IFN responsiveness. For tumors to be sufficiently infected by OVs to reach “reasonable” efficacy, some defects in tumor antiviral defenses are a prerequisite. Nevertheless, oncolytic viruses exert their efficacy not only by destructive replication in tumor cells but also by stimulating anti-tumor immune responses, and therefore overall efficacy of oncolytic viruses may be difficult to gauge based solely on capacity to replicate in cancer cells.
3.2. Oncolytic Virus Restriction by Innate Defenses
All oncolytic viruses tested to date display variable infectivity in different cancer cell lines. This variability is likely at least partly dependent on type I IFN as most oncolytic viruses are IFN-sensitive. IFN-secretion follows virus sensing by the cells via Toll-like receptors expressed on both the plasma membrane and in endocytotic vesicles, NOD-like receptors, STING, AIM2, NLP inflammosome and other danger sensing molecules, summarized elsewhere [93,94]. While all viruses carry an arsenal of counteracting molecules [69,95], no virus goes completely undetected. Even HSV amplicons, which are devoid of functional virus genes, induce a type I IFN response that mediates a STAT-1-dependent system-wide antiviral defense in mice within one hour of intravenous vector injection, resulting in significant reduction of transgene expression compared to expression in STAT-1 knockout animals [96].
During our own studies with attenuated SFV vector VA7 it became apparent that the dramatic and lasting therapeutic efficacy achieved following mere single intravenous (or intratumoral) injection of the virus in immunocompromised mice bearing human A2058 melanoma or U87 glioma xenografts [25,97] was not going to be recapitulated easily in immunocompetent animals [59,60,98]. The paradox in our studies was that while oncolytic SFV vector VA7 effectively replicated in and destroyed a variety of IFN-responsive cancer cell lines in vitro, it consistently failed to eradicate tumors generated from the same cell types in vivo, even if large doses of virus were injected directly into the tumor mass and even if the tumors were void of visible physical barriers (Figure 3). Conversely, only tumors generated from IFN-unresponsive cancer cells have seemed infectable in vivo by VA7 so far. In light of earlier findings by others, showing that type I IFN receptor knockout mice are highly susceptible to SFV infection and quickly succumb to multi-organ systemic infection [71], the parameters for oncolytic SFV efficacy seem clear: tumor cells must be defective in type I IFN response for the virus to be effective.
These findings are in line with emerging data from other groups, showing a remarkably strict dependence of oncolytic virus replication on defective type I IFN responsiveness, a dogma introduced at the turn of the 21st century [99]. For example, sarcomas and melanomas display differing permissiveness to oncolytic VSV, an obstacle which may be overcome by blocking type I IFN signaling [100,101]. In eight sarcoma cell lines, basal up-regulation of RIG-I and IFIT1 and rapid induction of STAT1 phosphorylation upon IFN I treatment correlated with resistance to oncolytic measles virus [102]. In other studies, human pancreatic and ovarian cancer cells display resistance against Rb-dependent oncolytic adenovirus, strongly correlating with intracellular levels of MxA, and acquired resistance to repeated oncolytic adenovirus injections in an intraperitoneal ovarian carcinoma model was associated with an increase in MxA as well as several other key ISGs in the tumors [103,104]. A study comparing normal human or mouse melanocytes to a panel of melanoma cells revealed strong correlation between permissiveness to oncolytic VSV and capacity to mount antiviral defense in response to type I IFN [105]. In this study, VSV-rp30, harboring two mutations in P and L genes, was superior to several other tested oncolytic VSV strains, but the dependence on defective IFN responsiveness was still retained. In yet another study, oncolytic VSV replicated in four out of twelve mesothelioma cell lines which were unable to mount antiviral defense upon IFN-beta pretreatment or to respond to infection by up-regulation of PKR, MxA or 2'5'-OAS mRNA [106]. In contrast, the non-permissive mesothelioma cells mounted antiviral defense, associated with PKR, MxA or 2'5'-OAS mRNA up-regulation, in response to infection or to exogenous type I IFN. The authors of this study further linked the observed pattern of virus resistance to clinical mesothelioma samples, where in a mesothelioma tissue array, significant variance in immunoreactivity against PKR-, p48- and/or IFNAR was observed, with only a few samples displaying lack of reactivity against all three components, arguing that most tumors in clinical settings would display at least some level of resistance to oncolytic VSV.
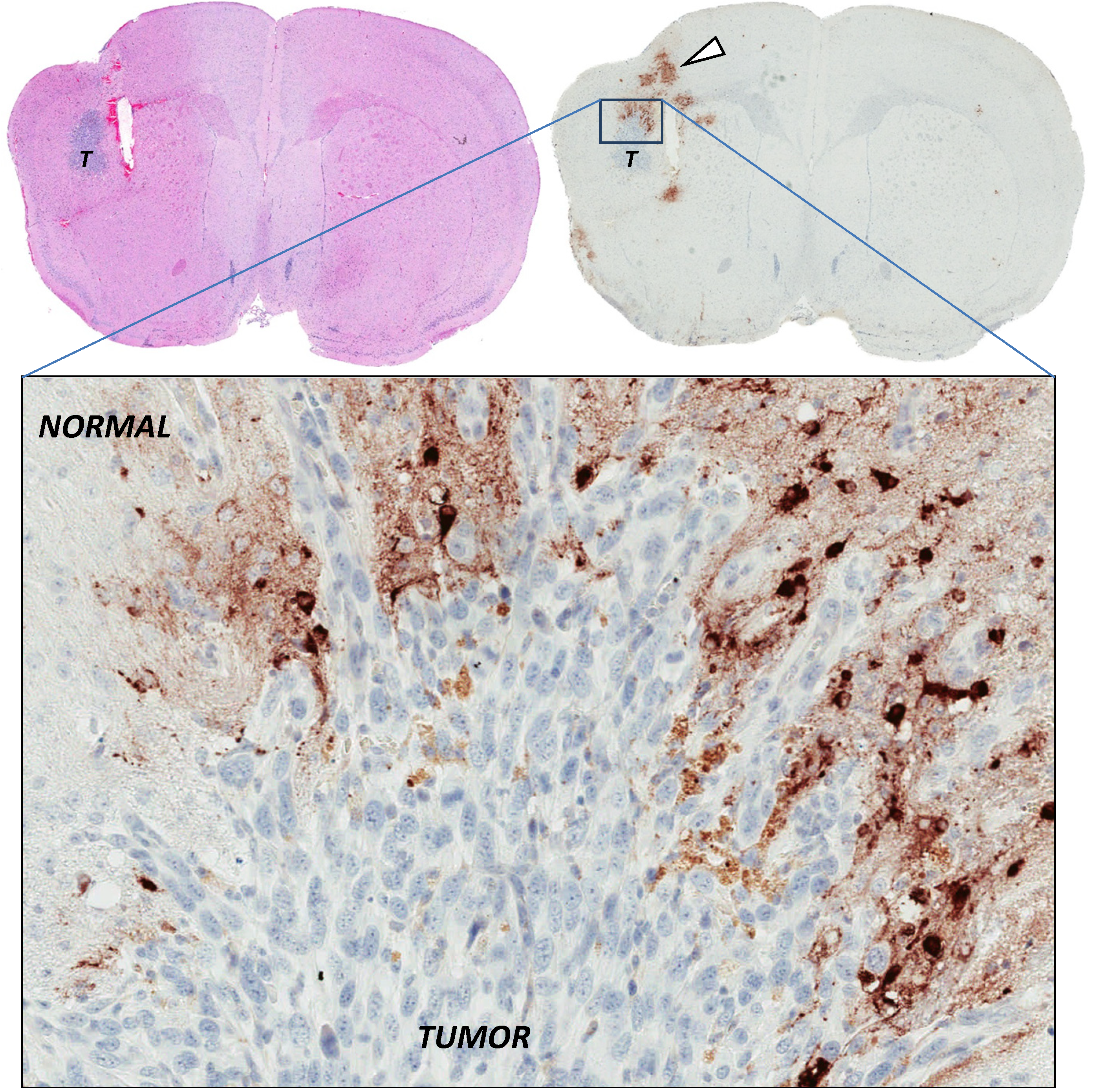
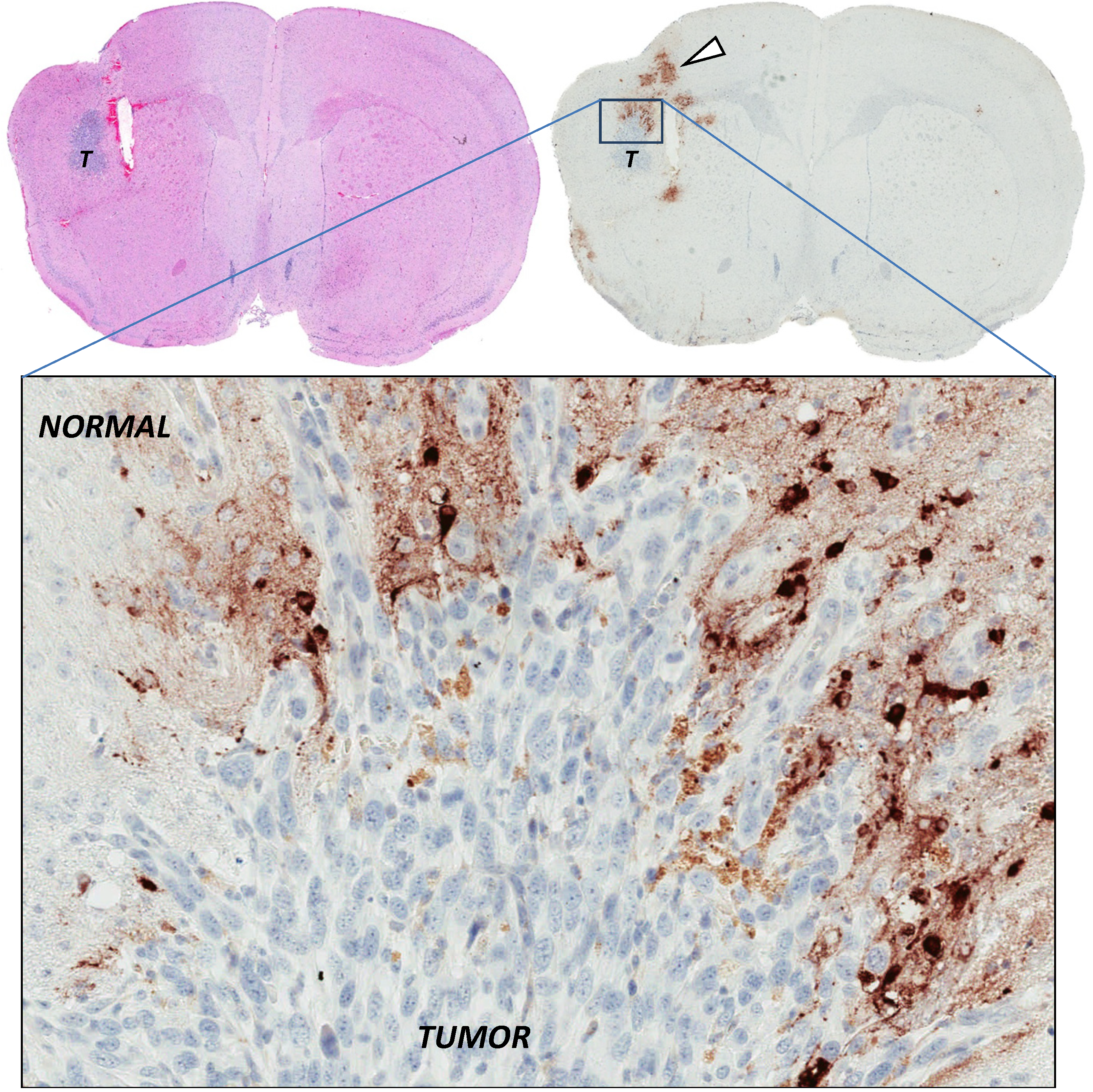
Figure 3.
Restricted infection by oncolytic virus in the absence of physical barriers. Intracranial syngeneic Balb/c mouse DBT gliomas (T) were injected into the same stereotactic coordinates with high-dose oncolytic Semliki Forest virus (SFV) vector VA7. Brains were sampled and stained for SFV antigens 24 h post virus injection (in brown), showing that SFV predominantly infects normal brain parenchymal cells rather than glioma cells. DBT tumors are homogeneous and void of thick extracellular matrix deposits, as seen in Figure 1, arguing that tumor cells resist virus infection by other intracellular means [60].
Figure 3.
Restricted infection by oncolytic virus in the absence of physical barriers. Intracranial syngeneic Balb/c mouse DBT gliomas (T) were injected into the same stereotactic coordinates with high-dose oncolytic Semliki Forest virus (SFV) vector VA7. Brains were sampled and stained for SFV antigens 24 h post virus injection (in brown), showing that SFV predominantly infects normal brain parenchymal cells rather than glioma cells. DBT tumors are homogeneous and void of thick extracellular matrix deposits, as seen in Figure 1, arguing that tumor cells resist virus infection by other intracellular means [60].
Oncolytic adenoviruses are often deleted for E1A, which normally binds Rb and prevents it from sequestering E2F, which results in the viruses being unable to replicate in normal cells. However, since adenovirus, as with all viruses, contains elements that antagonize antiviral defenses, such elements have also been targets for deletion to render the virus sensitive to innate antiviral defenses (which would be expected to remain intact only in normal cells). Cancer cells expressing Ras may down-regulate protein kinase R and inhibit type I IFN induction by interfering with RIG-I signaling [107,108]. Also, select interferon stimulated genes appear to be repressed by the Ras/MEK/ERK signaling cascade [109]. PKR activation shuts down E2F-dependent translation, including translation of virus messenger RNAs. Therefore, an adenovirus deleted for VA RNA, which antagonizes PKR, should be capable of replicating in Ras-overexpressing (tumor) cells. However, while this was seemingly the case [110], it was later discovered that Ras is not an obligatory determinant for PKR-inactivation and that virus replication was halted even in Ras overexpressing cells via functional PKR-mediated E2F phosphorylation, implying that the parameters of VA RNA-deleted adenovirus replication had to be revised [111]. Similar findings were obtained with oncolytic reovirus, against which Ras-independent tumor resistance emerged in vivo [112]. Thus, under some circumstances even tumor cells over-expressing Ras may respond to type I IFN or to stimulants of pattern recognition receptors, activating PKR and inhibiting virus translation. To complicate matters, MEK-inhibitors have been shown restore antiviral signaling capacity in Ras overexpressing tumors [108,109]. This implies that human tumors treated with MEK-inhibitors, such as Trametinib which was recently approved for treatment of BRAF-mutated melanoma, may be poor targets for oncolytic viruses as antiviral signaling capacity in such tumors may be restored.
Some oncolytic viruses, such as members of the poxvirus family, are seemingly resistant to the antiviral effects of type I IFNs in vitro and may be controlled by other cellular factors [113,114,115,116]. However, in most cases, sensitivity or resistance to type I IFN is assessed in vitro and using only one cytokine, which may mask synergies with other factors present in vivo. One possible explanation for the in vitro–in vivo discrepancy is the tumor micro-environment, which not only imposes physical barriers to oncolytic viruses but also alters intracellular antiviral defenses. For example, glioma cells were found in the brain to spontaneously secrete type I IFN, which conferred resistance against oncolytic HSV. Resistance was linked to ECM protein CCN1 interaction with glioma cell surface receptor alpha 6 beta 1 integrin, which activated interferon secretion [117]. It is tempting to speculate whether this or a similar ECM-glioma-interaction could have contributed to the resistance of syngeneic glioma cells we observed in vivo but not in vitro to both oncolytic SFV and vaccinia virus (FIG2; [60]). Also, while critical in many respects, antiviral signaling may occur without signaling via type I IFN receptor (IFNAR). For instance, upon infection of IFNAR KO mice with murine hepatitis virus (MHV), brain cells were found to have upregulated antiviral GTPase TGTP and IFITm1 and IFITm3, all of which have confirmed antiviral activity in vivo against a variety of viruses [118]. In these cases, IFN gamma and TNF alpha likely account for at least part of the observed ISG activity, where e.g., TGTP is upregulated by IFN-γ signaling and TNFα has been shown to induce ISG15 independently of IFNAR [119].
Interestingly, while most cancers are heterogeneous in antiviral defence signaling, one broad class of tumors in particular may display consistently low capacity to ward off oncolytic viruses; oncovirus-induced cancers. This is because oncoviruses, as all viruses, carry a complement of factors that abrogate antiviral sensors and effectors, which may result in tumors that are uniformly devoid of antiviral signaling. In an intriguing study, oncolytic VSV infected and destroyed human papilloma virus (HPV)-positive cervical carcinoma cells more effectively than HPV-negative head- and neck cancer cells due to HPV E6-mediated suppression of antiviral signaling [120]. HPV-positive or E6-expressing xenografts were efficiently eradicated from nude mice following VSV injection. The potential of this approach is that even oncolytic viruses that are extremely sensitive to type I IFN, such as attenuated SFV or M-mutated VSV, may function well against oncovirus-induced cancers where the oncovirus machinery ensures the lack of antiviral defense.
3.3. Exogenous Combination Therapy to Overcome Innate Defenses
While on first thought it would appear counterintuitive to antagonize the very factors that keep oncolytic viruses tumor-specific, emerging data shows that this can be accomplished in a safe way. One of the foremost strategies to increase oncolytic virus efficacy is to combine them with drugs that lower tumor antiviral defenses. In this regard, a primary target for interference is the prototypical type I IFN signaling cascade. While small molecular inhibitors against IFNAR are not available, activity of downstream signaling transducers may effectively be blocked. Human pancreatic cancer cells were shown to constitutively express high levels of MxA and OAS and to mount type I IFN-dependent resistance to oncolytic VSV. Resistance could be overcome by blocking activity of IFNAR-associated Janus kinase (JAK) 1 [100,113]. Similarly, JAK1/2 inhibitor ruxolitinib overcame type I IFN-dependent resistance of human SCC25 head and neck cancer cells against oncolytic VSV [121]. Ruxolitinib also successfully abrogated type I IFN-mediated antiviral effects elicited by macrophages [58], highlighting that this drug might also alleviate type I IFN-dependent tumor resistance to oncolytic viruses imposed by stromal cells. However, the obvious downside to using JAK1-inhibitors is their effects on critical host defense mechanisms, which may result in increased infection of normal cells. Moreover, JAK1-inhibitors may reduce anti-tumor immune responses elicited by oncolytic virotherapy. For example, ruxolitinib was shown to impair the capacity of dendritic cells promote CD8 T cell responses against model tumor antigens [122]. On the other hand, in this study clearance of adenovirus was delayed in ruxolitinib-treated normal animals, which makes it difficult to predict the overall impact of JAK1-inhibitors on tumor therapy where prolonged virus presence might translate to better therapeutic efficacy. Also, while the immune-stimulating and anti-tumor effects of type I and type II interferons may be adversely affected by JAK-inhibitors, central tumor-promoting cascades mediated by JAKs, such as IL-6-STAT3, may also be inhibited [123], yielding a difficult-to-predict net therapeutic outcome. At the moment, more studies are needed to establish the overall impact of interference with JAK1/2 signaling in cancer therapy.
Spurred by earlier reports of oncovirus reactivation and promoter enhancement by histone deacetylase (HDAC) inhibitors, this class of agents was assessed for capacity to enhance oncolytic virus potency by inhibiting antiviral responses. Indeed, such enhancement was observed with oncolytic VSV and SFV, and while it has been difficult to pinpoint the exact mechanisms that underlie HDAC-inhibitor enhancement of oncolytic virus replication in cancer cells, antiviral defenses in general are inhibited—for yet unknown reasons mainly in cancer cells and not in normal cells, which indirectly facilitates virus replication [124]. Interestingly, combination of HDAC inhibitors with conditionally replicating Δ24 E1A-deleted adenovirus gave improved virus replication rate and therapeutic efficacy in subcutaneous xenograft models only when the inhibitors were given before the virus, otherwise inhibition of replication was observed [125]. Similarly, HDAC inhibitors increased replication and oncolytic efficacy of HSV when given before the virus but not when given at the same time [126,127]. With nuclear DNA viruses, such as adenovirus and HSV, it is possible that HDAC inhibitors also alter virus genome accessibility to transcription factors, resulting in reduced replication or other adverse effects, implying that with these viruses HDAC inhibitors should be administered before the viruses.
Recently, it was found that HDAC inhibitor vorinostat activates NfκB signaling by facilitating hyperacetylation of the p65 subunit, which in turn activates cellular autophagy [128]. Autophagy was in another study shown to be important for VSV replication, presumably by inhibiting antiviral signaling. This is interesting, as a previous study revealed enhancement of VSV replication by inhibiting TNF-alpha-induced antiviral signaling mediated by p65 using NfκB inhibitors BMS-345541 or TPCA-1 [129]. In this study, neither inhibitor had an effect on STAT1 or STAT2 phosphorylation or their nuclear relocalization in response to type I IFN but the authors observed clear induction of ISG15, GBP1 and MX1 in response to type I IFN in U87 glioma cells, with the latter two being dependent on NfκB signaling. Taken together, NfκB pathway activation may result in different antiviral responses in different cancer types, and inhibition or enhancement of oncolytic viruses by NfκB inhibitors is dependent on other concomitant cellular mechanisms, such as autophagy.
Since HDAC inhibitors modulate promoter activity/accessibility, it is conceivable other epigenetic or promoter modifying agents could work in a similar manner to repress antiviral defense induction and enhance oncolytic viruses [130]. DNA demethylating agents 5-azacytidine (5-Aza) and decitabine synergized with oncolytic herpes simplex type 1 virus in glioma models in vitro and in vivo, as did HDAC inhibitor valproic acid (VPA), which was already shown before to work well with oncolytic HSV as well as several other oncolytic viruses [131]. Moreover, inhibition of cancer antiviral defenses, specifically RNAseL and PKR, resulted when cancer cells were exposed to sunitinib, a multi-tyrosine kinase inhibitor originally developed as an inhibitor of VEGF-R and PDGF-R signaling, yielding strong anti-tumor synergy with IFN-sensitive oncolytic VSV [132]. Since sunitinib is approved for renal cell carcinoma and imatinib-resistant gastric cancers, combination with oncolytic VSV constitutes a promising and clinically relevant approach warranting further investigation. Interstingly, sunitinib as well as several other receptor tyrosine kinase inhibitors displayed antiviral effects against polyomavirus BK [133], arguing that these compounds are not universal virus enhancers.
Cyclophosphamide (CPA) is a general immunosuppressant used to minimize Treg presence during immunotherapy and also oncolytic virotherapy. CPA may, however, also enhance replication and efficacy of oncolytic viruses by diminishing cytokine secretion by stromal cells, including type I IFN [134]. Another compound enhancing oncolytic virus efficacy by lowering cell responsiveness to type I IFN is triptolide, which acts downstream of IRF-3 activation [135]. Further, the kinase mammalian target of rapamycin (mTOR) enhances the antiviral effects of type I IFN by activation of its effector proteins 4E-BP, which binds and inactivates translation factor eIF4E, and S6K, which carries out cell-type specific signaling functions of type I IFN, such as activation of eukaryotic translation initiation factor 4B and promotion of ISG15 transcription. Consequently, rapamycin was able to enhance oncolytic VSV replication and anti-tumor efficacy by inhibiting type I IFN-mediated antiviral signaling via mTOR [136].
Several other compounds synergizing with oncolytic viruses have been identified via high-throughput drug library screening, many of which seem to act by antagonizing antiviral defenses. For instance, a novel virus sensitizer, Vse1, was found to greatly diminish antiviral effects of type I IFN against oncolytic VSV in several cancer cell types, and the drug also synergized with the virus in subcutaneous mouse tumor models [137]. For many of these new compounds, the mechanism of action is still poorly understood, necessitating extensive safety studies to exclude undesirable effects against normal cells.
Apart from classical type I IFN-induced antiviral defenses, which typically prevent virus translation and degrade virus genomes, other cellular machineries regulate oncolytic virus efficacy. Notably, strategies to increase cancer cell death in response to oncolytic viruses have been tested. In a recent study, a class of compounds called SMAC mimetics synergized in several cancer models with oncolytic VSV by removing cancer cell block to apoptosis in response to virus-induced type I IFN, TNF-α or TRAIL [138]. Interestingly, since virus infection-triggered cytokines act on nearby non-infected cells, SMAC mimetics may in combination with oncolytic viruses also cause significant bystander tumor-destruction. Normal cells are not affected by the IFN-SMAC mimetic synergy, providing an important safety aspect. In another study, blocking the ER stress response circuitry triggered by oncolytic rhabdovirus infection by a small molecular inhibitor of serine/threonine protein kinase and endoribonuclease IRE1α, which was identified in a genome-wide siRNA screen, greatly enhanced cytotoxicity via caspase-2-induced apoptosis and increased oncolytic efficacy in refractory tumor models in mice [139]. This effect was independent on induction or responsiveness to type I IFN. Conversely, inhibitors of nucleoside transporter-1 (ENT1) were discovered from a high-throughput screen of enhancers of oncolytic HSV, and to date such inhibitors have not been reported to alter cellular antiviral responses [140]. Other compounds, such as common chemotherapeutics, and their mechanisms synergizing with oncolytic viruses have been discussed elsewhere—for most of them the possible role of antiviral defense antagonism in enhancing oncolytic virus efficacy remains to be studied [141].
Thus, many compounds are available to interfere with tumor antiviral defenses. The outstanding question for most of them, however, seems to be why such compounds do not render normal cells sensitive to oncolytic viruses. Elucidation of the exact mechanisms of action and the differences between normal and cancer cells constitute a worthy goal for studies in the near future.
3.4. Virus Engineering and Combination to Overcome Innate Defenses
Already in the 1950s it became clear that using certain pathogenic wildtype viruses, such as West Nile virus and Bunyamwera virus, in cancer patients would result in off-target toxicity [142,143]. Characteristic for pathogenic viruses is a greater ability to circumvent or antagonize cellular innate antiviral defenses than attenuated strains. For example, unlike the prototypical oncolytic reovirus strain type 3 Dearing, the T1L strain of reovirus causes accumulation of IRF9 in the nucleus and inhibits activation of a select group of ISGs—yet because of this property the T1L strain is myocarditic unlike the oncolytic type 3 Dearing strain [78]. For these reasons, many oncolytic viruses used today, such as reovirus type 3 Dearing, oncolytic strains of Newcastle disease virus and M-mutated VSV have either a stronger capacity to induce or a weaker capacity to resist antiviral type I IFN than the corresponding wildtype strains [78,144,145].
An open question, however, is whether strain-specific or virus-specific differences that relate to specific components of the cellular antiviral machinery may be exploited for greater anti-tumor efficacy, i.e., if one virus fails, could another be used in its stead? While Ras expression was critical for oncolytic reovirus replication in cells overexpressing CUG2 oncogene, oncolytic vesicular stomatitis virus was unable to replicate in the same cells due to increased activation of STAT1 and OASL2 [146,147]. Thus, in these cells reovirus was not affected by STAT1 or OASL2, and it therefore remains interesting to see which other antiviral effectors may have been lacking in the CUG2-expressing cells and, conversely, how reovirus and VSV differ in terms of sensitivity to antiviral effectors. While several oncolytic viruses have been compared to each other in terms of replication rate and cytotoxicity, systematic studies with regard to tumor antiviral defenses have not been conducted.
As any attenuated virus is likely to replicate effectively only in its own niche (i.e., to be restricted by a specific set of ISGs), and as wild type strains may be too toxic for use in humans, a compromise may be achieved by incorporating select elements from wild type viruses into oncolytic virus backbones. Indeed, several chimeric and recombinant viruses have been generated that harbor specific elements of wild type strains or even of other unrelated viruses in order to better resist cancer antiviral defenses (Table 1). As an example of introducing wild type elements into attenuated viruses, Haralambieva reported on a measles virus Edmonston vaccine strain in which P gene was replaced with the counterpart from the wild type IC-B strain [148]. Later, Meng et al. included N, P, and L genes from wild type measles in Edmonston backbone, producing an enhanced but still safe oncolytic virus [149]. Edmonston vaccine virus induces a more robust IFN-response compared to wild type measles virus partly due to the weaker ability of V protein to suppress MDA5-mediated activation of IRF-3 and IFN-I induction [150,151]. In vivo, P-gene encoded proteins, particularly V, control the quality of the cytokine response to measles so that vaccine strains qualitatively induce a stronger inflammatory response compared to WT strains (where WT V protein suppresses multiple cytokine secretion). V protein short cytoplasmic tail has been shown to bind and inactivate STAT2, IRF7, MDA5 and the Rel homology domain of the NfκB subunit p65 but not of p50 [152]. Also, V causes LGP2 protein to bind and inactivate RIG-I [153], but it seemingly does not interfere with TICAM-1 (TRIF, multiple TLR adaptor, including TLR3)-mediated IFN-β induction. Thus, if cancer and normal cells would differ with regard to TICAM-1 or IRF-3, then such a virus could potentially target specifically cancer cells rather than normal cells. So far, the measles Edmonston-WT chimeras have proven safe in animals.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Chimeric and recombinant IFN-I-antagonistic viruses. Example chimeric and recombinant viruses with oncolytic potential designed to overcome antiviral defenses. Not all constructs have been tested as oncolytics. Also, note that the nomenclature of vaccinia virus soluble type I IFN scavenger is ambiguous in the literature; “B18R” and “B19R” are often used interchangeably. Here we use B19R to indicate the type I IFN scavenger, which is the official term for the molecule from the Western Reserve strain.
| Target virus | Target virus modifications | Donor virus | Donor gene(s) | Description | Reference |
|---|---|---|---|---|---|
| measles vaccine strain (Edmonston) | ΔP | measles wild type (IC-B) | P | wild type P gene product V is a stronger inhibitor of MDA5-mediated activation of IRF-3 and IFN-I than V from Edmonston strain | [148] |
| measles vaccine strain (Edmonston) | ΔN, P, L | measles wild type (IC-B) | N, P, L | compared to the construct above, addition of N and L created a chimera with stronger IFN antagonistic capacity | [149] |
| newcastle disease virus F3aa (lentogenic Hitchner B1) | F mutations conferring increased fusogenic capacity | Influenza A/Puerto Rico/8/1934 (PR8), H1N1 | NS1 (between P and M) | chimera showed superior oncolytic efficacy to parental virus | [154] |
| newcastle disease virus (mesogenic Beaudette C) | Influenza H5N1 or H1N1/09 | NS1 (between P and M) | while not tested as oncolytic viruses, these recombinants did display pathogenicity in chickens and increased capacity to replicate in human cells compared to parental virus | [155] | |
| vaccinia virus (Western reserve) | ΔE3L | Influenza | NS1 | This chimera has not yet been evaluated as an oncolytic agent. Vaccinia virus E3L is critical for replication in most cell types and for spread in normal mice by blocking ISG15—influenza NS1 partially restores the capacity to replicate in cells but the resulting chimera is still unable to spread in normal tissues in vivo | [156] |
| herpes simplex type 1 (Synco-2D) | γ34.5−/−, multiple mutations, expressing GALV under UL38 promoter | vaccinia virus | B19R | Vaccinia virus soluble type I IFN scavenger B19R facilitated replication and spread of oncolytic HSV. Oncolytic efficacy in animal models was increased | [157] |
| herpes simplex type 1 | γ34.5−/− | human cytomegalovirus | TRS1 or IRS1 | PKR-antagonists TRS1 and IRS1 conferred increased replication capacity to oncolytic HSV-1, yielding greater therapeutic efficacy in glioma models in mice | [158] |
| vesicular stomatitis virus | ΔΜ51 | vaccinia virus (Western reserve) | B19R | Superior ability to spread due to neutralization of paracrine type I IFN | [159] |
| maraba virus (MG1) | G protein (Q242R) and M protein (L123W) mutations | vaccinia virus (Western reserve) | B19R | Similar to the VSV recombinant but with the enhanced oncolytic capacity of the Maraba backbone. Virus was safe in mice | [159] |
As an example of heterologous virus constructs, lentogenic Newcastle disease virus engineered to express influenza virus PR8 (H1N1) NS1 showed enhanced oncolytic efficacy in vivo due to dampening of antiviral responses [154]. While such a virus did not display enhanced pathogenicity in that study, a mesogenic NDV virus harboring NS1 from influenza strain H1N1/09 did show increased pathogenicity in chickens as well as an increased ability to replicate in human cells [155], raising some cautionary warnings for future engineering of similar replicative viruses. As another example, vaccinia virus soluble type I IFN scavenger B19R (NOTE: B18R is often used in the literature) has been engineered into both oncolytic HSV and oncolytic rhabdoviruses, resulting in all cases in more effective therapy agents without loss of tumor specificity [157,159]. Oncolytic γ34.5 gene deleted HSV was complemented by two different PKR-antagonists from human cytomegalovirus, TRS1 and IRS1, generating a chimera capable of reaching wild type virus replication levels in cancer cells in vitro and in tumor models in vivo [158].
The selection of various viral IFN-I-antagonists that potentially could be used to augment attenuated oncolytic viruses is vast [69,95]. For example, increased replication of several viruses, including HIV-1, Sindbis virus and E1-deleted adenovirus in mammalian cells was observed when the cells were engineered to express vaccinia virus E3L protein, influenza A virus NS1 protein, ebola virus VP35 protein or HIV-1 Tat protein [160]. The choice of antagonist must primarily be made with safety in mind, but virus-specific and cell-specific differences in antagonist functions may potentially be exploited rationally when considering the overall combination. For example, vaccinia virus E3L is able to rescue VSV but not EMCV from exogenous IFN, whereas vaccinia virus K3L partially rescued EMCV but not VSV [161].
In anticipation of systematic, perhaps bioinformatically guided, “super-chimera” studies, two or more viruses that may complement each other’s shortcomings in antiviral defense antagonism have been considered. Notably, vaccinia virus was favorably combined with IFN-I-sensitive oncolytic VSV and SFV, yielding tumor-model-dependent increases in overall therapy efficacy that were dependent on vaccinia virus antagonism of type I IFN responses, which increased the replication of the IFN-sensitive viruses [37,60]. Such heterologous virotherapy approaches may also be used to generate immunological synergy, similar to heterologous prime-boost vaccination; anti-tumor immune responses may be increased due to targeting of the tumor by two different viruses (on two separate occasions), but anti-virus immune responses would be generated against different viruses each time, avoiding the problem of neutralizing immunity to and predominance of antivirus immune responses [162]. Some other ideas explored experimentally include engineering oncolytic viruses to express other oncolytic viruses. For example, the entire oncolytic parvovirus H-1 genome was placed under a regulatable promoter in oncolytic adenovirus, resulting in a more effective therapeutic entity than either virus alone without loss of tumor-specificity [163]. The genetic material of Semliki Forest virus replicons has been engineered into adenovirus and vaccinia virus backbones [164,165,166] and several other virus chimeras have been constructed [167]. However, the effects on tumor antiviral defenses of such a constructs remain to be studied.
4. Conclusions
The attribute “oncolytic” implies for a virus that it infects and, indeed, lyses the infected tumor. This property is primarily tested in vitro in cultured tumor cells. We have learned, however, that in the native tumor microenvironment in living hosts many viruses are no longer able to infect tumor cells or to kill them even if they manage to infect them. Tightly packed tumor cells and the network of supportive molecules of the extracellular matrix form a physical barrier to virus particle diffusion. It has also become evident that the tumor cells themselves may be much more capable of thwarting oncolytic virus advances than previously thought, with some tumor cells residing in a seemingly permanent non-permissive antiviral state. Tumors harbor multiple cell types in addition to the neoplastic cells, which may promote and propagate both physical and cellular virus resistance. As the efficacy of virotherapy in human cancer patients still falls shy of the achievements in animal models, it appears quite plausible that one or more of the barriers described in this review indeed constitute a real and formidable obstacle for oncolytic virus advancement into routine clinical use. Fortunately, some of the most difficult barriers have been identified, and a rapidly expanding arsenal of countermeasures is at our disposal. Our task is now to separate the wheat from the chaff and to systematically evaluate the proposed combination regimens that will yield the best results without compromising patient safety.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar]
- Polanska, U.M.; Orimo, A. Carcinoma-associated fibroblasts: Non-neoplastic tumour-promoting mesenchymal cells. J. Cell. Physiol. 2013, 228, 1651–1657. [Google Scholar] [CrossRef]
- Gritsenko, P.G.; Ilina, O.; Friedl, P. Interstitial guidance of cancer invasion. J. Pathol. 2012, 226, 185–199. [Google Scholar] [CrossRef]
- Adams, R.H.; Alitalo, K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat. Rev. Mol. Cell Biol. 2007, 8, 464–478. [Google Scholar] [CrossRef]
- Pickup, M.; Novitskiy, S.; Moses, H.L. The roles of TGFβ in the tumour microenvironment. Nat. Rev. Cancer 2013, 13, 788–799. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Karin, M. Inflammatory cytokines in cancer: Tumour necrosis factor and interleukin 6 take the stage. Ann. Rheum. Dis. 2011, 70, i104–i108. [Google Scholar] [CrossRef]
- Dranoff, G. Cytokines in cancer pathogenesis and cancer therapy. Nat. Rev. Cancer 2004, 4, 11–22. [Google Scholar] [CrossRef]
- Lippitz, B.E. Cytokine patterns in patients with cancer: A systematic review. Lancet Oncol. 2013, 14, e218–e228. [Google Scholar]
- Miest, T.S.; Cattaneo, R. New viruses for cancer therapy: Meeting clinical needs. Nat. Rev. Microbiol. 2014, 12, 23–34. [Google Scholar] [CrossRef]
- Nakashima, H.; Kaur, B.; Chiocca, E.A. Directing systemic oncolytic viral delivery to tumors via carrier cells. Cytokine Growth Factor Rev. 2010, 21, 119–126. [Google Scholar] [CrossRef]
- Willmon, C.; Harrington, K.; Kottke, T.; Prestwich, R.; Melcher, A.; Vile, R. Cell carriers for oncolytic viruses: Fed Ex for cancer therapy. Mol. Ther. 2009, 17, 1667–1676. [Google Scholar] [CrossRef]
- Alemany, R. Viruses in cancer treatment. Clin. Transl. Oncol. 2013, 15, 182–188. [Google Scholar] [CrossRef]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef]
- Tanaka, T.; Yamanaka, N.; Oriyama, T.; Furukawa, K.; Okamoto, E. Factors regulating tumor pressure in hepatocellular carcinoma and implications for tumor spread. Hepatology 1997, 26, 283–287. [Google Scholar] [CrossRef]
- Stohrer, M.; Boucher, Y.; Stangassinger, M.; Jain, R.K. Oncotic pressure in solid tumors is elevated. Cancer Res. 2000, 60, 4251–4255. [Google Scholar]
- Minchinton, A.I.; Tannock, I.F. Drug penetration in solid tumours. Nat. Rev. Cancer 2006, 6, 583–592. [Google Scholar] [CrossRef]
- Jain, R.K.; Tong, R.T.; Munn, L.L. Effect of vascular normalization by antiangiogenic therapy on interstitial hypertension, peritumor edema, and lymphatic metastasis: Insights from a mathematical model. Cancer Res. 2007, 67, 2729–2735. [Google Scholar] [CrossRef]
- Mok, W.; Boucher, Y.; Jain, R.K. Matrix metalloproteinases-1 and -8 improve the distribution and efficacy of an oncolytic virus. Cancer Res. 2007, 67, 10664–10668. [Google Scholar] [CrossRef]
- McKee, T.D.; Grandi, P.; Mok, W.; Alexandrakis, G.; Insin, N.; Zimmer, J.P.; Bawendi, M.G.; Boucher, Y.; Breakefield, X.O.; Jain, R.K. Degradation of fibrillar collagen in a human melanoma xenograft improves the efficacy of an oncolytic herpes simplex virus vector. Cancer Res. 2006, 66, 2509–2513. [Google Scholar] [CrossRef]
- Bilbao, R.; Bustos, M.; Alzuguren, P.; Pajares, M.J.; Drozdzik, M.; Qian, C.; Prieto, J. A blood-tumor barrier limits gene transfer to experimental liver cancer: The effect of vasoactive compounds. Gene Ther. 2000, 7, 1824–1832. [Google Scholar] [CrossRef]
- Kuriyama, N.; Kuriyama, H.; Julin, C.M.; Lamborn, K.; Israel, M.A. Pretreatment with protease is a useful experimental strategy for enhancing adenovirus-mediated cancer gene therapy. Hum. Gene Ther. 2000, 11, 2219–2230. [Google Scholar] [CrossRef]
- Schäfer, S.; Weibel, S.; Donat, U.; Zhang, Q.; Aguilar, R.J.; Chen, N.G.; Szalay, A.A. Vaccinia virus-mediated intra-tumoral expression of matrix metalloproteinase 9 enhances oncolysis of PC-3 xenograft tumors. BMC Cancer 2012, 12, 366. [Google Scholar]
- Hong, C.S.; Fellows, W.; Niranjan, A.; Alber, S.; Watkins, S.; Cohen, J.B.; Glorioso, J.C.; Grandi, P. Ectopic matrix metalloproteinase-9 expression in human brain tumor cells enhances oncolytic HSV vector infection. Gene Ther. 2010, 17, 1200–1205. [Google Scholar]
- Vähä-Koskela, M.J.; Kallio, J.P.; Jansson, L.C.; Heikkilä, J.E.; Zakhartchenko, V.A.; Kallajoki, M.A.; Kähäri, V.M.; Hinkkanen, A.E. Oncolytic capacity of attenuated replicative semliki forest virus in human melanoma xenografts in severe combined immunodeficient mice. Cancer Res. 2006, 66, 7185–7194. [Google Scholar] [CrossRef]
- Smith, E.; Breznik, J.; Lichty, B.D. Strategies to enhance viral penetration of solid tumors. Human Gene Ther. 2011, 22, 1053–1060. [Google Scholar] [CrossRef]
- Beyer, I.; Li, Z.; Persson, J.; Liu, Y.; van Rensburg, R.; Yumul, R.; Zhang, X.B.; Hung, M.C.; Lieber, A. Controlled extracellular matrix degradation in breast cancer tumors improves therapy by trastuzumab. Mol. Ther. 2011, 19, 479–489. [Google Scholar] [CrossRef]
- Ganesh, S.; Gonzalez Edick, M.; Idamakanti, N.; Abramova, M.; Vanroey, M.; Robinson, M.; Yun, C.O.; Jooss, K. Relaxin-expressing, fiber chimeric oncolytic adenovirus prolongs survival of tumor-bearing mice. Cancer Res. 2007, 67, 4399–4407. [Google Scholar] [CrossRef]
- Choi, I.K.; Strauss, R.; Richter, M.; Yun, C.O.; Lieber, A. Strategies to increase drug penetration in solid tumors. Front. Oncol. 2013, 3, 193. [Google Scholar]
- Eshun, F.K.; Currier, M.A.; Gillespie, R.A.; Fitzpatrick, J.L.; Baird, W.H.; Cripe, T.P. VEGF blockade decreases the tumor uptake of systemic oncolytic herpes virus but enhances therapeutic efficacy when given after virotherapy. Gene Ther. 2010, 17, 922–929. [Google Scholar]
- Thaci, B.; Ulasov, I.V.; Ahmed, A.U.; Ferguson, S.D.; Han, Y.; Lesniak, M.S. Anti-angiogenic therapy increases intratumoral adenovirus distribution by inducing collagen degradation. Gene Ther. 2013, 20, 318–327. [Google Scholar]
- Angarita, F.A.; Acuna, S.A.; Ottolino-Perry, K.; Zerhouni, S.; McCart, J.A. Mounting a strategic offense: Fighting tumor vasculature with oncolytic viruses. Trends Mol. Med. 2013, 19, 378–392. [Google Scholar] [CrossRef]
- Dingwell, K.S.; Brunetti, C.R.; Hendricks, R.L.; Tang, Q.; Tang, M.; Rainbow, A.J.; Johnson, D.C. Herpes simplex virus glycoproteins E and I facilitate cell-to-cell spread in vivo and across junctions of cultured cells. J. Virol. 1994, 68, 834–845. [Google Scholar]
- Rottner, K.; Stradal, T.E. Poxviruses taking a ride on actin: New users of known hardware. Cell Host Microbe 2009, 6, 497–499. [Google Scholar] [CrossRef]
- Smith, G.L.; Law, M. The exit of vaccinia virus from infected cells. Virus Res. 2004, 106, 189–197. [Google Scholar] [CrossRef]
- Guedan, S.; Grases, D.; Rojas, J.J.; Gros, A.; Vilardell, F.; Vile, R.; Mercade, E.; Cascallo, M.; Alemany, R. GALV expression enhances the therapeutic efficacy of an oncolytic adenovirus by inducing cell fusion and enhancing virus distribution. Gene Ther. 2012, 19, 1048–1057. [Google Scholar]
- Le Boeuf, F.; Diallo, J.S.; McCart, J.A.; Thorne, S.; Falls, T.; Stanford, M.; Kanji, F.; Auer, R.; Brown, C.W.; Lichty, B.D.; et al. Synergistic interaction between oncolytic viruses augments tumor killing. Mol. Ther. 2010, 18, 888–895. [Google Scholar] [CrossRef]
- Hoffmann, D.; Grunwald, T.; Kuate, S.; Wildner, O. Mechanistic analysis and comparison of viral fusogenic membrane proteins for their synergistic effects on chemotherapy. Cancer Biol. Ther. 2007, 6, 510–518. [Google Scholar] [CrossRef]
- Wang, H.; Liaw, Y.C.; Stone, D.; Kalyuzhniy, O.; Amiraslanov, I.; Tuve, S.; Verlinde, C.L.; Shayakhmetov, D.; Stehle, T.; Roffler, S.; et al. Identification of CD46 binding sites within the adenovirus serotype 35 fiber knob. J. Virol. 2007, 81, 12785–12792. [Google Scholar] [CrossRef]
- Wang, H.; Li, Z.Y.; Liu, Y.; Persson, J.; Beyer, I.; Moller, T.; Koyuncu, D.; Drescher, M.R.; Strauss, R.; Zhang, X.B.; et al. Desmoglein 2 is a receptor for adenovirus serotypes 3, 7, 11 and 14. Nat. Med. 2011, 17, 96–104. [Google Scholar]
- Coyne, C.B.; Bergelson, J.M. CAR: A virus receptor within the tight junction. Adv. Drug Deliv. Rev. 2005, 57, 869–882. [Google Scholar] [CrossRef]
- Cardone, J.; Al-Shouli, S.; Kemper, C. A novel role for CD46 in wound repair. Front. Immunol. 2011, 2, 28. [Google Scholar]
- Thorsteinsson, L.; O’Dowd, G.M.; Harrington, P.M.; Johnson, P.M. The complement regulatory proteins CD46 and CD59, but not CD55, are highly expressed by glandular epithelium of human breast and colorectal tumour tissues. APMIS 1998, 106, 869–878. [Google Scholar] [CrossRef]
- Nandi, S.; Ulasov, I.V.; Rolle, C.E.; Han, Y.; Lesniak, M.S. A chimeric adenovirus with an Ad 3 fiber knob modification augments glioma virotherapy. J. Gene Med. 2009, 11, 1005–1011. [Google Scholar]
- Martin, T.A.; Mason, M.D.; Jiang, W.G. Tight junctions in cancer metastasis. Front. Biosci. 2011, 16, 898–936. [Google Scholar] [CrossRef]
- Ding, L.; Lu, Z.; Lu, Q.; Chen, Y.H. The claudin family of proteins in human malignancy: A clinical perspective. Cancer Manag. Res. 2013, 5, 367–375. [Google Scholar]
- Beyer, I.; van Rensburg, R.; Strauss, R.; Li, Z.; Wang, H.; Persson, J.; Yumul, R.; Feng, Q.; Song, H.; Bartek, J.; et al. Epithelial junction opener JO-1 improves monoclonal antibody therapy of cancer. Cancer Res. 2011, 71, 7080–7090. [Google Scholar] [CrossRef]
- Beyer, I.; Cao, H.; Persson, J.; Song, H.; Richter, M.; Feng, Q.; Yumul, R.; van Rensburg, R.; Li, Z.; Berenson, R.; et al. Coadministration of epithelial junction opener JO-1 improves the efficacy and safety of chemotherapeutic drugs. Clin. Cancer Res. 2012, 18, 3340–3351. [Google Scholar] [CrossRef]
- Nagano, S.; Perentes, J.Y.; Jain, R.K.; Boucher, Y. Cancer cell death enhances the penetration and efficacy of oncolytic herpes simplex virus in tumors. Cancer Res. 2008, 68, 3795–3802. [Google Scholar]
- Heuberger, J.; Birchmeier, W. Interplay of cadherin-mediated cell adhesion and canonical Wnt signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a002915. [Google Scholar]
- Binder, C.; Hagemann, T.; Husen, B.; Schulz, M.; Einspanier, A. Relaxin enhances in vitro invasiveness of breast cancer cell lines by up-regulation of matrix metalloproteases. Mol. Hum. Reprod. 2002, 8, 789–796. [Google Scholar]
- Radestock, Y.; Hoang-Vu, C.; Hombach-Klonisch, S. Relaxin reduces xenograft tumour growth of human MDA-MB-231 breast cancer cells. Breast Cancer Res. 2008, 10, R71. [Google Scholar]
- Lavilla-Alonso, S.; Bauer, M.M.; Abo-Ramadan, U.; Ristimaki, A.; Halavaara, J.; Desmond, R.A.; Wang, D.; Escutenaire, S.; Ahtiainen, L.; Saksela, K.; et al. Macrophage metalloelastase (MME) as adjuvant for intra-tumoral injection of oncolytic adenovirus and its influence on metastases development. Cancer Gene Ther. 2012, 19, 126–134. [Google Scholar]
- Toth, K.; Djeha, H.; Ying, B.; Tollefson, A.E.; Kuppuswamy, M.; Doronin, K.; Krajcsi, P.; Lipinski, K.; Wrighton, C.J.; Wold, W.S. An oncolytic adenovirus vector combining enhanced cell-to-cell spreading, mediated by the ADP cytolytic protein, with selective replication in cancer cells with deregulated wnt signaling. Cancer Res. 2004, 64, 3638–3644. [Google Scholar] [CrossRef]
- De Palma, M.; Lewis, C.E. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell 2013, 23, 277–286. [Google Scholar] [CrossRef]
- Fulci, G.; Dmitrieva, N.; Gianni, D.; Fontana, E.J.; Pan, X.; Lu, Y.; Kaufman, C.S.; Kaur, B.; Lawler, S.E.; Lee, R.J.; et al. Depletion of peripheral macrophages and brain microglia increases brain tumor titers of oncolytic viruses. Cancer Res. 2007, 67, 9398–9406. [Google Scholar] [CrossRef]
- Lallemand, C.; Lebon, P.; Rizza, P.; Blanchard, B.; Tovey, M.G. Constitutive expression of specific interferon isotypes in peripheral blood leukocytes from normal individuals and in promonocytic U937 cells. J. Leukoc. Biol. 1996, 60, 137–146. [Google Scholar]
- Liu, Y.P.; Suksanpaisan, L.; Steele, M.B.; Russell, S.J.; Peng, K.W. Induction of antiviral genes by the tumor microenvironment confers resistance to virotherapy. Sci. Rep. 2013, 3, 2375. [Google Scholar]
- Ruotsalainen, J.; Martikainen, M.; Niittykoski, M.; Huhtala, T.; Aaltonen, T.; Heikkilä, J.; Bell, J.; Vähä-Koskela, M.; Hinkkanen, A. Interferon-beta sensitivity of tumor cells correlates with poor response to va7 virotherapy in mouse glioma models. Mol. Ther. 2012, 20, 1529–1539. [Google Scholar] [CrossRef]
- Vähä-Koskela, M.J.; Le Boeuf, F.; Lemay, C.; de Silva, N.; Diallo, J.S.; Cox, J.; Becker, M.; Choi, Y.; Ananth, A.; Sellers, C.; et al. Resistance to two heterologous neurotropic oncolytic viruses, Semliki Forest virus and vaccinia virus, in experimental glioma. J. Virol. 2013, 87, 2363–2366. [Google Scholar] [CrossRef]
- Giraldo, N.A.; Becht, E.; Remark, R.; Damotte, D.; Sautes-Fridman, C.; Fridman, W.H. The immune contexture of primary and metastatic human tumours. Curr. Opin. Immunol. 2014, 27C, 8–15. [Google Scholar]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef]
- Currier, M.A.; Eshun, F.K.; Sholl, A.; Chernoguz, A.; Crawford, K.; Divanovic, S.; Boon, L.; Goins, W.F.; Frischer, J.S.; Collins, M.H.; et al. VEGF blockade enables oncolytic cancer virotherapy in part by modulating intratumoral myeloid cells. Mol. Ther. 2013, 21, 1014–1023. [Google Scholar] [CrossRef]
- Burger, J.A.; Kipps, T.J. CXCR4: A key receptor in the crosstalk between tumor cells and their microenvironment. Blood 2006, 107, 1761–1767. [Google Scholar] [CrossRef]
- Gil, M.; Seshadri, M.; Komorowski, M.P.; Abrams, S.I.; Kozbor, D. Targeting CXCL12/CXCR4 signaling with oncolytic virotherapy disrupts tumor vasculature and inhibits breast cancer metastases. Proc. Natl. Acad. Sci. USA 2013, 110, E1291–E1300. [Google Scholar]
- Muthana, M.; Rodrigues, S.; Chen, Y.Y.; Welford, A.; Hughes, R.; Tazzyman, S.; Essand, M.; Morrow, F.; Lewis, C.E. Macrophage delivery of an oncolytic virus abolishes tumor regrowth and metastasis after chemotherapy or irradiation. Cancer Res. 2013, 73, 490–495. [Google Scholar] [CrossRef]
- Verheije, M.H.; Rottier, P.J. Retargeting of viruses to generate oncolytic agents. Adv. Virol. 2012, 2012, 798526. [Google Scholar]
- Power, A.T.; Bell, J.C. Taming the Trojan horse: Optimizing dynamic carrier cell/oncolytic virus systems for cancer biotherapy. Gene Ther. 2008, 15, 772–779. [Google Scholar] [CrossRef]
- Randall, R.E.; Goodbourn, S. Interferons and viruses: An interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 2008, 89, 1–47. [Google Scholar] [CrossRef]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Ann. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef]
- Muller, U.; Steinhoff, U.; Reis, L.F.; Hemmi, S.; Pavlovic, J.; Zinkernagel, R.M.; Aguet, M. Functional role of type I and type II interferons in antiviral defense. Science 1994, 264, 1918–1921. [Google Scholar]
- Fragkoudis, R.; Breakwell, L.; McKimmie, C.; Boyd, A.; Barry, G.; Kohl, A.; Merits, A.; Fazakerley, J.K. The type I interferon system protects mice from Semliki Forest virus by preventing widespread virus dissemination in extraneural tissues, but does not mediate the restricted replication of avirulent virus in central nervous system neurons. J. Gen. Virol. 2007, 88, 3373–3384. [Google Scholar] [CrossRef]
- Chapgier, A.; Wynn, R.F.; Jouanguy, E.; Filipe-Santos, O.; Zhang, S.; Feinberg, J.; Hawkins, K.; Casanova, J.L.; Arkwright, P.D. Human complete Stat-1 deficiency is associated with defective type I and II IFN responses in vitro but immunity to some low virulence viruses in vivo. J. Immunol. 2006, 176, 5078–5083. [Google Scholar]
- Sancho-Shimizu, V.; Perez de Diego, R.; Jouanguy, E.; Zhang, S.Y.; Casanova, J.L. Inborn errors of anti-viral interferon immunity in humans. Curr. Opin. Virol. 2011, 1, 487–496. [Google Scholar] [CrossRef]
- Bax, H.I.; Freeman, A.F.; Anderson, V.L.; Vesterhus, P.; Laerum, D.; Pittaluga, S.; Wilson, W.H.; Holland, S.M. B-cell lymphoma in a patient with complete interferon gamma receptor 1 deficiency. J. Clin. Immunol. 2013, 33, 1062–1066. [Google Scholar]
- Kawamoto, S.; Oritani, K.; Asada, H.; Takahashi, I.; Ishikawa, J.; Yoshida, H.; Yamada, M.; Ishida, N.; Ujiie, H.; Masaie, H.; et al. Antiviral activity of limitin against encephalomyocarditis virus, herpes simplex virus, and mouse hepatitis virus: Diverse requirements by limitin and alpha interferon for interferon regulatory factor 1. J. Virol. 2003, 77, 9622–9631. [Google Scholar] [CrossRef]
- Bianchi, M.E. DAMPs, PAMPs and alarmins: All we need to know about danger. J. Leukoc. Biol. 2007, 81, 1–5. [Google Scholar] [CrossRef]
- Zurney, J.; Kobayashi, T.; Holm, G.H.; Dermody, T.S.; Sherry, B. Reovirus mu2 protein inhibits interferon signaling through a novel mechanism involving nuclear accumulation of interferon regulatory factor 9. J. Virol. 2009, 83, 2178–2187. [Google Scholar] [CrossRef]
- Zurney, J.; Howard, K.E.; Sherry, B. Basal expression levels of IFNAR and Jak-STAT components are determinants of cell-type-specific differences in cardiac antiviral responses. J. Virol. 2007, 81, 13668–13680. [Google Scholar] [CrossRef]
- Dunn, G.P.; Koebel, C.M.; Schreiber, R.D. Interferons, immunity and cancer immunoediting. Nat. Rev. Immunol. 2006, 6, 836–848. [Google Scholar] [CrossRef]
- Dunn, G.P.; Bruce, A.T.; Sheehan, K.C.; Shankaran, V.; Uppaluri, R.; Bui, J.D.; Diamond, M.S.; Koebel, C.M.; Arthur, C.; White, J.M.; et al. A critical function for type I interferons in cancer immunoediting. Nat. Immunol. 2005, 6, 722–729. [Google Scholar]
- Sheehan, K.C.; Lai, K.S.; Dunn, G.P.; Bruce, A.T.; Diamond, M.S.; Heutel, J.D.; Dungo-Arthur, C.; Carrero, J.A.; White, J.M.; Hertzog, P.J.; et al. Blocking monoclonal antibodies specific for mouse IFN-alpha/beta receptor subunit 1 (IFNAR-1) from mice immunized by in vivo hydrodynamic transfection. J. Interferon Cytokine Res. 2006, 26, 804–819. [Google Scholar] [CrossRef]
- Saidi, R.F.; Williams, F.; Silberberg, B.; Mittal, V.K.; ReMine, S.G.; Jacobs, M.J. Expression of interferon receptors in pancreatic cancer: Identification of a novel prognostic factor. Surgery 2006, 139, 743–748. [Google Scholar] [CrossRef]
- Kondo, M.; Nagano, H.; Sakon, M.; Yamamoto, H.; Morimoto, O.; Arai, I.; Miyamoto, A.; Eguchi, H.; Dono, K.; Nakamori, S.; et al. Expression of interferon alpha/beta receptor in human hepatocellular carcinoma. Int. J. Oncol. 2000, 17, 83–88. [Google Scholar]
- Weichselbaum, R.R.; Ishwaran, H.; Yoon, T.; Nuyten, D.S.; Baker, S.W.; Khodarev, N.; Su, A.W.; Shaikh, A.Y.; Roach, P.; Kreike, B.; et al. An interferon-related gene signature for DNA damage resistance is a predictive marker for chemotherapy and radiation for breast cancer. Proc. Natl. Acad. Sci. USA 2008, 105, 18490–18495. [Google Scholar] [CrossRef]
- Khodarev, N.N.; Roach, P.; Pitroda, S.P.; Golden, D.W.; Bhayani, M.; Shao, M.Y.; Darga, T.E.; Beveridge, M.G.; Sood, R.F.; Sutton, H.G.; et al. STAT1 pathway mediates amplification of metastatic potential and resistance to therapy. PLoS One 2009, 4, e5821. [Google Scholar] [CrossRef]
- Khodarev, N.N.; Roizman, B.; Weichselbaum, R.R. Molecular pathways: interferon/stat1 pathway: Role in the tumor resistance to genotoxic stress and aggressive growth. Clin. Cancer Res. 2012, 18, 3015–3021. [Google Scholar] [CrossRef]
- Luszczek, W.; Cheriyath, V.; Mekhail, T.M.; Borden, E.C. Combinations of DNA methyltransferase and histone deacetylase inhibitors induce DNA damage in small cell lung cancer cells: Correlation of resistance with IFN-stimulated gene expression. Mol. Cancer Ther. 2010, 9, 2309–2321. [Google Scholar] [CrossRef]
- Cheon, H.; Yang, J.; Stark, G.R. The functions of signal transducers and activators of transcriptions 1 and 3 as cytokine-inducible proteins. J. Interferon Cytokine Res. 2011, 31, 33–40. [Google Scholar] [CrossRef]
- Wilson, S.S.; Wiens, M.E.; Smith, J.G. Antiviral mechanisms of human defensins. J. Mol. Biol. 2013, 425, 4965–4980. [Google Scholar] [CrossRef]
- Lundy, F.T.; Orr, D.F.; Gallagher, J.R.; Maxwell, P.; Shaw, C.; Napier, S.S.; Gerald Cowan, C.; Lamey, P.J.; Marley, J.J. Identification and overexpression of human neutrophil alpha-defensins (human neutrophil peptides 1, 2 and 3) in squamous cell carcinomas of the human tongue. Oral Oncol. 2004, 40, 139–144. [Google Scholar] [CrossRef]
- Lapteva, N.; Aldrich, M.; Rollins, L.; Ren, W.; Goltsova, T.; Chen, S.Y.; Huang, X.F. Attraction and activation of dendritic cells at the site of tumor elicits potent antitumor immunity. Mol. Ther. 2009, 17, 1626–1636. [Google Scholar] [CrossRef]
- Thompson, M.R.; Kaminski, J.J.; Kurt-Jones, E.A.; Fitzgerald, K.A. Pattern recognition receptors and the innate immune response to viral infection. Viruses 2011, 3, 920–940. [Google Scholar] [CrossRef]
- Takaoka, A.; Wang, Z.; Choi, M.K.; Yanai, H.; Negishi, H.; Ban, T.; Lu, Y.; Miyagishi, M.; Kodama, T.; Honda, K.; et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 2007, 448, 501–505. [Google Scholar] [CrossRef]
- Haller, O.; Kochs, G.; Weber, F. The interferon response circuit: Induction and suppression by pathogenic viruses. Virology 2006, 344, 119–130. [Google Scholar] [CrossRef]
- Suzuki, M.; Chiocca, E.A.; Saeki, Y. Early STAT1 activation after systemic delivery of HSV amplicon vectors suppresses transcription of the vector-encoded transgene. Mol. Ther. 2007, 15, 2017–2026. [Google Scholar] [CrossRef]
- Heikkilä, J.E.; Vähä-Koskela, M.J.; Ruotsalainen, J.J.; Martikainen, M.W.; Stanford, M.M.; McCart, J.A.; Bell, J.C.; Hinkkanen, A.E. Intravenously administered alphavirus vector VA7 eradicates orthotopic human glioma xenografts in nude mice. PLoS One 2010, 5, e8603. [Google Scholar]
- Määttä, A.M.; Liimatainen, T.; Wahlfors, T.; Wirth, T.; Vähä-Koskela, M.; Jansson, L.; Valonen, P.; Hakkinen, K.; Rautsi, O.; Pellinen, R.; et al. Evaluation of cancer virotherapy with attenuated replicative Semliki forest virus in different rodent tumor models. Int. J. Cancer 2007, 121, 863–870. [Google Scholar] [CrossRef]
- Stojdl, D.F.; Lichty, B.; Knowles, S.; Marius, R.; Atkins, H.; Sonenberg, N.; Bell, J.C. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat. Med. 2000, 6, 821–825. [Google Scholar]
- Paglino, J.C.; van den Pol, A.N. Vesicular stomatitis virus has extensive oncolytic activity against human sarcomas: Rare resistance is overcome by blocking interferon pathways. J. Virol. 2011, 85, 9346–9358. [Google Scholar] [CrossRef]
- Blackham, A.U.; Northrup, S.A.; Willingham, M.; D’Agostino, R.B., Jr.; Lyles, D.S.; Stewart, J.H.T. Variation in susceptibility of human malignant melanomas to oncolytic vesicular stomatitis virus. Surgery 2013, 153, 333–343. [Google Scholar] [CrossRef]
- Berchtold, S.; Lampe, J.; Weiland, T.; Smirnow, I.; Schleicher, S.; Handgretinger, R.; Kopp, H.G.; Reiser, J.; Stubenrauch, F.; Mayer, N.; et al. Innate immune defense defines susceptibility of sarcoma cells to measles vaccine virus-based oncolysis. J. Virol. 2013, 87, 3484–3501. [Google Scholar] [CrossRef]
- Monsurro, V.; Beghelli, S.; Wang, R.; Barbi, S.; Coin, S.; di Pasquale, G.; Bersani, S.; Castellucci, M.; Sorio, C.; Eleuteri, S.; et al. Anti-viral state segregates two molecular phenotypes of pancreatic adenocarcinoma: Potential relevance for adenoviral gene therapy. J. Transl. Med. 2010, 8, 10. [Google Scholar] [CrossRef]
- Liikanen, I.; Monsurro, V.; Ahtiainen, L.; Raki, M.; Hakkarainen, T.; Diaconu, I.; Escutenaire, S.; Hemminki, O.; Dias, J.D.; Cerullo, V.; et al. Induction of interferon pathways mediates in vivo resistance to oncolytic adenovirus. Mol. Ther. 2011, 19, 1858–1866. [Google Scholar] [CrossRef]
- Wollmann, G.; Davis, J.N.; Bosenberg, M.W.; van den Pol, A.N. Vesicular stomatitis virus variants selectively infect and kill human melanomas but not normal melanocytes. J. Virol. 2013, 87, 6644–6659. [Google Scholar] [CrossRef]
- Saloura, V.; Wang, L.C.; Fridlender, Z.G.; Sun, J.; Cheng, G.; Kapoor, V.; Sterman, D.H.; Harty, R.N.; Okumura, A.; Barber, G.N.; et al. Evaluation of an attenuated vesicular stomatitis virus (vsv) vector expressing interferon-beta for use in malignant pleural mesothelioma: heterogeneity in interferon-responsiveness defines potential efficacy. Hum. Gene Ther. 2010, 21, 51–64. [Google Scholar]
- Shmulevitz, M.; Pan, L.Z.; Garant, K.; Pan, D.; Lee, P.W. Oncogenic Ras promotes reovirus spread by suppressing IFN-beta production through negative regulation of RIG-I signaling. Cancer Res. 2010, 70, 4912–4921. [Google Scholar] [CrossRef]
- Battcock, S.M.; Collier, T.W.; Zu, D.; Hirasawa, K. Negative regulation of the alpha interferon-induced antiviral response by the Ras/Raf/MEK pathway. J. Virol. 2006, 80, 4422–4430. [Google Scholar] [CrossRef]
- Christian, S.L.; Zu, D.; Licursi, M.; Komatsu, Y.; Pongnopparat, T.; Codner, D.A.; Hirasawa, K. Suppression of IFN-induced transcription underlies IFN defects generated by activated Ras/MEK in human cancer cells. PLoS One 2012, 7, e44267. [Google Scholar]
- Cascallo, M.; Capella, G.; Mazo, A.; Alemany, R. Ras-dependent oncolysis with an adenovirus VAI mutant. Cancer Res. 2003, 63, 5544–5550. [Google Scholar]
- Schumann, M.; Dobbelstein, M. Activating Ras mutations fail to ensure efficient replication of adenovirus mutants lacking VA-RNA. Cell Cycle 2006, 5, 315–321. [Google Scholar] [CrossRef]
- Kim, M.; Egan, C.; Alain, T.; Urbanski, S.J.; Lee, P.W.; Forsyth, P.A.; Johnston, R.N. Acquired resistance to reoviral oncolysis in Ras-transformed fibrosarcoma cells. Oncogene 2007, 26, 4124–4134. [Google Scholar] [CrossRef]
- Moerdyk-Schauwecker, M.; Shah, N.R.; Murphy, A.M.; Hastie, E.; Mukherjee, P.; Grdzelishvili, V.Z. Resistance of pancreatic cancer cells to oncolytic vesicular stomatitis virus: Role of type I interferon signaling. Virology 2013, 436, 221–234. [Google Scholar]
- Evgin, L.; Vähä-Koskela, M.; Rintoul, J.; Falls, T.; Le, B.F.; Barrett, J.W.; Bell, J.C.; Stanford, M.M. Potent oncolytic activity of raccoonpox virus in the absence of natural pathogenicity. Mol. Ther. 2010, 18, 896–902. [Google Scholar] [CrossRef]
- Stanford, M.M.; Breitbach, C.J.; Bell, J.C.; McFadden, G. Innate immunity, tumor microenvironment and oncolytic virus therapy: Friends or foes? Curr. Opin. Mol. Ther. 2008, 10, 32–37. [Google Scholar]
- Zemp, F.J.; McKenzie, B.A.; Lun, X.; Maxwell, L.; Reilly, K.M.; McFadden, G.; Yong, V.W.; Forsyth, P.A. Resistance to oncolytic myxoma virus therapy in nf1−/−/trp53−/− syngeneic mouse glioma models is independent of anti-viral type-I interferon. PLoS One 2013, 8, e65801. [Google Scholar]
- Haseley, A.; Boone, S.; Wojton, J.; Yu, L.; Yoo, J.Y.; Yu, J.; Kurozumi, K.; Glorioso, J.C.; Caligiuri, M.A.; Kaur, B. Extracellular matrix protein CCN1 limits oncolytic efficacy in glioma. Cancer Res. 2012, 72, 1353–1362. [Google Scholar] [CrossRef]
- Raaben, M.; Groot Koerkamp, M.J.; Rottier, P.J.; de Haan, C.A. Type I interferon receptor-independent and -dependent host transcriptional responses to mouse hepatitis coronavirus infection in vivo. BMC Genomics 2009, 10, 350. [Google Scholar] [CrossRef]
- Chairatvit, K.; Wongnoppavich, A.; Choonate, S. Up-regulation of interferon-stimulated gene15 and its conjugates by tumor necrosis factor-alpha via type I interferon-dependent and -independent pathways. Mol. Cell. Biochem. 2012, 368, 195–201. [Google Scholar] [CrossRef]
- Le Boeuf, F.; Niknejad, N.; Wang, J.; Auer, R.; Weberpals, J.I.; Bell, J.C.; Dimitroulakos, J. Sensitivity of cervical carcinoma cells to vesicular stomatitis virus-induced oncolysis: Potential role of human papilloma virus infection. Int. J. Cancer 2012, 131, E204–E215. [Google Scholar] [CrossRef]
- Escobar-Zarate, D.; Liu, Y.P.; Suksanpaisan, L.; Russell, S.J.; Peng, K.W. Overcoming cancer cell resistance to VSV oncolysis with JAK1/2 inhibitors. Cancer Gene Ther. 2013, 20, 582–589. [Google Scholar]
- Heine, A.; Held, S.A.; Daecke, S.N.; Wallner, S.; Yajnanarayana, S.P.; Kurts, C.; Wolf, D.; Brossart, P. The JAK-inhibitor ruxolitinib impairs dendritic cell function in vitro and in vivo. Blood 2013, 122, 1192–1202. [Google Scholar] [CrossRef]
- Looyenga, B.D.; Hutchings, D.; Cherni, I.; Kingsley, C.; Weiss, G.J.; Mackeigan, J.P. STAT3 is activated by JAK2 independent of key oncogenic driver mutations in non-small cell lung carcinoma. PLoS One 2012, 7, e30820. [Google Scholar]
- Nguyen, T.L.; Abdelbary, H.; Arguello, M.; Breitbach, C.; Leveille, S.; Diallo, J.S.; Yasmeen, A.; Bismar, T.A.; Kirn, D.; Falls, T.; et al. Chemical targeting of the innate antiviral response by histone deacetylase inhibitors renders refractory cancers sensitive to viral oncolysis. Proc. Natl. Acad. Sci. USA 2008, 105, 14981–14986. [Google Scholar] [CrossRef]
- Kim, D.R.; Park, M.Y.; Lim, H.J.; Park, J.S.; Cho, Y.J.; Lee, S.W.; Yoon, H.I.; Lee, J.H.; Kim, Y.S.; Lee, C.T. Combination therapy of conditionally replicating adenovirus and histone deacetylase inhibitors. Int. J. Mol. Med. 2012, 29, 218–224. [Google Scholar]
- Otsuki, A.; Patel, A.; Kasai, K.; Suzuki, M.; Kurozumi, K.; Chiocca, E.A.; Saeki, Y. Histone deacetylase inhibitors augment antitumor efficacy of herpes-based oncolytic viruses. Mol. Ther. 2008, 16, 1546–1555. [Google Scholar] [CrossRef]
- Katsura, T.; Iwai, S.; Ota, Y.; Shimizu, H.; Ikuta, K.; Yura, Y. The effects of trichostatin A on the oncolytic ability of herpes simplex virus for oral squamous cell carcinoma cells. Cancer Gene Ther. 2009, 16, 237–245. [Google Scholar]
- Shulak, L.; Beljanski, V.; Chiang, C.; Dutta, S.M.; van Grevenynghe, J.; Belgnaoui, S.M.; Nguyen, T.L.; di Lenardo, T.; Semmes, O.J.; Lin, R.; et al. Histone deacetylase inhibitors potentiate vesicular stomatitis virus oncolysis in prostate cancer cells by modulating nf-kappab-dependent autophagy. J. Virol. 2014, 88, 2927–2940. [Google Scholar] [CrossRef]
- Du, Z.; Whitt, M.A.; Baumann, J.; Garner, J.M.; Morton, C.L.; Davidoff, A.M.; Pfeffer, L.M. Inhibition of type I interferon-mediated antiviral action in human glioma cells by the IKK inhibitors BMS-345541 and TPCA-1. J. Interferon Cytokine Res. 2012, 32, 368–377. [Google Scholar] [CrossRef]
- Forbes, N.E.; Abdelbary, H.; Lupien, M.; Bell, J.C.; Diallo, J.S. Exploiting tumor epigenetics to improve oncolytic virotherapy. Front. Genet. 2013, 4, 184. [Google Scholar]
- Okemoto, K.; Kasai, K.; Wagner, B.; Haseley, A.; Meisen, H.; Bolyard, C.; Mo, X.; Wehr, A.; Lehman, A.; Fernandez, S.; et al. DNA demethylating agents synergize with oncolytic HSV1 against malignant gliomas. Clin. Cancer. 2013, 19, 5952–5959. [Google Scholar] [CrossRef]
- Jha, B.K.; Dong, B.; Nguyen, C.T.; Polyakova, I.; Silverman, R.H. Suppression of antiviral innate immunity by sunitinib enhances oncolytic virotherapy. Mol. Ther. 2013, 21, 1749–1757. [Google Scholar] [CrossRef]
- Randhawa, P.S.; Farasati, N.A.; Huang, Y.; Mapara, M.Y.; Shapiro, R. Viral drug sensitivity testing using quantitative PCR: Effect of tyrosine kinase inhibitors on polyomavirus BK replication. Am. J. Clin. Pathol. 2010, 134, 916–920. [Google Scholar] [CrossRef]
- Wakimoto, H.; Fulci, G.; Tyminski, E.; Chiocca, E.A. Altered expression of antiviral cytokine mRNAs associated with cyclophosphamide’s enhancement of viral oncolysis. Gene Ther. 2004, 11, 214–223. [Google Scholar]
- Ben Yebdri, F.; van Grevenynghe, J.; Tang, V.A.; Goulet, M.L.; Wu, J.H.; Stojdl, D.F.; Hiscott, J.; Lin, R. Triptolide-mediated inhibition of interferon signaling enhances vesicular stomatitis virus-based oncolysis. Mol. Ther. 2013, 21, 2043–2053. [Google Scholar] [CrossRef]
- Alain, T.; Lun, X.; Martineau, Y.; Sean, P.; Pulendran, B.; Petroulakis, E.; Zemp, F.J.; Lemay, C.G.; Roy, D.; Bell, J.C.; et al. Vesicular stomatitis virus oncolysis is potentiated by impairing mTORC1-dependent type I IFN production. Proc. Natl. Acad. Sci. USA 2010, 107, 1576–1581. [Google Scholar] [CrossRef]
- Diallo, J.S.; Le, B.F.; Lai, F.; Cox, J.; Vaha-Koskela, M.; Abdelbary, H.; MacTavish, H.; Waite, K.; Falls, T.; Wang, J.; et al. A high-throughput pharmacoviral approach identifies novel oncolytic virus sensitizers. Mol. Ther. 2010, 18, 1123–1129. [Google Scholar] [CrossRef]
- Beug, S.T.; Tang, V.A.; Lacasse, E.C.; Cheung, H.H.; Beauregard, C.E.; Brun, J.; Nuyens, J.P.; Earl, N.; St-Jean, M.; Holbrook, J.; et al. Smac mimetics and innate immune stimuli synergize to promote tumor death. Nat. Biotechnol. 2014, 32, 182–190. [Google Scholar] [CrossRef]
- Mahoney, D.J.; Lefebvre, C.; Allan, K.; Brun, J.; Sanaei, C.A.; Baird, S.; Pearce, N.; Gronberg, S.; Wilson, B.; Prakesh, M.; et al. Virus-tumor interactome screen reveals ER stress response can reprogram resistant cancers for oncolytic virus-triggered caspase-2 cell death. Cancer Cell 2011, 20, 443–456. [Google Scholar]
- Passer, B.J.; Cheema, T.; Zhou, B.; Wakimoto, H.; Zaupa, C.; Razmjoo, M.; Sarte, J.; Wu, S.; Wu, C.L.; Noah, J.W.; et al. Identification of the ENT1 antagonists dipyridamole and dilazep as amplifiers of oncolytic herpes simplex virus-1 replication. Cancer Res. 2010, 70, 3890–3895. [Google Scholar] [CrossRef]
- Wennier, S.T.; Liu, J.; McFadden, G. Bugs and drugs: Oncolytic virotherapy in combination with chemotherapy. Curr. Pharm. Biotechnol. 2012, 13, 1817–1833. [Google Scholar] [CrossRef]
- Newman, W.; Southam, C.M. Virus treatment in advanced cancer; a pathological study of fifty-seven cases. Cancer 1954, 7, 106–118. [Google Scholar] [CrossRef]
- Southam, C.M.; Hilleman, M.R.; Werner, J.H. Pathogenicity and oncolytic capacity of RI virus strain RI-67 in man. J. Lab. Clin. Med. 1956, 47, 573–582. [Google Scholar]
- Elankumaran, S.; Chavan, V.; Qiao, D.; Shobana, R.; Moorkanat, G.; Biswas, M.; Samal, S.K. Type I interferon-sensitive recombinant newcastle disease virus for oncolytic virotherapy. J. Virol. 2010, 84, 3835–3844. [Google Scholar] [CrossRef]
- Stojdl, D.F.; Lichty, B.D.; tenOever, B.R.; Paterson, J.M.; Power, A.T.; Knowles, S.; Marius, R.; Reynard, J.; Poliquin, L.; Atkins, H.; et al. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell 2003, 4, 263–275. [Google Scholar]
- Malilas, W.; Koh, S.S.; Srisuttee, R.; Boonying, W.; Cho, I.R.; Jeong, C.S.; Johnston, R.N.; Chung, Y.H. Cancer upregulated gene 2, a novel oncogene, confers resistance to oncolytic vesicular stomatitis virus through STAT1-OASL2 signaling. Cancer Gene Ther. 2013, 20, 125–132. [Google Scholar]
- Park, E.H.; Park, E.H.; Cho, I.R.; Srisuttee, R.; Min, H.J.; Oh, M.J.; Jeong, Y.J.; Jhun, B.H.; Johnston, R.N.; Lee, S.; et al. CUG2, a novel oncogene confers reoviral replication through Ras and p38 signaling pathway. Cancer Gene Ther. 2010, 17, 307–314. [Google Scholar]
- Haralambieva, I.; Iankov, I.; Hasegawa, K.; Harvey, M.; Russell, S.J.; Peng, K.W. Engineering oncolytic measles virus to circumvent the intracellular innate immune response. Mol. Ther. 2007, 15, 588–597. [Google Scholar] [CrossRef]
- Meng, X.; Nakamura, T.; Okazaki, T.; Inoue, H.; Takahashi, A.; Miyamoto, S.; Sakaguchi, G.; Eto, M.; Naito, S.; Takeda, M.; et al. Enhanced antitumor effects of an engineered measles virus Edmonston strain expressing the wild-type N, P, L genes on human renal cell carcinoma. Mol. Ther. 2010, 18, 544–551. [Google Scholar] [CrossRef]
- Takaki, H.; Watanabe, Y.; Shingai, M.; Oshiumi, H.; Matsumoto, M.; Seya, T. Strain-to-strain difference of V protein of measles virus affects MDA5-mediated IFN-beta-inducing potential. Mol. Immunol. 2011, 48, 497–504. [Google Scholar]
- Shingai, M.; Ebihara, T.; Begum, N.A.; Kato, A.; Honma, T.; Matsumoto, K.; Saito, H.; Ogura, H.; Matsumoto, M.; Seya, T. Differential type I IFN-inducing abilities of wild-type versus vaccine strains of measles virus. J. Immunol. 2007, 179, 6123–6133. [Google Scholar]
- Schuhmann, K.M.; Pfaller, C.K.; Conzelmann, K.K. The measles virus V protein binds to p65 (RelA) to suppress NF-kappaB activity. J. Virol. 2011, 85, 3162–3171. [Google Scholar] [CrossRef]
- Childs, K.; Randall, R.; Goodbourn, S. Paramyxovirus V proteins interact with the RNA Helicase LGP2 to inhibit RIG-I-dependent interferon induction. J. Virol. 2012, 86, 3411–3421. [Google Scholar] [CrossRef]
- Zamarin, D.; Martinez-Sobrido, L.; Kelly, K.; Mansour, M.; Sheng, G.; Vigil, A.; Garcia-Sastre, A.; Palese, P.; Fong, Y. Enhancement of oncolytic properties of recombinant newcastle disease virus through antagonism of cellular innate immune responses. Mol. Ther. 2009, 17, 697–706. [Google Scholar] [CrossRef]
- Kim, S.H.; Samal, S.K. Inhibition of host innate immune responses and pathogenicity of recombinant Newcastle disease viruses expressing NS1 genes of influenza A viruses. J. Gen. Virol. 2010, 91, 1996–2001. [Google Scholar] [CrossRef]
- Guerra, S.; Abaitua, F.; Martinez-Sobrido, L.; Esteban, M.; Garcia-Sastre, A.; Rodriguez, D. Host-range restriction of vaccinia virus E3L deletion mutant can be overcome in vitro, but not in vivo, by expression of the influenza virus NS1 protein. PLoS One 2011, 6, e28677. [Google Scholar]
- Fu, X.; Rivera, A.; Tao, L.; Zhang, X. Incorporation of the B18R gene of vaccinia virus into an oncolytic herpes simplex virus improves antitumor activity. Mol. Ther. 2012, 20, 1871–1881. [Google Scholar] [CrossRef]
- Shah, A.C.; Parker, J.N.; Gillespie, G.Y.; Lakeman, F.D.; Meleth, S.; Markert, J.M.; Cassady, K.A. Enhanced antiglioma activity of chimeric HCMV/HSV-1 oncolytic viruses. Gene Ther. 2007, 14, 1045–1054. [Google Scholar]
- Le Boeuf, F.; Batenchuk, C.; Vähä-Koskela, M.; Breton, S.; Roy, D.; Lemay, C.; Cox, J.; Abdelbary, H.; Falls, T.; Waghray, G.; et al. Model-based rational design of an oncolytic virus with improved therapeutic potential. Nat. Commun. 2013, 4, 1974. [Google Scholar]
- De Vries, W.; Haasnoot, J.; van der, V.; van Montfort, T.; Zorgdrager, F.; Paxton, W.; Cornelissen, M.; van Kuppeveld, F.; de Haan, P.; Berkhout, B. Increased virus replication in mammalian cells by blocking intracellular innate defense responses. Gene Ther. 2008, 15, 545–552. [Google Scholar]
- Shors, S.T.; Beattie, E.; Paoletti, E.; Tartaglia, J.; Jacobs, B.L. Role of the vaccinia virus E3L and K3L gene products in rescue of VSV and EMCV from the effects of IFN-alpha. J. Interferon Cytokine Res. 1998, 18, 721–729. [Google Scholar] [CrossRef]
- Bridle, B.W.; Chen, L.; Lemay, C.G.; Diallo, J.S.; Pol, J.; Nguyen, A.; Capretta, A.; He, R.; Bramson, J.L.; Bell, J.C.; et al. HDAC inhibition suppresses primary immune responses, enhances secondary immune responses, and abrogates autoimmunity during tumor immunotherapy. Mol. Ther. 2013, 21, 887–894. [Google Scholar]
- El-Andaloussi, N.; Bonifati, S.; Kaufmann, J.K.; Mailly, L.; Daeffler, L.; Deryckere, F.; Nettelbeck, D.M.; Rommelaere, J.; Marchini, A. Generation of an adenovirus-parvovirus chimera with enhanced oncolytic potential. J. Virol. 2012, 86, 10418–10431. [Google Scholar] [CrossRef]
- Sanchez-Puig, J.M.; Lorenzo, M.M.; Blasco, R. A vaccinia virus recombinant transcribing an alphavirus replicon and expressing alphavirus structural proteins leads to packaging of alphavirus infectious single cycle particles. PLoS One 2013, 8, e75574. [Google Scholar] [CrossRef]
- Yang, Y.; Xiao, F.; Lu, Z.; Li, Z.; Zuo, H.; Zhang, Q.; Li, Q.; Wang, H.; Wang, L.S. Development of a novel adenovirus-alphavirus hybrid vector with RNA replicon features for malignant hematopoietic cell transduction. Cancer Gene Ther. 2013, 20, 429–436. [Google Scholar]
- Guan, M.; Rodriguez-Madoz, J.R.; Alzuguren, P.; Gomar, C.; Kramer, M.G.; Kochanek, S.; Prieto, J.; Smerdou, C.; Qian, C. Increased efficacy and safety in the treatment of experimental liver cancer with a novel adenovirus-alphavirus hybrid vector. Cancer Res. 2006, 66, 1620–1629. [Google Scholar] [CrossRef]
- Kaufmann, J.K.; Nettelbeck, D.M. Virus chimeras for gene therapy, vaccination, and oncolysis: Adenoviruses and beyond. Trends Mol. Med. 2012, 18, 365–376. [Google Scholar] [CrossRef]
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Vähä-Koskela, M.; Hinkkanen, A. Tumor Restrictions to Oncolytic Virus. Biomedicines 2014, 2, 163-194. https://doi.org/10.3390/biomedicines2020163
AMA Style
Vähä-Koskela M, Hinkkanen A. Tumor Restrictions to Oncolytic Virus. Biomedicines. 2014; 2(2):163-194. https://doi.org/10.3390/biomedicines2020163
Chicago/Turabian StyleVähä-Koskela, Markus, and Ari Hinkkanen. 2014. "Tumor Restrictions to Oncolytic Virus" Biomedicines 2, no. 2: 163-194. https://doi.org/10.3390/biomedicines2020163