
Complementary Approaches to Existing Target Based Drug Discovery for Identifying Novel Drug Targets

Abstract
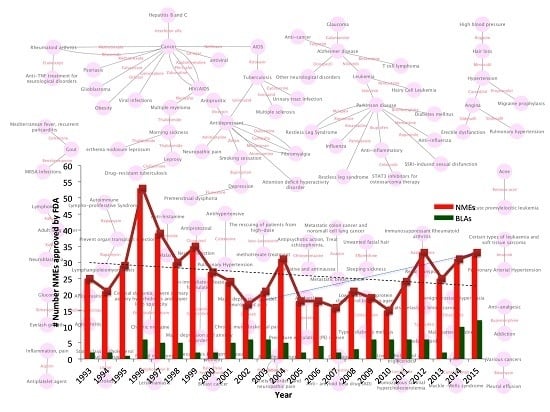
:
1. Introduction
1.1. Target-Based Drug Design
1.2. Target Validation
1.3. Druggability
2. Challenges in Pharma and Possible Solutions
3. Complementary Approaches
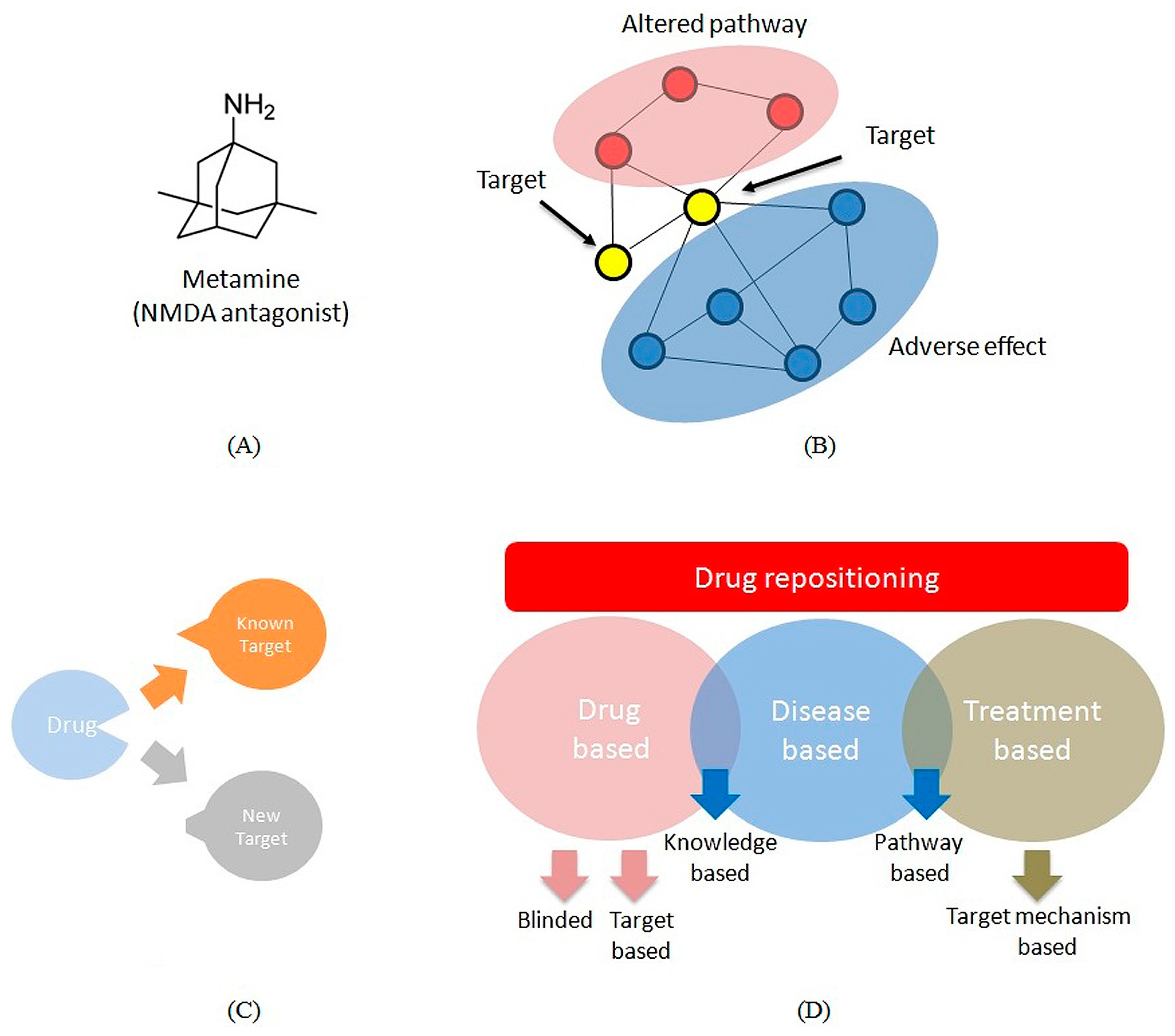
3.1. Multi-Target Drugs
3.2. Repositioning
3.3. Molecular Imaging
3.4. Translational Research
3.5. Human Model Organism
4. Lack of Innovation or Need of an Alternative Approach
5. Conclusions
Supplementary Materials
Author Contributions
Conflicts of Interest
References
- FDA. Innovation and Stagnation: Challenge and Opportunity on the Critical Path to New Medical Products; FDA White Paper; Food and Drug Administration: Silver Spring, MD, USA, 2004. [Google Scholar]
- Caskey, C.T. The drug development crisis: Efficiency and safety. Annu. Rev. Med. 2007, 58, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.M.; Mytelka, D.S.; Dunwiddie, C.T.; Persinger, C.C.; Munos, B.H.; Lindborg, S.R.; Schacht, A.L. How to improve R&D productivity: The pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discov. 2010, 9, 203–214. [Google Scholar] [PubMed]
- Brown, D.; Superti-Furga, G. Rediscovering the sweet spot in drug discovery. Drug Discov. Today 2003, 8, 1067–1077. [Google Scholar] [CrossRef]
- Kola, I.; Landis, J. Can the pharmaceutical industry reduce attrition rates? Nat. Rev. Drug Discov. 2004, 3, 711–715. [Google Scholar] [CrossRef] [PubMed]
- Scannell, J.W.; Blanckley, A.; Boldon, H.; Warrington, B. Diagnosing the decline in pharmaceutical R&D efficiency. Nat. Rev. Drug Discov. 2012, 11, 191–200. [Google Scholar] [PubMed]
- DiMasi, J.A.; Feldman, L.; Seckler, A.; Wilson, A. Trends in risks associated with new drug development: Success rates for investigational drugs. Clin. Pharmacol. Ther. 2010, 87, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Hay, M.; Thomas, D.W.; Craighead, J.L.; Economides, C.; Rosenthal, J. Clinical development success rates for investigational drugs. Nat. Biotechnol. 2014, 32, 40–51. [Google Scholar] [CrossRef] [PubMed]
- CSDD. Cost to Develop and Win Marketing Approval for a New Drug Is $2.6 Billion; Tufts Center for the Study of Drug Development: Boston, MA, USA, 18 November 2014. [Google Scholar]
- Van den Haak, M.A. Industry Success Rates 2004; CMR Report 04-234R; CMR International: Surrey, UK, 2004. [Google Scholar]
- Swinney, D.C.; Anthony, J. How were new medicines discovered? Nat. Rev. Drug Discov. 2011, 10, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Eder, J.; Sedrani, R.; Wiesmann, C. The discovery of first-in-class drugs: Origins and evolution. Nat. Rev. Drug Discov. 2014, 13, 577–587. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.; Gill, J. Rethinking innovation in pharmaceutical R&D. J. Commer. Biotechnol. 2005, 12, 45–49. [Google Scholar]
- Matheson, D.; Loring, B. Hitting the target and missing the point. Nurs. N. Z. 2011, 17, 18–19. [Google Scholar] [PubMed]
- Roti, G.; Stegmaier, K. Genetic and proteomic approaches to identify cancer drug targets. Br. J. Cancer 2012, 106, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Sams-Dodd, F. Target-based drug discovery: Is something wrong? Drug Discov. Today 2005, 10, 139–147. [Google Scholar] [CrossRef]
- Sams-Dodd, F. Drug discovery: Selecting the optimal approach. Drug Discov. Today 2006, 11, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Ecker, D.J.; Griffey, R.H. RNA as a small-molecule drug target: Doubling the value of genomics. Drug Discov. Today 1999, 4, 420–429. [Google Scholar] [CrossRef]
- Singh, S.; Malik, B.K.; Sharma, D.K. Molecular drug targets and structure based drug design: A holistic approach. Bioinformation 2006, 1, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Huynen, M.A.; Diaz-Lazcoz, Y.; Bork, P. Differential genome display. Trends Genet. 1997, 13, 389–390. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Drysdale, M.J.; Lentzen, G.; Matassova, N.; Murchie, A.I.; Aboul-Ela, F.; Afshar, M. RNA as a drug target. Prog. Med. Chem. 2002, 39, 73–119. [Google Scholar] [PubMed]
- Sakharkar, K.R.; Sakharkar, M.K.; Chow, V.T. Biocomputational strategies for microbial drug target identification. Methods Mol. Med. 2008, 142, 1–9. [Google Scholar] [PubMed]
- Read, T.D.; Gill, S.R.; Tettelin, H.; Dougherty, B.A. Finding drug targets in microbial genomes. Drug Discov. Today 2001, 6, 887–892. [Google Scholar] [CrossRef]
- Plunkett, G.; Rose, D.J.; Durfee, T.J.; Blattner, F.R. Sequence of Shiga toxin 2 phage 933W from Escherichia coli O157:H7: Shiga toxin as a phage late-gene product. J. Bacteriol. 1999, 181, 1767–1778. [Google Scholar] [PubMed]
- Kostich, M.; English, J.; Madison, V.; Gheyas, F.; Wang, L.; Qiu, P.; Greene, J.; Laz, T.M. Human members of the eukaryotic protein kinase family. Genome Biol. 2002, 3. [Google Scholar] [CrossRef]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed]
- Sioud, M. Main approaches to target discovery and validation. Methods Mol. Biol. 2007, 360, 1–12. [Google Scholar] [PubMed]
- Schartl, M. Beyond the Zebrafish: Diverse fish species for modeling human disease. Dis. Models Mech. 2014, 7, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Smith, C. Drug target validation: Hitting the target. Nature 2003, 422. [Google Scholar] [CrossRef] [PubMed]
- Justice, M.J.; Siracusa, L.D.; Stewart, A.F. Technical approaches for mouse models of human disease. Dis. Models Mech. 2011, 4, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Klupa, T.; Skupien, J.; Malecki, M.T. Monogenic models: What have the single gene disorders taught us? Curr. Diabetes Rep. 2012, 12, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Jeffery, C.J. Multifunctional proteins: Examples of gene sharing. Ann. Med. 2003, 35, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.W.; Stefani, M.; dos Remedios, C.G.; Charleston, M.A. Differential variability analysis of gene expression and its application to human diseases. Bioinformatics 2008, 24, i390–i398. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D. Applications of antisense and siRNAs during preclinical drug development. Drug Discov. Today 2002, 7, 912–917. [Google Scholar] [CrossRef]
- Hopkins, A.L.; Groom, C.R. The druggable genome. Nat. Rev. Drug Discov. 2002, 1, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Gonzaga-Jauregui, C.; Lupski, J.R.; Gibbs, R.A. Human genome sequencing in health and disease. Annu. Rev. Med. 2012, 63, 35–61. [Google Scholar] [CrossRef] [PubMed]
- Betz, U.A. How many genomics targets can a portfolio afford? Drug Discov. Today 2005, 10, 1057–1063. [Google Scholar] [CrossRef]
- Dipple, K.M.; McCabe, E.R. Modifier genes convert “simple” Mendelian disorders to complex traits. Mol. Genet. Metab. 2000, 71, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Collier, R. Drug development cost estimates hard to swallow. CMAJ 2009, 180, 279–280. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.; Sitzmann, M.; Pugliese, A.; Nicklaus, M.C. Software and resources for computational medicinal chemistry. Future Med. Chem. 2011, 3, 1057–1085. [Google Scholar] [CrossRef] [PubMed]
- Hasson, S.A.; Inglese, J. Innovation in academic chemical screening: Filling the gaps in chemical biology. Curr. Opin. Chem. Biol. 2013, 17, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Conley, J.M.; Brand, C.S.; Bogard, A.S.; Pratt, E.P.; Xu, R.; Hockerman, G.H.; Ostrom, R.S.; Dessauer, C.W.; Watts, V.J. Development of a high-throughput screening paradigm for the discovery of small-molecule modulators of adenylyl cyclase: Identification of an adenylyl cyclase 2 inhibitor. J. Pharmacol. Exp. Ther. 2013, 347, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Fridkin, M.; Youdim, M. From single target to multitarget/network therapeutics in Alzheimer’s therapy. Pharmaceuticals 2014, 7, 113–135. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.J.; Pan, W.; Hu, Y.J.; Wang, Y.T. Multi-target drugs: The trend of drug research and development. PLoS ONE 2012, 7, e40262. [Google Scholar] [CrossRef] [PubMed]
- Csermely, P. Strong links are important, but weak links stabilize them. Trends Biochem. Sci. 2004, 29, 331–334. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.; Wong, S.T. Toward better drug repositioning: Prioritizing and integrating existing methods into efficient pipelines. Drug Discov. Today 2014, 19, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Novac, N. Challenges and opportunities of drug repositioning. Trends Pharmacol. Sci. 2013, 34, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Hemphill, T.A. The NIH promotes drug repurposing and rescue. Res. Technol. Manag. 2012, 55, 6. [Google Scholar]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Southall, N.; Wang, Y.; Yasgar, A.; Shinn, P.; Jadhav, A.; Nguyen, D.T.; Austin, C.P. The NCGC pharmaceutical collection: A comprehensive resource of clinically approved drugs enabling repurposing and chemical genomics. Sci. Transl. Med. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Chong, C.R.; Chen, X.; Shi, L.; Liu, J.O.; Sullivan, D.J. A clinical drug library screen identifies astemizole as an antimalarial agent. Nat. Chem. Biol. 2006, 2, 415–416. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT00570908 (accessed on 17 November 2016).
- Sirota, M.; Dudley, J.T.; Kim, J.; Chiang, A.P.; Morgan, A.A.; Sweet-Cordero, A.; Sage, J.; Butte, A.J. Discovery and preclinical validation of drug indications using compendia of public gene expression data. Sci. Transl. Med. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.; Fu, C.; Zhao, H.; Cui, K.; Chang, J.; Wong, S.T. A novel method of transcriptional response analysis to facilitate drug repositioning for cancer therapy. Cancer Res. 2012, 72, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Russell, W.M.S.; Burch, R.L. The Principles of Humane Experimental Technique; Methuen: London, UK, 1959. [Google Scholar]
- Massoud, T.F.; Gambhir, S.S. Molecular imaging in living subjects: Seeing fundamental biological processes in a new light. Genes Dev. 2003, 17, 545–580. [Google Scholar] [CrossRef] [PubMed]
- Cunha, L.; Horvath, I.; Ferreira, S.; Lemos, J.; Costa, P.; Vieira, D.; Veres, D.S.; Szigeti, K.; Summavielle, T.; Mathe, D.; et al. Preclinical imaging: An essential ally in modern biosciences. Mol. Diagn. Ther. 2014, 18, 153–173. [Google Scholar] [CrossRef] [PubMed]
- Cunha, L.; Szigeti, K.; Mathe, D.; Metello, L.F. The role of molecular imaging in modern drug development. Drug Discov. Today 2014, 19, 936–948. [Google Scholar] [CrossRef] [PubMed]
- Mankoff, S.P.; Brander, C.; Ferrone, S.; Marincola, F.M. Lost in Translation: Obstacles to Translational Medicine. J. Transl. Med. 2004, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, J.E. The promise of translational physiology. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G1127–G1128. [Google Scholar] [PubMed]
- Lotsch, J.; Geisslinger, G. Bedside-to-bench pharmacology: A complementary concept to translational pharmacology. Clin. Pharmacol. Ther. 2010, 87, 647–649. [Google Scholar] [CrossRef] [PubMed]
- Alabaster, V. The In Vivo Pharmacology Training Group. The fall and rise of in vivo pharmacology. Trends Pharmacol. Sci. 2002, 23, 13–18. [Google Scholar] [CrossRef]
- Bowes, J.; Brown, A.; Hamon, J.; Jarolimek, W.; Sridhar, A.; Waldron, G.; Whitebread, S. Reducing safety-related drug attrition: The use of in vitro pharmacological profiling. Nat. Rev. Drug Discov. 2012, 11, 909–922. [Google Scholar] [CrossRef] [PubMed]
- Sherer, T.B. Money without collaboration won’t bring cures. Nat. Med. 2013, 19. [Google Scholar] [CrossRef] [PubMed]
- Cook, D.; Brown, D.; Alexander, R.; March, R.; Morgan, P.; Satterthwaite, G.; Pangalos, M.N. Lessons learned from the fate of AstraZeneca’s drug pipeline: A five-dimensional framework. Nat. Rev. Drug Discov. 2014, 13, 419–431. [Google Scholar] [CrossRef] [PubMed]
- Kislinger, T.; Gramolini, A.O. Proteome analysis of mouse model systems: A tool to model human disease and for the investigation of tissue-specific biology. J. Proteom. 2010, 73, 2205–2218. [Google Scholar] [CrossRef] [PubMed]
- Pound, P.; Ebrahim, S.; Sandercock, P.; Bracken, M.B.; Roberts, I. Where is the evidence that animal research benefits humans? BMJ 2004, 328, 514–517. [Google Scholar] [CrossRef] [PubMed]
- Horrobin, D.F. Modern biomedical research: An internally self-consistent universe with little contact with medical reality? Nat. Rev. Drug Discov. 2003, 2, 151–154. [Google Scholar] [CrossRef] [PubMed]
- Littman, B.H.; Williams, S.A. The ultimate model organism: Progress in experimental medicine. Nat. Rev. Drug Discov. 2005, 4, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Wall, R.J.; Shani, M. Are animal models as good as we think? Theriogenology 2008, 69, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Gonzalez, F.; Alten, R.H.; Bensen, W.G.; Brown, J.P.; Sibley, J.T.; Dougados, M.; Bombardieri, S.; Durez, P.; Ortiz, P.; de-Miquel, G.; et al. Clinical trial of a leucotriene B4 receptor antagonist, BIIL 284, in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2007, 66, 628–632. [Google Scholar] [CrossRef] [PubMed]
- Tarazi, F.I.; Zhang, K.; Baldessarini, R.J. Dopamine D4 receptors: Beyond schizophrenia. J. Recept. Signal Transduct. Res. 2004, 24, 131–147. [Google Scholar] [CrossRef] [PubMed]
- Greaves, P.; Williams, A.; Eve, M. First dose of potential new medicines to humans: How animals help. Nat. Rev. Drug Discov. 2004, 3, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Greek, R.; Menache, A. Systematic reviews of animal models: Methodology versus epistemology. Int. J. Med. Sci. 2013, 10, 206–221. [Google Scholar] [CrossRef] [PubMed]
- Knobloch, J.; Shaughnessy, J.D., Jr.; Ruther, U. Thalidomide induces limb deformities by perturbing the Bmp/Dkk1/Wnt signaling pathway. FASEB J. 2007, 21, 1410–1421. [Google Scholar] [CrossRef] [PubMed]
- Epstein, R.A. Regulatory paternalism in the market for drugs: Lessons from Vioxx and Celebrex. Yale J. Health Policy Law Ethics 2005, 5, 741–770. [Google Scholar] [PubMed]
- Seok, J.; Warren, H.S.; Cuenca, A.G.; Mindrinos, M.N.; Baker, H.V.; Xu, W.; Richards, D.R.; McDonald-Smith, G.P.; Gao, H.; Hennessy, L.; et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc. Natl. Acad. Sci. USA 2013, 110, 3507–3512. [Google Scholar] [CrossRef] [PubMed]
- Brock, A.; Goh, H.T.; Yang, B.; Lu, Y.; Li, H.; Loh, Y.H. Cellular reprogramming: A new technology frontier in pharmaceutical research. Pharm. Res. 2012, 29, 35–52. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Studer, L. Induced pluripotent stem cell technology for the study of human disease. Nat. Methods 2010, 7, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Mack, D.L.; Guan, X.; Wagoner, A.; Walker, S.J.; Childers, M.K. Disease-in-a-dish: The contribution of patient-specific induced pluripotent stem cell technology to regenerative rehabilitation. Am. J. Phys. Med. Rehabil. 2014, 93 (Suppl. 3), S155–S168. [Google Scholar] [CrossRef] [PubMed]
- Raitano, S.; Ordovas, L.; De Muynck, L.; Guo, W.; Espuny-Camacho, I.; Geraerts, M.; Khurana, S.; Vanuytsel, K.; Toth, B.I.; Voets, T.; et al. Restoration of progranulin expression rescues cortical neuron generation in an induced pluripotent stem cell model of frontotemporal dementia. Stem Cell Rep. 2015, 4, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Kumler, I.; Tuxen, M.K.; Nielsen, D.L. A systematic review of dual targeting in HER2-positive breast cancer. Cancer Treat. Rev. 2014, 40, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Bollag, G.; Tsai, J.; Zhang, J.; Zhang, C.; Ibrahim, P.; Nolop, K.; Hirth, P. Vemurafenib: The first drug approved for BRAF-mutant cancer. Nat. Rev. Drug Discov. 2012, 11, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Schmid, E.F.; Smith, D.A. Keynote review: Is declining innovation in the pharmaceutical industry a myth? Drug Discov. Today 2005, 10, 1031–1039. [Google Scholar] [CrossRef]
- Enna, S.J.; Williams, M. The decreased number of new drug approvals (NDAs) has been a topic of considerable debate over the past decade. Preface. Adv. Pharmacol. 2009, 57. [Google Scholar] [CrossRef]
- Stack, R.S.; Harrington, R.A. Biomedical innovation: A risky business at risk. Sci. Transl. Med. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Garnier, J.P. Rebuilding the R&D engine in big pharma. Harv. Bus. Rev. 2008, 86, 68–70. [Google Scholar] [PubMed]
- Owens, P.K.; Raddad, E.; Miller, J.W.; Stille, J.R.; Olovich, K.G.; Smith, N.V.; Jones, R.S.; Scherer, J.C. A decade of innovation in pharmaceutical R&D: The Chorus model. Nat. Rev. Drug Discov. 2015, 14, 17–28. [Google Scholar] [PubMed]
- Peck, R.W.; Lendrem, D.W.; Grant, I.; Lendrem, B.C.; Isaacs, J.D. Why is it hard to terminate failing projects in pharmaceutical R&D? Nat. Rev. Drug Discov. 2015, 14, 663–664. [Google Scholar] [PubMed]
- Mullard, A. 2014 FDA drug approvals. Nat. Rev. Drug Discov. 2015, 14, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. EMA recommended 39 new drug approvals last year. Nat. Rev. Drug Discov. 2016, 15, 77. [Google Scholar] [CrossRef]
- Sekhon, B.S. Repositioning Drugs and Biologics; Retargeting Old/Existing for Potential New Therapeutic Applications. J. Pharm. Educ. Res. 2013, 4, 1–15. [Google Scholar]
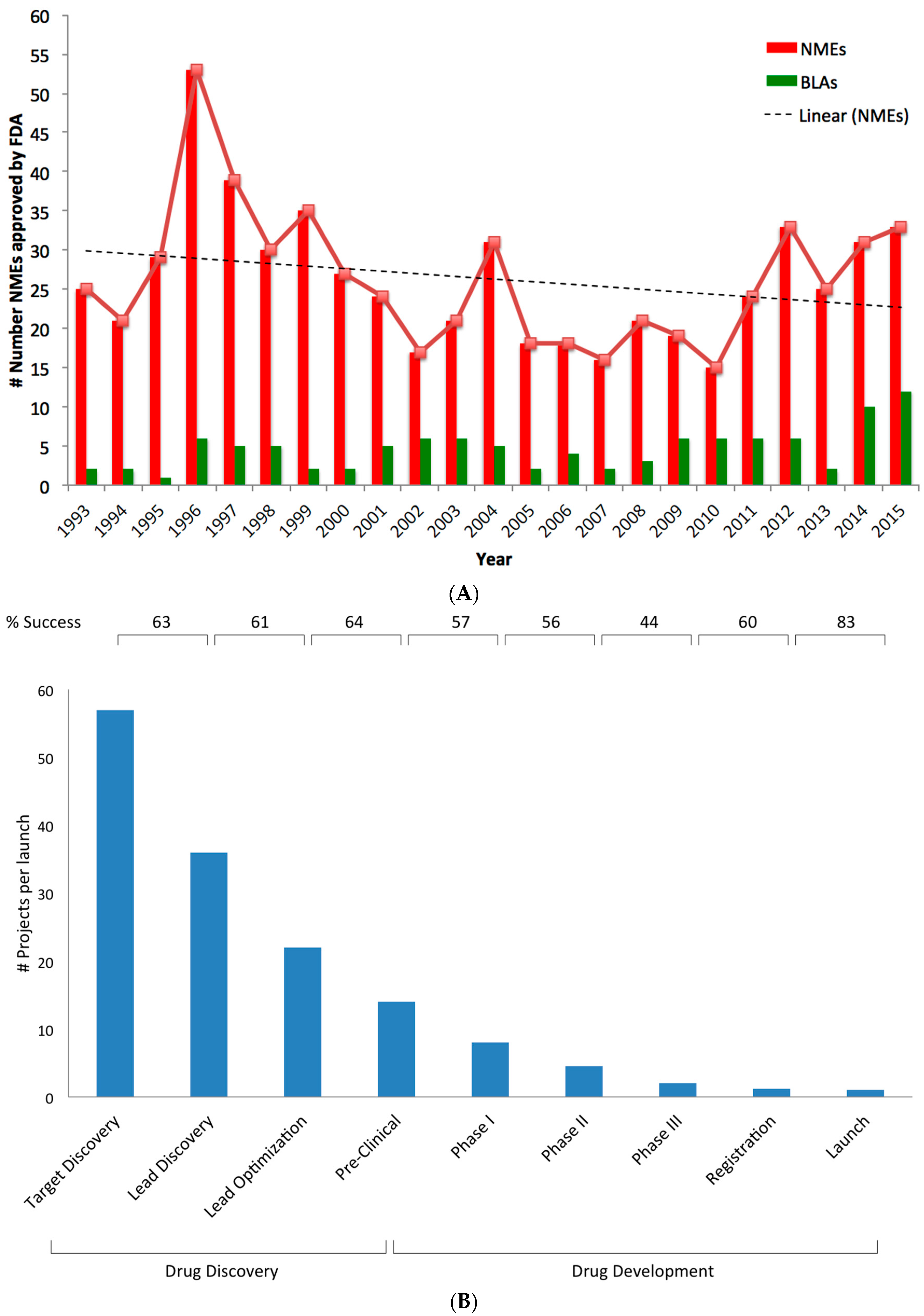
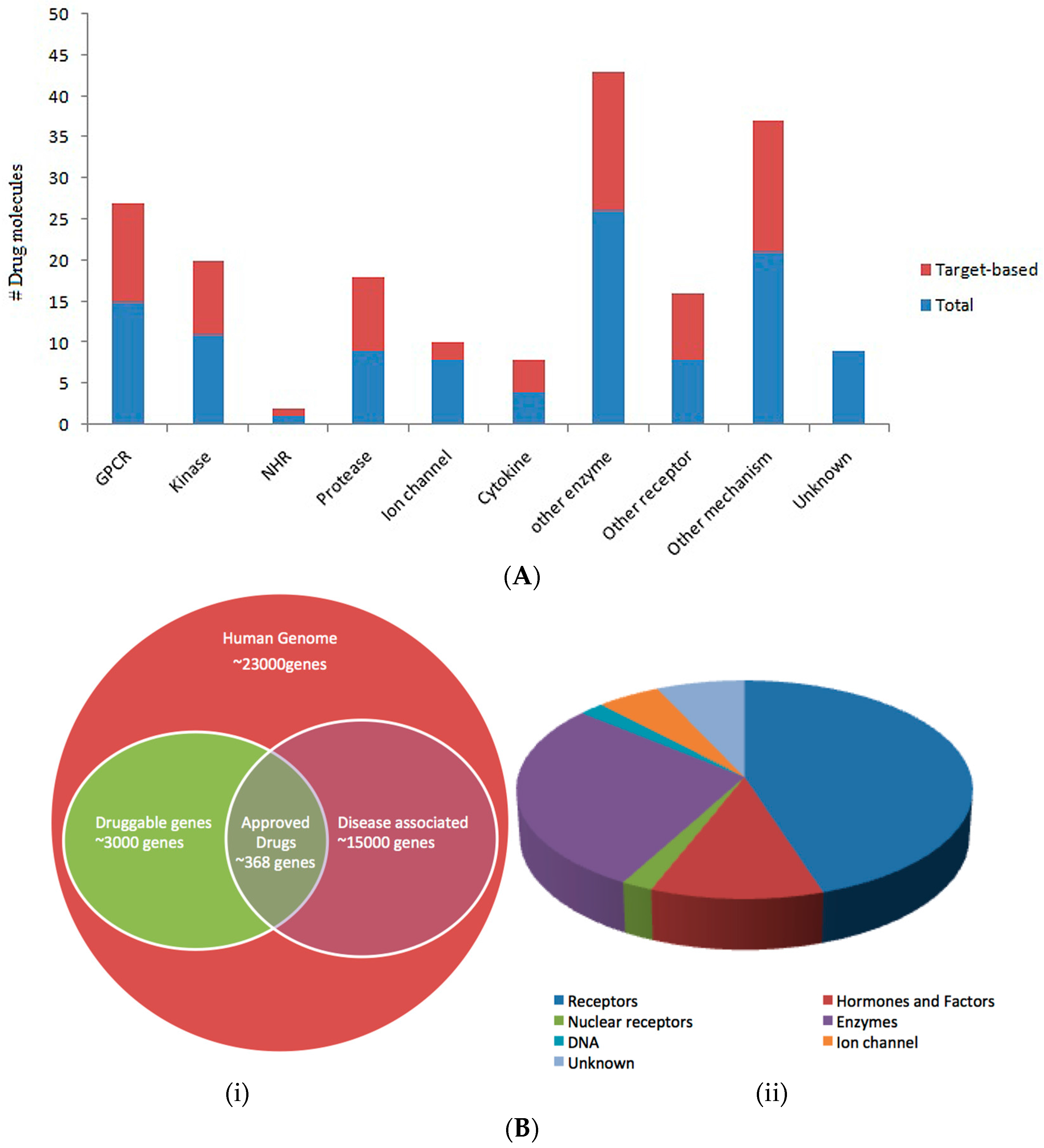
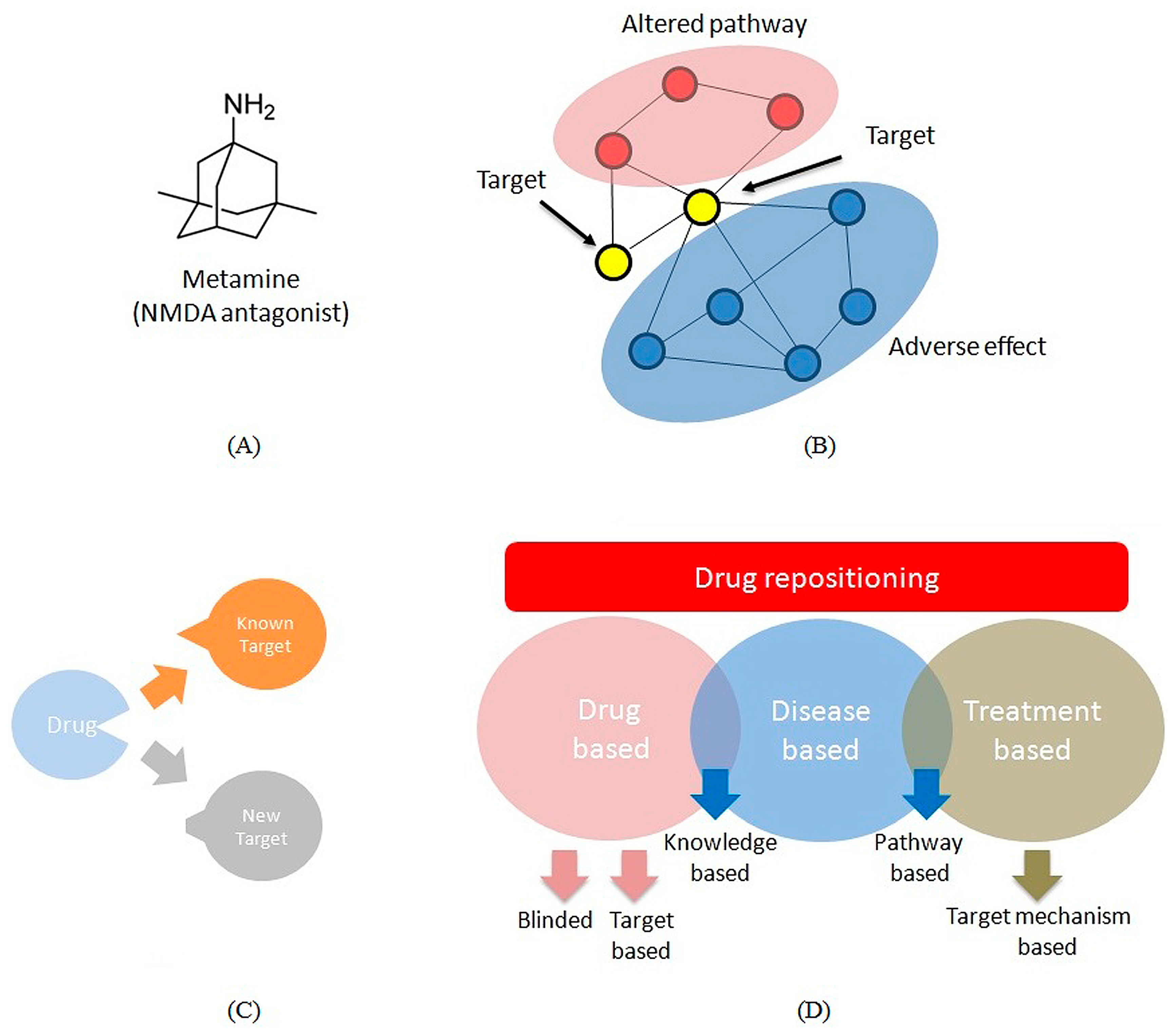
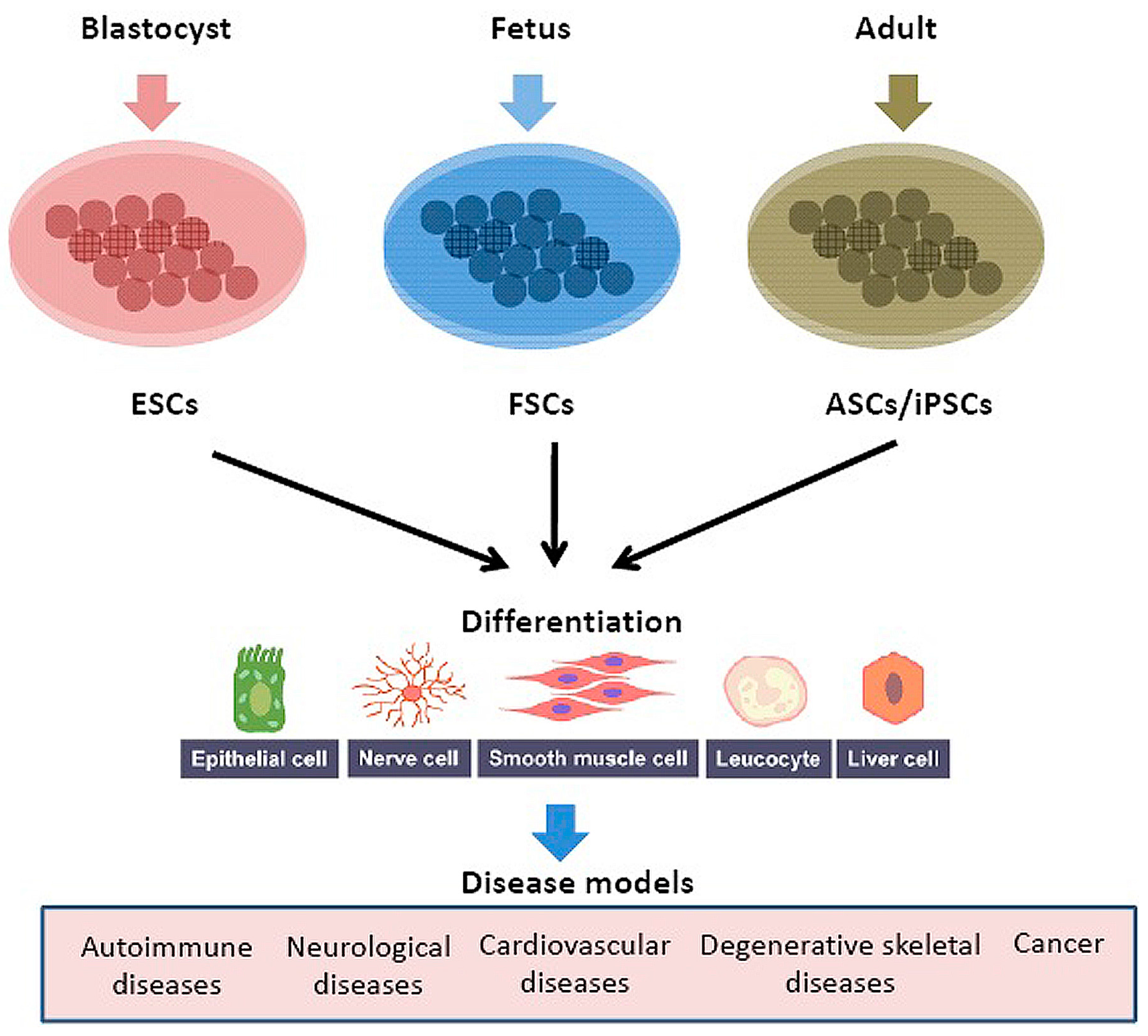
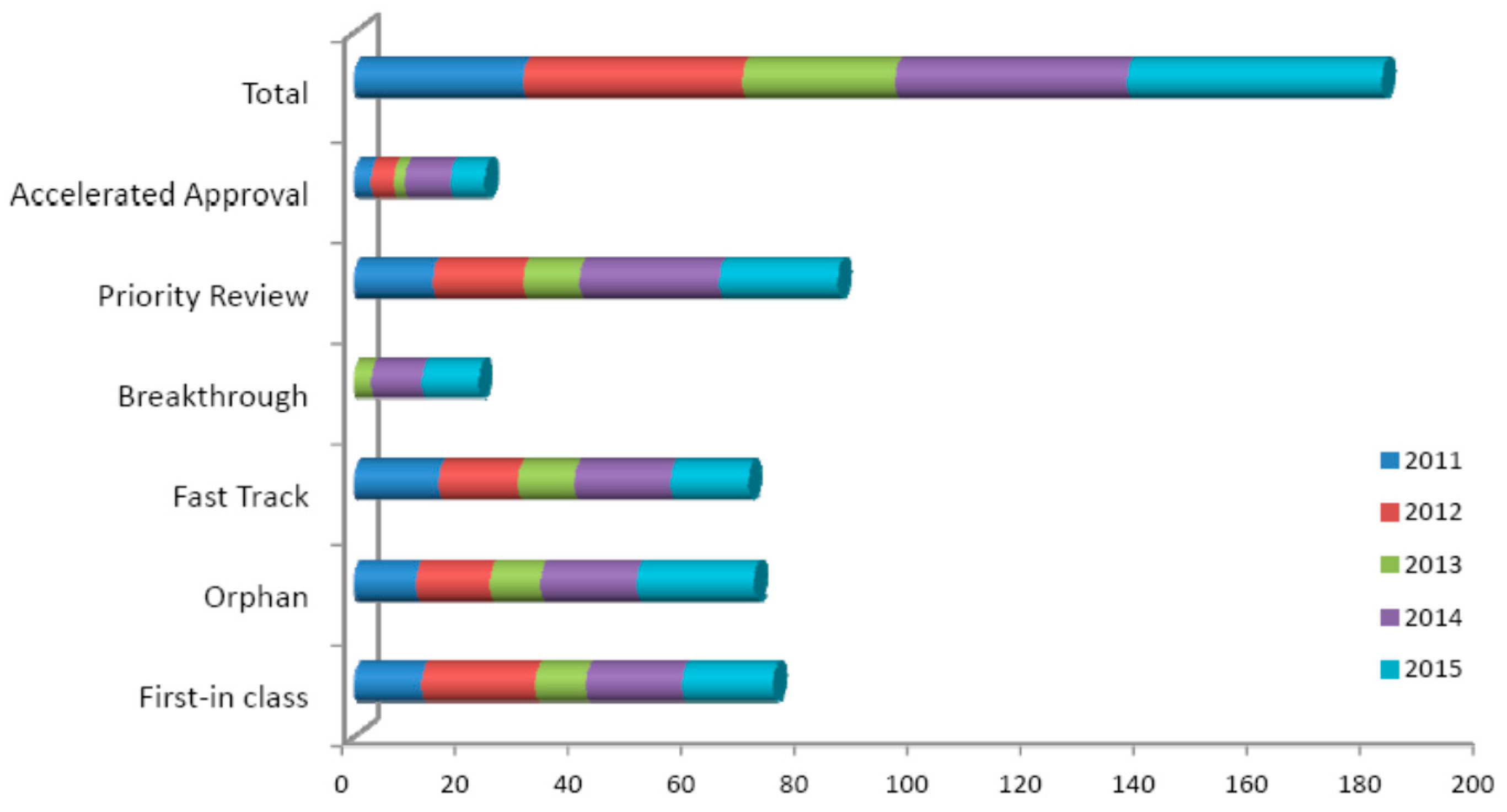

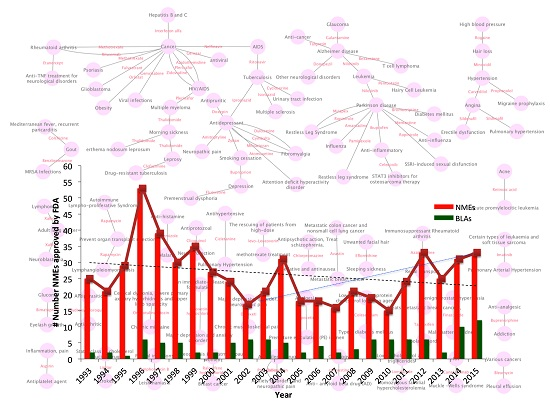{kind=link}
{kind=link}
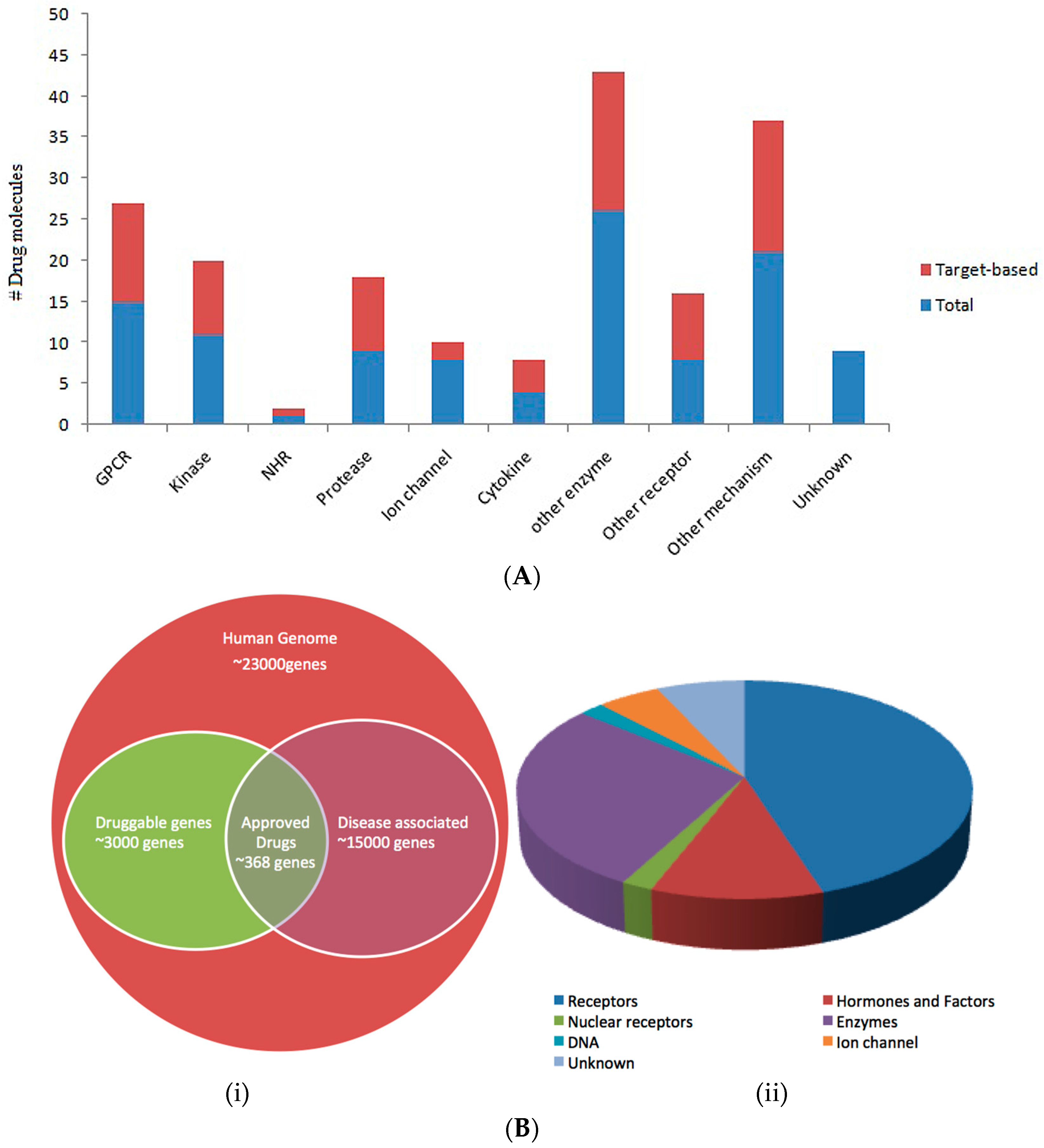{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Imaging | Sensitivity | Probe | Applications |
|---|---|---|---|
| Positron-emission tomography (PET) | pm | Positron emitting radiotracer | Cancers, heart disease, gastrointestinal, endocrine, neurological disorders, any abnormalities within the body |
| 15O, 11C, 18F, 76Br | |||
| Single-photon-emission tomography (SPECT) | pm | Gamma rays emitting radiotracer | Cancers, dopamine transporters (DAT), neurological diseases, pharmacokinetics and pharmacodynamics |
| 123I, 99mTc | |||
| Magnetic resonance imaging (MRI) | µm | Para- or superpara-magnetic radiotracer | Cancers, bleeding, injury, blood vessel diseases, or infection, cellular metabolism |
| Optical imaging (OI) | pm | Fluorophores | Gene expression |
| Drug | Disease | Influential Biomarker |
|---|---|---|
| Lynparza (olaparib) | Ovarian cancer | BRCA |
| Vimizim (elosulfase alpha) | Mucopolysaccharidosis Type IV | Type A or B |
| Cyrazma (ramucirumab) | Gastric or gastro-esophageal junction adenocarcinoma or non-small cell lung cancer (NSCLC) | EGFR or ALK |
| Zykadia (ceritinib) | Gastric or gastro-esophageal junction adenocarcinoma or non-small cell lung cancer (NSCLC) | ALK |
| Beleodaq (belinostat) | Peripheral T-cell lymphoma | UGT1A1 |
| Cerdelga (eliglustat) | Gaucher disease type 1 | CYP2D6 |
| Harvoni (ledipasvir and sofosbuvir) | Chronic hepatitis C infection | Genotype 1 |
| Viekira Pak (ombitasvir, paritaprevir, and ritonavir; dasabuvir) | Chronic hepatitis C infection | Genotype 1 |
| Blincyto (blinatumomab) | B-cell precursor acute lymphoblastic leukemia (ALL) | Philadelphia chromosome biomarker |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vasaikar, S.; Bhatia, P.; Bhatia, P.G.; Chu Yaiw, K. Complementary Approaches to Existing Target Based Drug Discovery for Identifying Novel Drug Targets. Biomedicines 2016, 4, 27. https://doi.org/10.3390/biomedicines4040027
Vasaikar S, Bhatia P, Bhatia PG, Chu Yaiw K. Complementary Approaches to Existing Target Based Drug Discovery for Identifying Novel Drug Targets. Biomedicines. 2016; 4(4):27. https://doi.org/10.3390/biomedicines4040027
Chicago/Turabian StyleVasaikar, Suhas, Pooja Bhatia, Partap G. Bhatia, and Koon Chu Yaiw. 2016. "Complementary Approaches to Existing Target Based Drug Discovery for Identifying Novel Drug Targets" Biomedicines 4, no. 4: 27. https://doi.org/10.3390/biomedicines4040027