
Viroimmunotherapy for Colorectal Cancer: Clinical Studies


Beckman Research Institute, City of Hope National Medical Center, Duarte 91010, CA, USA
*
Author to whom correspondence should be addressed.
Biomedicines 2017, 5(1), 11; https://doi.org/10.3390/biomedicines5010011
Submission received: 1 February 2017
/
Accepted: 2 March 2017
/
Published: 10 March 2017
(This article belongs to the Special Issue Oncolytic Viruses as a Novel Form of Immunotherapy for Cancer)
Abstract
:Colorectal cancer is a leading cause of cancer incidence and death. Therapies for those with unresectable or recurrent disease are not considered curative at present. More effective and less toxic therapies are desperately needed. Historically, the immune system was thought to be an enemy to oncolytic viral therapy. Thinking that oncolysis would be the only mechanism for cell death, oncolytic virologists theorized that immune clearance was a detriment to oncolysis. Recent advances in our understanding of the tumor microenvironment, and the interplay of tumor survival and a patient’s immune system have called into question our understanding of both arenas. It remains unclear what combination of restrictions or enhancements of innate and/or cell-mediated immunity can yield the highest likelihood of viral efficacy. This article reviews the variety of mechanisms explored for viruses such as immunotherapy for colorectal cancer.
1. Introduction
The human immune system has evolved for eons to respond to exogenous infectious organisms like bacteria, fungi, and viruses. Likewise, organisms like viruses have evolved to evade host immune systems. Cancer cells, while historically poorly understood, have a robust ability to evade immune cell destruction. Recently, researchers in the silos of oncolytic viral therapy and cancer immunotherapy have begun to merge the principles of their respective fields, and seek to understand how oncolytic viruses can incite and/or train the host immune system against tumors. Colorectal cancer is the third most common cancer and the third leading cause of cancer death in the United States [1]. Despite enhanced survival across all stages with recent improvements in targeted chemotherapy and aggressive surgical management, patients suffering distant metastases still have a dismal survival of under 15% [1]. Novel therapies are desperately needed to prevent and treat primary colorectal cancer and metastases. This article explores the complex interplay between the available data regarding viroimmunotherapy for colorectal cancer.
Early in the evolution of the field, oncolytic viral therapy researchers focused on oncolysis as the most predominant form of tumor cell killing. Thus, initial efforts were aimed at immunosuppression to prevent viral clearance and allow for increased viral oncolysis. However, as virotherapy and immunotherapy have evolved, investigators have begun to elucidate the complex interactions of viruses, the immune system, and the tumor microenvironment. Unfortunately, given the complexities at hand, the literature contains conflicting information and theories wherein expectations fail to match up with reality [2,3]. For instance, one would expect that viral efficacy in immunodeficient animal models would be dampened in immunocompetent models, and while some data would support this theory [4,5], other authors have shown that this does not always prove to be true [6]. Moreover, one would expect that some component of oncolysis or viral infection would be needed to initiate an immune response, yet elegant data wherein viral agents are unable to infect certain tumor models still achieve anti-tumor effects by initiating a natural killer response [7]. It is likely that some combination of immunosuppression to block viral clearance, facilitation of innate immune activity against tumor cells, and augmentation of humoral immunity to facilitate long term anti-tumor immunity will ultimately yield success. Investigators have taken many different angles to address these questions. From viruses as cancer vaccines to engineered immune cells in combination with viral therapies, there are a number of different approaches that can shed light on some of the fundamental principles of viroimmunotherapy. This article reviews and contextualizes the available preclinical and clinical data regarding immunotherapy and viral immunotherapy for colorectal cancer.
2. Cancer Vaccines
The context of oncolytic viral immunotherapy cannot be fully understood without understanding the groundwork laid in the standard immunotherapy community up to this point. Recent studies have pointed out that the response of a tumor to immunotherapy depends on the immunogenicity of the tumor [8]. Certain types of cancer such as metastatic melanoma and renal cell carcinoma are thought to be highly immunogenic based on the following: (i) occasional spontaneous regression [9,10]; (ii) improved survival associated with infiltrating T-lymphocytes [11,12]; (iii) response to non-antigen specific immunotherapies such as interferon-α, interleukin-2, and anti-cytotoxic T-lymphocyte-associated antigen 4 (CTLA4) [13,14]; (iv) higher incidence of these malignancies in immune-suppressed individuals [13,15]; and (v) presence of tumor-associated antigens and human leukocyte antigen (HLA)-restricted epitopes within these antigens [16,17]. Until recently, colorectal cancer was thought to have low immunogenicity, and was considered a poor candidate for immunotherapy [18]. However, recent studies have shown that the genetic and epigenetic changes contributing to the development of colorectal cancer also result into the formation of neo-antigens that are recognised by the immune system [19,20,21]. These neo-antigens, tumor-associated antigen (TAA), or tumor-specific antigen (TSA), have been shown to elicit anti-tumor immune response. Furthermore, tumor infiltration by immune cells has been shown to correlate with better prognosis in colorectal cancer (CRC) patients [22]. These findings suggest that CRC could be an excellent target for immunotherapy.
The ultimate goal of a therapeutic cancer vaccine is to eliminate the existing tumor and prevent cancer recurrence. Cancer vaccines are inherently difficult to develop because they require an appropriate antigen target, and a robust understanding of immune response and how to manipulate it [23]. Thus, it is assumed that each tumor or tumor cell possess a uniform quality, and that an immune system trained against a single antigen will accomplish eradication. In theory, by infecting cancer cells and targeting immunity against infected cells, the virus-mediated immunity subverts the need for tumor-specific antigen. However, since it is highly unlikely that every tumor cell will get infected with the virus, the use of virus alone would not be sufficient to eliminate tumors. Investigators have tried a variety of vaccine strategies including tumor cells, peptides, dendritic cells, DNA, and viral vector-based vaccines [24].
3. Virus Infected Autologous Tumor Cell Vaccines
One interesting combination of principles came from the employment of virus-infected irradiated tumor cells as autologous cancer vaccine. Schlag et al. used colorectal cancer cells isolated from liver metastases for the vaccination purpose [25]. In this study, cells harvested from resected metastasized tumors were growth-arrested through irradiation. The cells were then infected with the Newcastle disease virus (NDV) and were injected into patients, a total of five times in two week intervals. This study found that the vaccination was well tolerated and there was a reduction in the rate of disease recurrence in vaccine-treated patients compared to the control group. A similar study by Ockert et al. compared the anti-tumor efficacy of autologous cancer cells that were infected with either NDV or bacillus Calmette-Guerin (BCG) [26]. In this study, the NDV-infected autologous cancer cells showed better anti-tumor response than the BCG-infected cells. The authors concluded that the presence of virus-encoded antigens on the surface of the infected cancer cells could have resulted in stronger anti-tumor immune response. Furthermore, the virus itself can stimulate different immune cells including CD4+ T cells, natural killer (NK) cells, and macrophages, all of which can mount an anti-tumor response [26]. Although these initial studies using NDV as adjuvant in autologous cancer cell vaccinations for colorectal cancer showed promising results, a recently completed large scale randomized trial failed to demonstrate the ability of such cancer vaccines to improve overall survival of CRC patients [27].
4. Viral Vector-Based Vaccines
Viruses are the most commonly used vectors for vaccination as they are inherently immunogenic [28,29]. Many types of viruses have the ability to directly infect dendritic cells (DC), the professional antigen presenters. Direct infection of DC by a virus engineered to express TSA or TAA could allow for enhanced presentation of tumor antigens to the T and B cells that could efficiently target the cancer cells [29,30]. Because of the ability of cancer cells to undergo ‘immune-edition’, they may escape the immune cells targeted to a single antigen [24,31]. Therefore, it is desirable to express multiple TAA and TSAs from the vector in order to minimize the risk of immune escape. However, every viral vector has a limited cloning capacity; some viruses may not be able to accommodate the gene for more than one antigen depending on the size of the antigen-encoding gene [32]. Therefore, viral vector should be selected based on several factors including but not limited to cloning capacity, immunogenicity, and pre-existing immunity.
Many viral vector-based therapeutic vaccines have been evaluated for colorectal cancer in pre-clinical studies, and several of them have been studied in different phases of clinical trial. These studies have used a variety of viruses including; poxvirus (vaccinia virus, canarypox virus, fowlpox virus, avipoxvirus, and modified virus Ankara); adenovirus; adeno-associated virus; and retroviruses [33]. These studies have explored many different proteins that are either exclusively or pre-dominantly expressed on colorectal cancer cells (CRC), as potential tumor-specific antigens. The most commonly targeted tumor antigens for therapeutic vaccinations of CRC include carcinoembryonic antigen (CEA), epithelial glycoprotien (Ep-CAM), and guanylyl cyclase 2C (GUCY2C) [34]. Table 1 shows a list of viral vectors encoding different tumor antigens that have been studied in clinical settings as therapeutic vaccines for CRC. Despite showing excellent safety profiles, these viral vectors have fallen short of showing meaningful anti-tumor effects in large scale clinical trials [35,36]. Failure of these vaccines may have been due, in part, to immune-edition by tumor cells as most of the viral vectors were engineered to encode only a single tumor antigen. Furthermore, the vectors completely relied on the immune system to eliminate tumors, as they were not intended for direct killing of cancer cells.
5. Oncolytic Virus
Unlike viral vector-based vaccines, oncolytic viruses (OVs) are designed to directly kill cancer cells by the virtue of their selective replication in cancer cells. Oncolytic viruses are thought to exert their anti-neoplastic activities through a variety of ways. While the exact mechanism of oncolysis differs from virus to virus, and even for the same virus depending on the structure and encoded transgene, there are some common mechanisms employed by most oncolytic viruses to achieve an anti-neoplastic effect. First, replication of many different viruses in a cancer cell can induce lysis of the cell [37]. Second, oncolytic viruses could induce specific and non-specific anti-tumor immunity that can aid to the overall efficacy of the virus. Although the role of immune system has been a matter of debate for a long time in oncolytic virotherapy, recent advancements suggest that the immune system plays a favorable role [38,39]. Oncolytic viruses are often constructed to encode a transgene that can further enhance the anti-tumor effect. A variety of transgenes ranging from immune-stimulatory genes to pro-apoptotic genes have been inserted into different oncolytic viruses to enhance their anti-tumor efficacy. For example, the immune-stimulatory genes IL-2, IL-4, IL-12, and granulocyte macrophage colony-stimulating factor (GM-CSF), and pro-apoptotic genes such as tumor necrosis factor α, p53, and tumor necrosis factor related apoptosis inducing ligands have been studied as therapeutic genes in different oncolytic viruses [40,41,42,43,44,45].
Cell death caused by direct replication of oncolytic viruses is complex and does not clearly fit into anyone of the traditional modes of cell death such as apoptosis, necrosis, or autophagy [46]. This is partly because oncolytic viruses are thought to hijack the cell death machinery, allowing death to occur only when cellular resources have been fully exploited for maximal production of progeny viruses [46]. Several studies have shown that cell deaths caused by oncolytic viruses are “immunogenic” [47,48,49]. A recent study by Tomoki et al. has shown that an oncolytic adenovirus causes immunogenic cell death in CT26 cells, and that the resulting lysate could protect immune-competent mice from tumor formation upon re-challenge with CT26 cells [50]. Several oncolytic viruses in combination with chemotherapeutic agents are currently being evaluated in clinical trials for the treatment of CRC. For example, the oncolytic vaccinia virus Jx-549 in combination with irinotecan has recently completed phase I trial in patients with CRC; the result of this study is yet to be published (NCT01394939). Likewise, Reolysin, an oncolytic reovirus, is also being evaluated in combination with irinotecan, leucovorin, 5-fluorouracil, and bevacizumab for the treatment of K-RAS mutant metastatic CRC (NCT01274624).
6. Combination of Oncolytic Viruses with Immune Checkpoint Inhibitors
Among many immune checkpoint proteins, programmed death 1 (PD-1) and CTLA4 are primarily employed by cancer cells to dampen anti-tumor immune response [51,52]. Consequently, inhibitors of CTLA4 and PD-1 have been shown to exert effective anti-tumor activity against a variety of malignancies in preclinical and clinical studies [51,53]. Ipilimumab, a monoclonal antibody against CTLA4 was the first checkpoint inhibitor to receive FDA approval [54]. It was approved in 2011 for the treatment of melanoma. Since then, several other checkpoint inhibitors have been approved, and many others are awaiting approval for the treatment of different malignancies [54,55]. Unfortunately, checkpoint inhibitors have not shown robust clinical efficacy in the treatment of colorectal cancer [56,57]. A recently completed phase II trial reported that tremelimumab, a fully humanised antibody against CTLA4, was unable to achieve objective responses in metastatic colorectal cancer patients [58]. Out of 45 evaluable patients, only one patient showed partial response, while the remaining patients all had progressive disease. Similarly, antibodies targeting the interaction between PD-1 and its ligand PD-L1 have shown little therapeutic benefits in unselected colorectal cancer patients [59]. Interestingly, PD-1 inhibitors have shown encouraging results against a small fraction of CRC patients with microsatellite unstable tumors [60]. Approximately 15% of all colorectal cancer cases have microsatellite instability, and previous studies have shown that microsatellite instability makes the tumor more immunogenic [61,62,63].
Recent studies have indicated that immune-activation is an important aspect in determining the anti-tumor efficacy of oncolytic viruses [39]. However, tumors often suppress the anti-tumor immune response especially through the checkpoint axis [64]. Therefore, it seems logical that the combination of the oncolytic virus with checkpoint inhibitors would result in more effective treatment of cancer. One of the earliest studies combining an oncolytic virus with checkpoint inhibitors was the combination of an oncolytic vesicular stomatitis virus (VSV) with anti-CTLA4 [65]. In this study, the combination resulted in complete regression of breast tumors in 80% of mice, while the combination of the same oncolytic virus with antibody against TGF-β or IL-10 resulted in complete tumor regression in less than 20% mice. Likewise, in 2014, a study by Zamarain et al. showed that the overall anti-tumor efficacy of an oncolytic NDV could be enhanced by combination with checkpoint inhibitors [66]. In this study, the authors used mouse models of melanoma (B16 cells) and colon cancer (MC38 cells) to investigate the therapeutic efficacy of an oncolytic NDV in combination with an anti-CTLA4 antibody. A combination of intra-tumoral injections of the oncolytic virus and systemically delivered anti-CTLA4 was found to be superior in controlling both injected and un-injected tumors compared to treatment with either the virus or anti-CTLA4 alone. The therapeutic effect of the combination therapy was dependent on tumor infiltration by CD8+ and CD4+ T cells, NK cells, and levels of type I interferon, and it was less dependent on the sensitivity of the cancer cells to NDV-mediated lysis. This study once again highlighted the importance of the immune system in the success of oncolytic viruses. Another study by Rojas et al. demonstrated that the combination of an oncolytic vaccinia virus with a CTLA4 inhibitor enhances anti-tumor response in mouse models of colon (MC38 cells) and renal (RENCA cells) cancer [67]. These pre-clinical studies have provided a strong rationale for testing the combination of oncolytic viruses with checkpoint inhibitors in clinical settings. Indeed, several early stage clinical trials are ongoing to evaluate the combination of oncolytic viruses with checkpoint (mainly PD-1 and CTLA4) inhibitors in a wide range of solid tumors. One such combination, that includes colorectal cancer patients, is the combination of enadenotucirev (a chimeric adenovirus) with nivolumab (anti-PD-1 antibody) in phase I clinical trial (NCT02636036).
7. Combination of Oncolytic Viruses with T Cells Expressing Chimeric Antigen Receptor
Like oncolytic viruses, T-cells engineered to express chimeric antigen receptors (CAR-T), are a novel class of experimental therapeutics. In preclinical and clinical studies CAR-T cells have shown very exciting results, especially in the treatment of hematological malignancies [77,78]. In contrast, for hematological malignancies, the effect of T cells in solid cancer has been modest, at best [79,80]. The poor performance of CAR-T cells in solid tumors is thought to be due, in part, to the immune-suppressed microenvironment in solid tumors and sub-optimal migration of the CAR-T cells to the tumors [81]. In order to ensure that high numbers of CAR-T cells reach target tumors, usually very high numbers of (up to 10 billion) CAR-T cells are injected into patients [81,82]. Such high doses of these cells often result into serious toxicities and could even be fatal. For instance, a colon cancer patient with metastatic lesions in their lungs and liver was injected intravenously with 1 × 1010 CAR-T cells expressing receptor for ERBB2 (an antigen over-expressed on many solid cancers including colon cancer). The patient suffered respiratory distress and cytokine storm, and ultimately succumbed to the treatment-related toxicities [82]. Similarly, in a separate study, three colorectal cancer patients were injected with 2–4 × 108 CAR-T cells targeted to CEA; while only one patient showed partial response, all three patients experienced severe inflammatory colitis [83]. Despite having immense therapeutic potential, as evidenced from the trials with hematological malignancies, the usage of CAR expressing T-cells (CAR-T) cells in the treatment of solid cancers such as colorectal cancer has been plagued by the toxicities associated with high doses of these cells [80,81]. One way to bypass the severe toxicities would be to combine CAR-T cells with other therapeutics, ones that could potentially mitigate factors limiting the efficacy of CAR-T cells and allow achievement of meaningful anti-tumor response with low numbers of T cells.
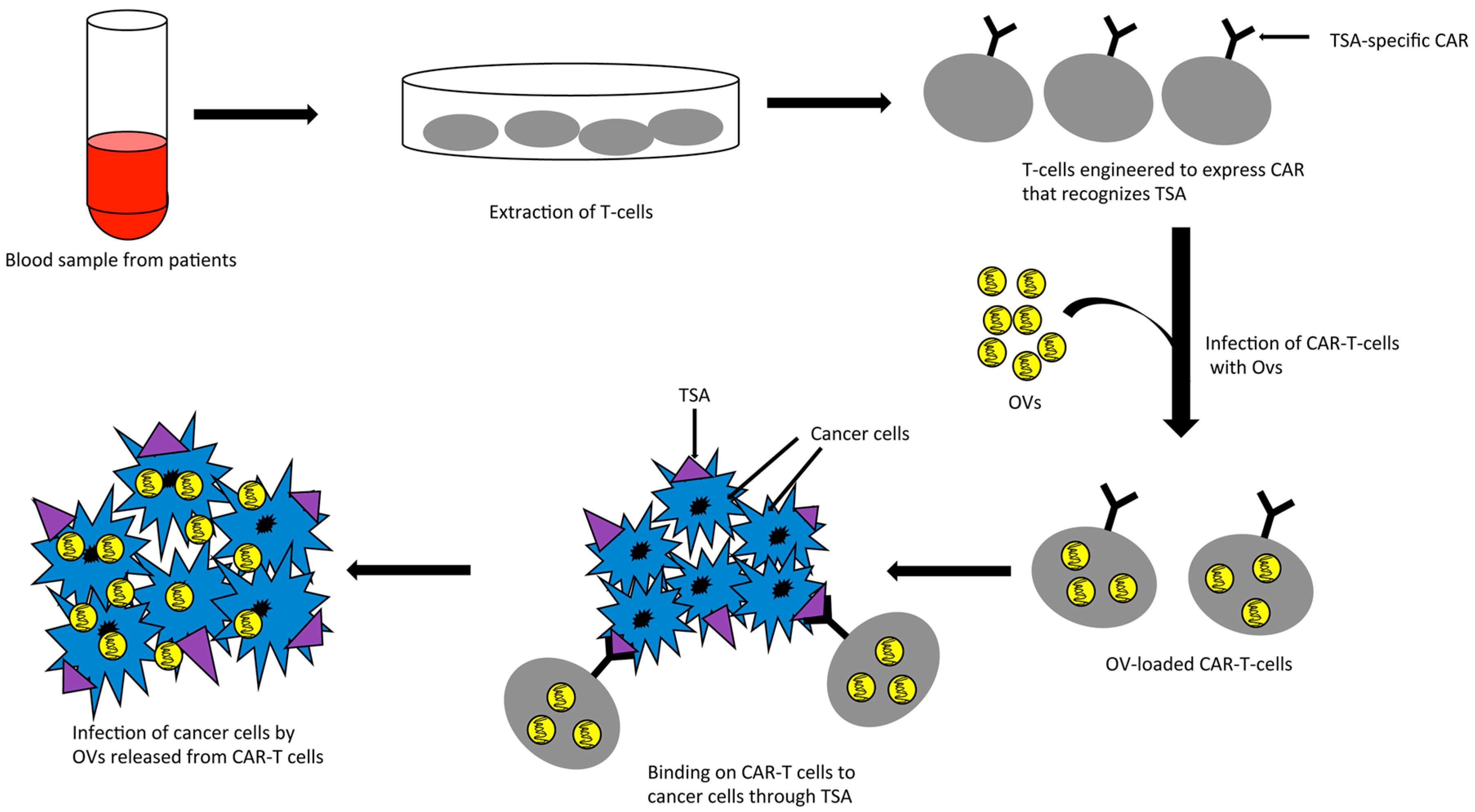
Interestingly, while the optimal anti-tumor activities of oncolytic viruses, especially in the face of preexisting antiviral immunity, are often limited by induction of immunity, the anti-tumor activities of CAR-T cells are often limited by an immune-suppressed environment within solid tumors. The opposing roles of the immune system on these two therapeutic agents suggest that a combination of CAR-T cells and oncolytic viruses may be complementary, and use the opposing roles of the immune system to their advantage. Indeed, a combination of an oncolytic adenovirus, encoding the cytokines IL-15 and RANTES, with CAR-T cells targeted to GD2 antigen has shown better anti-tumor efficacy than either of the treatments alone in a mouse model of neuroblastoma [84]. Another elegant strategy of combining oncolytic virus with CAR-T cells is the “Trojan horse” approach in which the CAR-T cells could be used as a carrier to safely deliver oncolytic viruses to the target tumors where both the virus and the T cells could wage the war against tumor cells (Figure 1). The use of CAR-T cells should protect the virus from antibody-mediated neutralization and anti-viral cellular immunity in the blood stream. The feasibility of such an approach was first demonstrated by Thorne et al. in a study where they used “cytokine induced killer” (CIK) cells as the carrier for delivering an oncolytic vaccinia virus to tumors through systemic routes in a mouse model [85]. Although they did not use CAR-T cells in their study, the concept of their study could be extrapolated for CAR-T cells as CIKs show inherent characteristics of tumor homing and cancer cell killing [86]. In line with this, a recent study by VanSeggelen et al. shows that oncolytic viruses with either RNA or DNA genome can be loaded onto CAR-T cells and efficiently transferred to tumor cells [87]. The anti-tumor effect of these novel combinations of oncolytic virus and CAR-T cells is yet to be evaluated in case of colorectal cancer.
8. Immune Analysis in Clinical Trials Examining Oncolytic Virus Versus Colorectal Cancer
Many oncolytic viral clinical trials have taken place in recent years. Few of them have been focused exclusively on colorectal cancer, and even fewer have gathered comprehensive if any information regarding immune response to viral therapy outside of the aforementioned trials utilizing viral vaccine vectors. Table 2 details trials of oncolytic virus versus colorectal cancer. While some of the more heterogeneous trials did include occasional colorectal cancer patients, their results did not include colorectal patients as an isolated group and have thus been excluded from this discussion. Much of our understanding of immune response to viral therapy is speculative. Because equipoise is hard to prove in patients with resectable diseases, most oncolytic viral trials involve patients who have failed multiple lines of traditional therapies and are not surgical candidates. For this reason, it is difficult to justify any tissue procurement. We are thus left with serum markers of immune reactivity and speculation.
In 2006, Kemeny and Fong detailed their experience injecting NV1020, an herpes simplex virus-1 (HSV-1) vector, via hepatic arterial infusion pumps. They found that fluctuation in IL-2, IFN-γ, and TNF-α were minor and did not exhibit a consistent pattern in relation to virus administration. Furthermore, CD4+ and CD8+ ratios varied inconsistently and in minor ways [88]. They saw impressive disease stability and responses in the highest dosing groups, but unfortunately, we do not glean much information in the way of immune response from this trial because tissue biopsies were not routinely performed.
Park et al. released their findings following administration of JX-549, an oncolytic and immunotherapeutic vaccinia virus expressing GM-CSF and β-galactosidase [89]. They found that GM-CSF was induced acutely after each infusion, though it was unclear if this was an endogenous expression, or an early expression resulting from viral infection [89]. The dose escalation involved four total injections. Eleven of the 15 trial patients completed all four planned injections. The remaining four patients left the trial early, secondary to disease progression. Interestingly, they found that certain collections of cytokines were elevated after the first doses, and lowered after the second doses (IL-6, IL-8, IL-18, macrophage inflammatory protein-1α, monocyte chemoattractant protein-1, MIP-1β, and TNF-α). This in contrast to a separate group which were higher after cycles 2–4 than after cycle one (IL-2, IL-10, IFN-γ). While there did appear to be dose-dependent tumor size stability in this study, no tumor tissue was obtained. That being said, this is one of the more comprehensive cytokine profile analyses and may serve as a foundation for later understanding of immune responses to vaccinia therapy [89].
Balint et al. evaluated a CEA-targeting adenovirus vaccine and specifically sought to analyze immunogenicity. They specifically examined cytolytic T cell responses, T-regulatory (Treg) and T-effector cell ratios in the context of HLA-A2 status. They noted a dose-dependent increase in CEA-specific cell-mediated immunity (CMI) with the majority of patients in the highest dose cohort experiencing CMI as indicated by high levels of IFN-γ secreting spot-forming cells [90]. They also showed significantly elevated granzyme B secretion post-immunization compared to baseline samples. This did not appear to change based on presence of HLA-A2. It does not appear that there was a consistent change in Treg to T-effector cell ratios over the treatment course [90]. They further noted that a high level of cell-mediated immunity as measured by IFN-γ secretion was required before detection of activated T cells.
Of note, in 2002, Pecora et al. administered PV701, a Newcastle Disease Virus to 79 patients, of whom 23 had colorectal cancer. They reported cytokine data for what they called 10 representative patients who were given one or more doses of 12 × 109 or 24 × 109 PFU/m2. In these patients, they measured IFN-α, IFN-β, IFN-γ, IL-6, and TNF-α and they did not stratify their results by disease type. They did however observe that IFN-α was the predominant cytokine produced and that detectable increases in all but IFN-β were seen within 6 h of dosing, usually peaking around 20 h and returning to baseline 2–3 days after dosing. Conversely, they reported that IFN-β was only detectable 20 h after dosing [91]. This is another demonstration that the focus of early oncolytic therapies during clinical trials was not on the complex interplay of immune mediators. Thus, it is difficult to draw meaningful conclusions about viroimmunotherapy from these data.
Calvo et al. have preliminarily reported data from their phase I dose-escalating study of an oncolytic Ad11/Ad3 chimeric group B adenovirus given intravenously to patients with metastatic epithelial tumors [92]. They report results from 34 patients, 26 of whom have colorectal disease. The virus was administered 3 days in a row as a single cycle. They report significant increase in cytokines (TNF, IFN, IL6, IL12) occurring on days in higher dosing groups, that attenuates with day 3 and 5 dosing and prolonged infusion duration. Further expanded results are awaited, but thus far no major immunologic conclusions have been published. Another trial looking at a reovirus vector combined with FOLFIRI and bevacizumab in FOLFIRI-naïve patients with KRAS mutant metastatic colorectal cancer is also being administered, and the oncolytic viral community eagerly awaits these results.
9. Conclusions
Oncolytic viroimmunotherapy for colorectal cancer is a rapidly evolving concept bridging two rapidly evolving fields. It is likely that some combination of the therapies listed above will ultimately be employed. However, in the meantime it is important for researchers in the field to more comprehensively address cytokine changes in the context of innate and cellular immunity. Designing trials that facilitate treated tumor procurement would be ideal correlates for the serum analyses. The coming decade is likely to bring a wealth of new information. It will be important for immunologists, virologists, oncologists, and surgeons to work closely together to advance the field and improve survival for colorectal cancer patients.
Acknowledgments
The authors would like to thank Sang-In Kim for his assistance creating the graphics for Figure 1.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Siegel, R.; Desantis, C.; Jemal, A. Colorectal cancer statistics, 2014. CA Cancer J. Clin. 2014, 64, 104–117. [Google Scholar] [CrossRef] [PubMed]
- Melcher, A.; Parato, K.; Rooney, C.M.; Bell, J.C. Thunder and lightning: Immunotherapy and oncolytic viruses collide. Mol. Ther. J. Am. Soc. Gene Ther. 2011, 19, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Tsun, A.; Miao, X.N.; Wang, C.M.; Yu, D.C. Oncolytic Immunotherapy for Treatment of Cancer; Zhang, S., Ed.; Springer: Dordrecht, The Netherlands, 2016. [Google Scholar]
- Fulci, G.; Breymann, L.; Gianni, D.; Kurozomi, K.; Rhee, S.S.; Yu, J.; Kaur, B.; Louis, D.N.; Weissleder, R.; Caligiuri, M.A.; et al. Cyclophosphamide enhances glioma virotherapy by inhibiting innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12873–12878. [Google Scholar] [CrossRef] [PubMed]
- Lun, X.Q.; Jang, J.H.; Tang, N.; Deng, H.; Head, R.; Bell, J.C.; Stojdl, D.F.; Nutt, C.L.; Senger, D.L.; Forsyth, P.A.; et al. Efficacy of systemically administered oncolytic vaccinia virotherapy for malignant gliomas is enhanced by combination therapy with rapamycin or cyclophosphamide. Clin. Cancer Res. 2009, 15, 2777–2788. [Google Scholar] [CrossRef]
- Parato, K.A.; Senger, D.; Forsyth, P.A.; Bell, J.C. Recent progress in the battle between oncolytic viruses and tumours. Nat. Rev. Cancer 2005, 5, 965–976. [Google Scholar] [CrossRef] [PubMed]
- Prestwich, R.J.; Errington, F.; Diaz, R.M.; Pandha, H.S.; Harrington, K.J.; Melcher, A.A.; Vile, R.G. The case of oncolytic viruses versus the immune system: Waiting on the judgment of Solomon. Hum. Gene Ther. 2009, 20, 1119–1132. [Google Scholar] [CrossRef] [PubMed]
- Blankenstein, T.; Coulie, P.G.; Gilboa, E.; Jaffee, E.M. The determinants of tumour immunogenicity. Nat. Rev. Cancer 2012, 12, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Komenaka, I.; Hoerig, H.; Kaufman, H.L. Immunotherapy for melanoma. Clin. Dermatol. 2004, 22, 251–265. [Google Scholar] [CrossRef] [PubMed]
- Janiszewska, A.D.; Poletajew, S.; Wasiutynski, A. Spontaneous regression of renal cell carcinoma. Contemp. Oncol. (Pozn) 2013, 17, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Day, C.L., Jr.; Sober, A.J.; Kopf, A.W.; Lew, R.A.; Mihm, M.C., Jr.; Hennessey, P.; Golomb, F.M.; Harris, M.N.; Gumport, S.L.; Raker, J.W.; et al. A prognostic model for clinical stage I melanoma of the upper extremity. The importance of anatomic subsites in predicting recurrent disease. Ann. Surg. 1981, 193, 436–440. [Google Scholar] [CrossRef] [PubMed]
- Belldegrun, A.; Muul, L.M.; Rosenberg, S.A. Interleukin 2 expanded tumor-infiltrating lymphocytes in human renal cell cancer: Isolation, characterization, and antitumor activity. Cancer Res. 1988, 48, 206–214. [Google Scholar] [PubMed]
- Itsumi, M.; Tatsugami, K. Immunotherapy for renal cell carcinoma. Clin. Dev. Immunol. 2010, 2010. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L. Vaccines for melanoma and renal cell carcinoma. Semin. Oncol. 2012, 39, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.A.; Page, D.L.; Holt, J.T. Identification of genes expressed in premalignant breast disease by microscopy-directed cloning. Proc. Natl. Acad. Sci. USA 1994, 91, 9257–9261. [Google Scholar] [CrossRef] [PubMed]
- Bernhard, H.; Maeurer, M.J.; Jager, E.; Wolfel, T.; Schneider, J.; Karbach, J.; Seliger, B.; Huber, C.; Storkus, W.S.; Lotze, M.T.; et al. Recognition of human renal cell carcinoma and melanoma by HLA-A2-restricted cytotoxic T lymphocytes is mediated by shared peptide epitopes and up-regulated by interferon-γ. Scand. J. Immunol. 1996, 44, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A. Development of cancer immunotherapies based on identification of the genes encoding cancer regression antigens. J. Natl. Cancer Inst. 1996, 88, 1635–1644. [Google Scholar] [CrossRef] [PubMed]
- De Vries, N.L.; Swets, M.; Vahrmeijer, A.L.; Hokland, M.; Kuppen, P.J. The Immunogenicity of Colorectal Cancer in Relation to Tumor Development and Treatment. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Kloor, M.; Michel, S.; von Knebel Doeberitz, M. Immune evasion of microsatellite unstable colorectal cancers. Int. J. Cancer 2010, 127, 1001–1010. [Google Scholar] [CrossRef] [PubMed]
- Dalerba, P.; Maccalli, C.; Casati, C.; Castelli, C.; Parmiani, G. Immunology and immunotherapy of colorectal cancer. Crit. Rev. Oncol. Hematol. 2003, 46, 33–57. [Google Scholar] [CrossRef]
- Jochems, C.; Schlom, J. Tumor-infiltrating immune cells and prognosis: The potential link between conventional cancer therapy and immunity. Exp. Biol. Med. 2011, 236, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Van der Burg, S.H.; Arens, R.; Ossendorp, F.; van Hall, T.; Melief, C.J. Vaccines for established cancer: Overcoming the challenges posed by immune evasion. Nat. Rev. Cancer 2016, 16, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Manjili, M.H.; Subjeck, J.R.; Sarkar, D.; Fisher, P.B.; Wang, X.Y. Therapeutic cancer vaccines: Past, present, and future. Adv. Cancer Res. 2013, 119, 421–475. [Google Scholar] [PubMed]
- Schlag, P.; Manasterski, M.; Gerneth, T.; Hohenberger, P.; Dueck, M.; Herfarth, C.; Liebrich, W.; Schirrmacher, V. Active specific immunotherapy with Newcastle-disease-virus-modified autologous tumor cells following resection of liver metastases in colorectal cancer. First evaluation of clinical response of a phase II-trial. Cancer Immunol. Immunother. CII 1992, 35, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Ockert, D.; Schirrmacher, V.; Beck, N.; Stoelben, E.; Ahlert, T.; Flechtenmacher, J.; Hagmüller, E.; Buchcik, R.; Nagel, M.; Saeger, H.D. Newcastle disease virus-infected intact autologous tumor cell vaccine for adjuvant active specific immunotherapy of resected colorectal carcinoma. Clin. Cancer Res. 1996, 2, 21–28. [Google Scholar] [PubMed]
- Schulze, T.; Kemmner, W.; Weitz, J.; Wernecke, K.D.; Schirrmacher, V.; Schlag, P.M. Efficiency of adjuvant active specific immunization with Newcastle disease virus modified tumor cells in colorectal cancer patients following resection of liver metastases: Results of a prospective randomized trial. Cancer Immunol. Immunother. CII 2009, 58, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Ura, T.; Okuda, K.; Shimada, M. Developments in Viral Vector-Based Vaccines. Vaccines (Basel) 2014, 2, 624–641. [Google Scholar] [CrossRef] [PubMed]
- Larocca, C.; Schlom, J. Viral vector-based therapeutic cancer vaccines. Cancer J. 2011, 17, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Moss, B. Genetically engineered poxviruses for recombinant gene expression, vaccination, and safety. Proc. Natl. Acad. Sci. USA 1996, 93, 11341–11348. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.; Emi, M.; Tanabe, K. Cancer immunoediting from immune surveillance to immune escape. Immunology 2007, 121, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kay, M.A.; Glorioso, J.C.; Naldini, L. Viral vectors for gene therapy: The art of turning infectious agents into vehicles of therapeutics. Nat. Med. 2001, 7, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Xiang, B.; Snook, A.E.; Magee, M.S.; Waldman, S.A. Colorectal cancer immunotherapy. Discov. Med. 2013, 15, 301–308. [Google Scholar] [PubMed]
- Rahma, O.E.; Khleif, S.N. Therapeutic vaccines for gastrointestinal cancers. Gastroenterol. Hepatol. (N Y) 2011, 7, 517–564. [Google Scholar]
- Lynch, D.; Murphy, A. The emerging role of immunotherapy in colorectal cancer. Ann. Transl. Med. 2016, 4, 305. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.L.; Gulley, J.L.; Arlen, P.M.; Beetham, P.K.; Tsang, K.Y.; Slack, R.; Hodge, J.W.; Doren, S.; Grosenbach, D.W.; Hwang, J.; et al. Phase I study of sequential vaccinations with fowlpox-CEA(6D)-TRICOM alone and sequentially with vaccinia-CEA(6D)-TRICOM, with and without granulocyte-macrophage colony-stimulating factor, in patients with carcinoembryonic antigen-expressing carcinomas. J. Clin. Oncol. 2005, 23, 720–731. [Google Scholar] [CrossRef] [PubMed]
- Mullen, J.T.; Tanabe, K.K. Viral oncolysis. Oncologist 2002, 7, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef] [PubMed]
- Lichty, B.D.; Breitbach, C.J.; Stojdl, D.F.; Bell, J.C. Going viral with cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Andreansky, S.; He, B.; van Cott, J.; McGhee, J.; Markert, J.M.; Gillespie, G.Y.; Roizman, B.; Whitley, R.J. Treatment of intracranial gliomas in immunocompetent mice using herpes simplex viruses that express murine interleukins. Gene Ther. 1998, 5, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.N.; Gillespie, G.Y.; Love, C.E.; Randall, S.; Whitley, R.J.; Markert, J.M. Engineered herpes simplex virus expressing IL-12 in the treatment of experimental murine brain tumors. Proc. Natl. Acad. Sci. USA 2000, 97, 2208–2213. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Oh, J.Y.; Park, B.H.; Lee, D.E.; Kim, J.S.; Park, H.E.; Roh, M.S.; Je, J.E.; Yoon, J.H.; Thorne, S.H.; et al. Systemic armed oncolytic and immunologic therapy for cancer with JX-594, a targeted poxvirus expressing GM-CSF. Mol. Ther. 2006, 14, 361–370. [Google Scholar] [CrossRef]
- Han, Z.Q.; Assenberg, M.; Liu, B.L.; Wang, Y.B.; Simpson, G.; Thomas, S.; Coffin, R.S. Development of a second-generation oncolytic Herpes simplex virus expressing TNFα for cancer therapy. J. Gene Med. 2007, 9, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Heiber, J.F.; Barber, G.N. Vesicular stomatitis virus expressing tumor suppressor p53 is a highly attenuated, potent oncolytic agent. J. Virol. 2011, 85, 10440–10450. [Google Scholar] [CrossRef] [PubMed]
- Bai, F.L.; Yu, Y.H.; Tian, H.; Ren, G.P.; Wang, H.; Zhou, B.; Han, X.H.; Yu, Q.Z.; Li, D.S. Genetically engineered Newcastle disease virus expressing interleukin-2 and TNF-related apoptosis-inducing ligand for cancer therapy. Cancer Biol. Ther. 2014, 15, 1226–1238. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.J.; Peng, K.W.; Bell, J.C. Oncolytic virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef] [PubMed]
- Takasu, A.; Masui, A.; Hamada, M.; Imai, T.; Iwai, S.; Yura, Y. Immunogenic cell death by oncolytic herpes simplex virus type 1 in squamous cell carcinoma cells. Cancer Gene Ther. 2016, 23, 107–113. [Google Scholar] [CrossRef]
- Angelova, A.L.; Grekova, S.P.; Heller, A.; Kuhlmann, O.; Soyka, E.; Giese, T.; Aprahamian, M.; Bour, G.; Rüffer, S.; Cziepluch, C.; et al. Complementary induction of immunogenic cell death by oncolytic parvovirus H-1PV and gemcitabine in pancreatic cancer. J. Virol. 2014, 88, 5263–5276. [Google Scholar] [CrossRef] [PubMed]
- Workenhe, S.T.; Mossman, K.L. Oncolytic virotherapy and immunogenic cancer cell death: Sharpening the sword for improved cancer treatment strategies. Mol. Ther. 2014, 22, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Yamano, T.; Kubo, S.; Fukumoto, M.; Yano, A.; Mawatari-Furukawa, Y.; Okamura, H.; Tomita, N. Whole cell vaccination using immunogenic cell death by an oncolytic adenovirus is effective against a colorectal cancer model. Mol. Ther. Oncol. 2016, 3, 16031. [Google Scholar] [CrossRef] [PubMed]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Intlekofer, A.M.; Thompson, C.B. At the bench: Preclinical rationale for CTLA-4 and PD-1 blockade as cancer immunotherapy. J. Leukoc. Biol. 2013, 94, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Boutros, C.; Tarhini, A.; Routier, E.; Lambotte, O.; Ladurie, F.L.; Carbonnel, F.; Izzeddine, H.; Marabelle, A.; Champiat, S.; Berdelou, A.; et al. Safety profiles of anti-CTLA-4 and anti-PD-1 antibodies alone and in combination. Nat. Rev. Clin. Oncol. 2016, 13, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Marincola, F.M. 2015: The Year of Anti-PD-1/PD-L1s Against Melanoma and Beyond. EBioMedicine 2015, 2, 92–93. [Google Scholar] [CrossRef] [PubMed]
- West, H.J. JAMA Oncology Patient Page. Immune Checkpoint Inhibitors. JAMA Oncol. 2015, 1, 115. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.Y.; Gore, I.; Fong, L.; Venook, A.; Beck, S.B.; Dorazio, P.; Criscitiello, P.J.; Healey, D.I.; Huang, B.; Gomez-Navarro, J.; et al. Phase II study of the anti-cytotoxic T-lymphocyte-associated antigen 4 monoclonal antibody, tremelimumab, in patients with refractory metastatic colorectal cancer. J. Clin. Oncol. 2010, 28, 3485–3490. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.P.; Sharma, P.K.; Krishnan, G.; Lockhart, A.C. Immune checkpoints and immunotherapy for colorectal cancer. Gastroenterol. Rep. (Oxf) 2015, 3, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Boland, C.R.; Goel, A. Microsatellite instability in colorectal cancer. Gastroenterology 2010, 138, 2073–2087. [Google Scholar] [CrossRef] [PubMed]
- Banerjea, A.; Ahmed, S.; Hands, R.E.; Huang, F.; Han, X.; Shaw, P.M.; Feakins, R.; Bustin, S.A.; Dorudi, S. Colorectal cancers with microsatellite instability display mRNA expression signatures characteristic of increased immunogenicity. Mol. Cancer. 2004, 3, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilar, E.; Gruber, S.B. Microsatellite instability in colorectal cancer-the stable evidence. Nat. Rev. Clin. Oncol. 2010, 7, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Farkona, S.; Diamandis, E.P.; Blasutig, I.M. Cancer immunotherapy: The beginning of the end of cancer? BMC Med. 2016, 14. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Whitaker-Dowling, P.; Griffin, J.A.; Barmada, M.A.; Bergman, I. Recombinant vesicular stomatitis virus targeted to Her2/neu combined with anti-CTLA4 antibody eliminates implanted mammary tumors. Cancer Gene Ther. 2009, 16, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Zamarin, D.; Holmgaard, R.B.; Subudhi, S.K.; Park, J.S.; Mansour, M.; Palese, P.; Merghoub, T.; Wolchok, J.D.; Allison, J.P. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci. Transl. Med. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Rojas, J.J.; Sampath, P.; Hou, W.; Thorne, S.H. Defining Effective Combinations of Immune Checkpoint Blockade and Oncolytic Virotherapy. Clin. Cancer Res. 2015, 21, 5543–5551. [Google Scholar] [CrossRef] [PubMed]
- Sobol, R.E.; Shawler, D.L.; Carson, C.; Van Beveren, C.; Mercola, D.; Fakhrai, H.; Garrett, M.A.; Barone, R.; Goldfarb, P.; Bartholomew, R.M.; et al. Interleukin 2 gene therapy of colorectal carcinoma with autologous irradiated tumor cells and genetically engineered fibroblasts: A Phase I study. Clin. Cancer Res. 1999, 5, 2359–2365. [Google Scholar] [PubMed]
- Conry, R.M.; Khazaeli, M.B.; Saleh, M.N.; Allen, K.O.; Barlow, D.L.; Moore, S.E.; Craig, D.; Arani, R.B.; Schlom, J.; LoBuglio, A.F. Phase I trial of a recombinant vaccinia virus encoding carcinoembryonic antigen in metastatic adenocarcinoma: Comparison of intradermal versus subcutaneous administration. Clin. Cancer Res. 1999, 5, 2330–2337. [Google Scholar] [PubMed]
- Morse, M.A.; Chaudhry, A.; Gabitzsch, E.S.; Hobeika, A.C.; Osada, T.; Clay, T.M.; Amalfitano, A.; Burnett, B.K.; Devi, G.R.; Hsu, D.S.; et al. Novel adenoviral vector induces T-cell responses despite anti-adenoviral neutralizing antibodies in colorectal cancer patients. Cancer Immunol. Immunother. CII 2013, 62, 1293–1301. [Google Scholar] [CrossRef] [PubMed]
- Neidhart, J.; Allen, K.O.; Barlow, D.L.; Carpenter, M.; Shaw, D.R.; Triozzi, P.L.; Conry, R.M. Immunization of colorectal cancer patients with recombinant baculovirus-derived KSA (Ep-CAM) formulated with monophosphoryl lipid A in liposomal emulsion, with and without granulocyte-macrophage colony-stimulating factor. Vaccine 2004, 22, 773–780. [Google Scholar] [CrossRef] [PubMed]
- Ullenhag, G.J.; Frodin, J.E.; Mosolits, S.; Kiaii, S.; Hassan, M.; Bonnet, M.C.; Moingeon, P.; Mellstedt, H.; Rabbani, H. Immunization of colorectal carcinoma patients with a recombinant canarypox virus expressing the tumor antigen Ep-CAM/KSA (ALVAC-KSA) and granulocyte macrophage colony—Stimulating factor induced a tumor-specific cellular immune response. Clin. Cancer Res. 2003, 9, 2447–2456. [Google Scholar] [PubMed]
- Von Mehren, M.; Arlen, P.; Tsang, K.Y.; Rogatko, A.; Meropol, N.; Cooper, H.S.; Davey, M.; McLaughlin, S.; Schlom, J.; Weiner, L.M. Pilot study of a dual gene recombinant avipox vaccine containing both carcinoembryonic antigen (CEA) and B7.1 transgenes in patients with recurrent CEA-expressing adenocarcinomas. Clin. Cancer Res. 2000, 6, 2219–2228. [Google Scholar] [PubMed]
- Marshall, J.L.; Hoyer, R.J.; Toomey, M.A.; Faraguna, K.; Chang, P.; Richmond, E.; Pedicano, J.E.; Gehan, E.; Peck, R.A.; Arlen, P.; et al. Phase I study in advanced cancer patients of a diversified prime-and-boost vaccination protocol using recombinant vaccinia virus and recombinant nonreplicating avipox virus to elicit anti-carcinoembryonic antigen immune responses. J. Clin. Oncol. 2000, 18, 3964–3973. [Google Scholar] [CrossRef] [PubMed]
- Hodge, J.W.; Grosenbach, D.W.; Aarts, W.M.; Poole, D.J.; Schlom, J. Vaccine therapy of established tumors in the absence of autoimmunity. Clin. Cancer Res. 2003, 9, 1837–1849. [Google Scholar] [PubMed]
- Gulley, J.L.; Arlen, P.M.; Tsang, K.Y.; Yokokawa, J.; Palena, C.; Poole, D.J.; Remondo, C.; Cereda, V.; Jones, J.L.; Pazdur, M.P.; et al. Pilot study of vaccination with recombinant CEA-MUC-1-TRICOM poxviral-based vaccines in patients with metastatic carcinoma. Clin. Cancer Res. 2008, 14, 3060–3069. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Teachey, D.T.; Porter, D.L.; Grupp, S.A. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood 2015, 125, 4017–4023. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Wang, Y.; Lu, X.; Han, W. Chimeric Antigen Receptors Modified T-Cells for Cancer Therapy. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef] [PubMed]
- Kakarla, S.; Gottschalk, S. CAR T cells for solid tumors: Armed and ready to go? Cancer J. 2014, 20, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Abken, H. Adoptive therapy with CAR redirected T cells: The challenges in targeting solid tumors. Immunotherapy 2015, 7, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Yu, D.; Essand, M. Prospects to improve chimeric antigen receptor T-cell therapy for solid tumors. Immunotherapy 2016, 8, 1355–1361. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Parkhurst, M.R.; Yang, J.C.; Langan, R.C.; Dudley, M.E.; Nathan, D.A.; Feldman, S.A.; Davis, J.L.; Morgan, R.A.; Merino, M.J.; Sherry, R.M.; et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol. Ther. 2011, 19, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Nishio, N.; Diaconu, I.; Liu, H.; Cerullo, V.; Caruana, I.; Hoyos, V.; Bouchier-Hayes, L.; Savoldo, B.; Dotti, G. Armed oncolytic virus enhances immune functions of chimeric antigen receptor-modified T cells in solid tumors. Cancer Res. 2014, 74, 5195–5205. [Google Scholar] [CrossRef] [PubMed]
- Thorne, S.H.; Negrin, R.S.; Contag, C.H. Synergistic antitumor effects of immune cell-viral biotherapy. Science 2006, 311, 1780–1784. [Google Scholar] [CrossRef] [PubMed]
- Edinger, M.; Cao, Y.A.; Verneris, M.R.; Bachmann, M.H.; Contag, C.H.; Negrin, R.S. Revealing lymphoma growth and the efficacy of immune cell therapies using in vivo bioluminescence imaging. Blood 2003, 101, 640–648. [Google Scholar] [CrossRef] [PubMed]
- VanSeggelen, H.; Tantalo, D.G.; Afsahi, A.; Hammill, J.A.; Bramson, J.L. Chimeric antigen receptor-engineered T cells as oncolytic virus carriers. Mol. Ther. Oncol. 2015, 2. [Google Scholar] [CrossRef] [PubMed]
- Kemeny, N.; Brown, K.; Covey, A.; Kim, T.; Bhargava, A.; Brody, L.; Guilfoyle, B.; Haag, N.P.; Karrasch, M.; Glasschroeder, B.; et al. Phase, I.; open-label, dose-escalating study of a genetically engineered herpes simplex virus, NV1020, in subjects with metastatic colorectal carcinoma to the liver. Hum. Gene Ther. 2006, 17, 1214–1224. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Breitbach, C.J.; Lee, J.; Park, J.O.; Lim, H.Y.; Kang, W.K.; Moon, A.; Mun, J.H.; Sommermann, E.M.; Avidal, L.M.; et al. Phase 1b Trial of Biweekly Intravenous Pexa-Vec (JX-594), an Oncolytic and Immunotherapeutic Vaccinia Virus in Colorectal Cancer. Mol. Ther. J. Am. Soc. Gene Ther. 2015, 23, 1532–1540. [Google Scholar] [CrossRef] [PubMed]
- Balint, J.P.; Gabitzsch, E.S.; Rice, A.; Latchman, Y.; Xu, Y.; Messerschmidt, G.L.; Chaudhry, A.; Morse, M.A.; Jones, F.R. Extended evaluation of a phase 1/2 trial on dosing, safety, immunogenicity, and overall survival after immunizations with an advanced-generation Ad5 [E1-, E2b-]-CEA(6D) vaccine in late-stage colorectal cancer. Cancer Immunol. Immunother. CII 2015, 64, 977–987. [Google Scholar] [CrossRef] [PubMed]
- Pecora, A.L.; Rizvi, N.; Cohen, G.I.; Meropol, N.J.; Sterman, D.; Marshall, J.L.; Goldberg, S.; Gross, P.; O’neil, J.D.; Groene, W.S.; et al. Phase I trial of intravenous administration of PV701, an oncolytic virus, in patients with advanced solid cancers. J. Clin. Oncol. 2002, 20, 2251–2266. [Google Scholar] [CrossRef] [PubMed]
- Calvo, E.; Gil-Martin, M.; Machiels, J.-P.; Rottey, S.; Cubillo, A.; Salazar, R.; Mardjuadi, F.I.; Geboes, K.P.; Ellis, C.; Beadle, J.W.; et al. (Eds.) A first-in-class, first-in-human phase I study of enadenotucirev, an oncolytic Ad11/Ad3 chimeric group B adenovirus, administered intravenously in patients with metastatic epithelial tumors. In Proceedings of the 2014 ASCO Annual meeting, Chicago, IL, USA, 30 May–3 June 2014.
Figure 1.
CAR-T cells as carrier for oncolytic viruses. TSA: tumor specific antigen; CAR: chimeric antigen receptor; OVs: oncolytic viruses; CAR-T: CAR expressing T-cells.
Figure 1.
CAR-T cells as carrier for oncolytic viruses. TSA: tumor specific antigen; CAR: chimeric antigen receptor; OVs: oncolytic viruses; CAR-T: CAR expressing T-cells.

{kind=link}
Table 1.
Clinical trials that are using viruses as direct vaccines or as vectors in the preparation of autologous cell-based vaccines.
| Virus | Treatment Type | Transgene (Tumor Antigen or Cytokine) | Phase of Trial | Outcome | Immune Response | References |
|---|---|---|---|---|---|---|
| Retrovirus | Therapeutic vaccination | IL-2 | I | No objective response demonstrated | Tumor-specific CTL | [68] |
| Vaccinia virus | Therapeutic vaccination | CEA | I | No objective response | Not reported | [69] |
| Adenovirus | Therapeutic vaccination | CEA | I | Increased overall survival | CEA-specific immunity | [70] |
| Adenovirus | Therapeutic vaccination | GUCY2C | I | Not published | GUCY2C-specific antibody and T-cell responses | NCT01972737 |
| Baculovirus | Therapeutic vaccination | Ep-CAM | I | Not published | Ep-CAM-specific cellular immune response | [71] |
| Canarypox virus | Therapeutic vaccination | Ep-CAM | I | Not published | Ep-CAM-specific cellular immune response | [72] |
| Avipox virus | Therapeutic vaccination | CEA, B7-1 | Pilot | Stable disease in some patients | CEA-specific CTL | [73] |
| Vaccinia + Avipox virus | Therapeutic vaccination | CEA | I | No objective anti-tumor response | Antibody against CEA | [74] |
| Vaccinia + Fowlpox | Therapeutic vaccination | CEA, B7-1, ICAM-1, LFA-3 | I | Stable disease in some patients | CEA-specific CTL | [75,76] |
| Vaccinia virus | Oncolytic virotherapy | GM-CSF | I | Not published | Not published | NCT01394939 |
| Herpes simplex virus | Oncolytic virotherapy | None | I | Not published | Not published | NCT00149396 |
| Adenovirus | Oncolytic virotherapy | None | I | Not published | Not published | NCT02028442 |
IL: interleukin; CEA: carcinoembryonic antigen; GUCY2C: guanylyl cylase C; Ep-CAM: epithelial cell adhesion molecule; ICAM-1: intercellular adhesion molecule; LFA: lymphocyte function-associated antigen; GM-CSF: granulocyte macrophage colony-stimulating factor.
| Authors & Year | Vector | Phase | N | Delivery | Results | Adverse Effects | Immune Investigations |
|---|---|---|---|---|---|---|---|
| Kemeny & Fong 2006 [88] NCT00149396 | NV1020 HSV+ GMCSF | I | 12 | 3 × 106 3 × 107 1 × 108 | GGT rise, diarrhea, elev WBC | TNF-α, IL-2, IL-1, IFN-γ, CD4+/CD8+ ratio | |
| Calvo 2014 [92] NCT02028442 | Ad11/ad3 Enadenotucirev | I/II | 161 | 1 × 1010–6 × 1012 | No survival data reported yet | Flu-like sx, elevated GGT | Elevated TNF, IFN, IL-6, and IL-12 on Day 1 after higher doses |
| Park SH 2015 [89] NCT01380600 | JX-594 tk attenuated Vaccinia | Ib | 15 | Up to 4 IV q14 days Dose 1 × 106 pfu/kg, 1 × 107, 3 × 107 | 67% stable disease | Pox skin lesions Flu like symptoms | IL-2, IL-6, IL-8, IL-10, IL-18, MIP-1α, MCP-1, MIP-1β, and TNF-α |
| Balint 2015 [90] NCT02028442 | A11/Ad3 group B adenovirus | I/II | 32 | 1 × 109 q3 weeks × 3 1 × 1010 q3 weeks × 3 1 × 1011 q3 weeks × 3 5 × 1011 q3 weeks × 3 | No objective ant-tumor responses; Median survival 13mos in optimal tx grp | Injection site rxn Fever, flu-like symptoms | Cytolytic T cell responses IFN-γ TNF-α |
| NCT01274624 | Reolysin + Folfiri + avastin in Folfiri naïve KRAS mutants | I | 12 | No data reported yet Due Fall 2017 | |||
| NCT02636036 | Ad11/Ad3 Enadenotucirev + Anti-PD-1 | I | Study completion June 2019 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Chaurasiya, S.; Warner, S. Viroimmunotherapy for Colorectal Cancer: Clinical Studies. Biomedicines 2017, 5, 11. https://doi.org/10.3390/biomedicines5010011
AMA Style
Chaurasiya S, Warner S. Viroimmunotherapy for Colorectal Cancer: Clinical Studies. Biomedicines. 2017; 5(1):11. https://doi.org/10.3390/biomedicines5010011
Chicago/Turabian StyleChaurasiya, Shyambabu, and Susanne Warner. 2017. "Viroimmunotherapy for Colorectal Cancer: Clinical Studies" Biomedicines 5, no. 1: 11. https://doi.org/10.3390/biomedicines5010011
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.