T-Cell Manipulation Strategies to Prevent Graft-Versus-Host Disease in Haploidentical Stem Cell Transplantation


John van Geest Cancer Research Centre, Nottingham Trent University, Clifton Lane NG11 8NS, UK
*
Author to whom correspondence should be addressed.
Biomedicines 2017, 5(2), 33; https://doi.org/10.3390/biomedicines5020033
Submission received: 8 April 2017
/
Revised: 16 June 2017
/
Accepted: 19 June 2017
/
Published: 21 June 2017
(This article belongs to the Special Issue Cell Therapy for the Treatment of GVHD)
Abstract
:Allogeneic haematopoietic stem cell transplantation (HSCT) from an human leukocyte antigen (HLA)-identical donor can be curative for eligible patients with non-malignant and malignant haematological disorders. HSCT from alternative donor sources, such as HLA-mismatched haploidentical donors, is increasingly considered as a viable therapeutic option for patients lacking HLA-matched donors. Initial attempts at haploidentical HSCT were associated with vigorous bidirectional alloreactivity, leading to unacceptably high rates of graft rejection and graft-versus-host disease (GVHD). More recently, new approaches for mitigating harmful T-cell alloreactivity that mediates GVHD, while preserving the function of tumour-reactive natural killer (NK) cells and γδ T cells, have led to markedly improved clinical outcomes, and are successfully being implemented in the clinic. This article will provide an update on in vitro strategies and in vivo approaches aimed at preventing GVHD by selectively manipulating key components of the adaptive immune response, such as T-cell receptor (TCR)-αβ T cells and CD45RA-expressing naive T cells.
1. Introduction
Although human leukocyte antigen (HLA)-matched related donors (MRD) remain the preferred source of stem cells for allogeneic haematopoietic stem cell transplantation (HSCT), only 25% of the patients will locate a fully HLA-matched sibling [1]. Also, the identification of a complete HLA-matched unrelated donor (MUD) for HSCT remains a challenge, despite the availability of large international donor registries. Alternative donor sources are increasingly being considered and include HLA partially-matched or haploidentical family donors, i.e., related donors who share with the patient a single identical copy of chromosome 6, containing the HLA loci [2].
The failure of engraftment and graft-versus-host disease (GVHD) are major hurdles to the success of allogeneic HSCT. Graft rejection can be attributed to major disparities in HLA loci between the donor and recipient, resulting in an undesired immune response mounted by the host’s immune system. GVHD is the consequence of donor immune cells attacking the host, leading to extensive tissue damage and, in many instances, life-threatening complications and patient death [3]. Clinical GVHD is conventionally classified as acute GVHD (if occurring within the first 100 days after HSCT) and chronic GVHD (which occurs after day 100 post-HSCT). Immune suppressive treatments are a cornerstone in GVHD prophylaxis, as reviewed elsewhere [3]. Novel approaches to preserve the beneficial anti-leukaemia effects of donor T cells without inducing detrimental GVH responses are needed to maximise the therapeutic potential of allogeneic HSCT. Despite the complexities relating to the mechanistic understanding of GVHD and its treatment [4], allogeneic HSCT remains a highly successful and curative modality for haematological malignancies and inherited or acquired non-malignant blood disorders, as well as for an expanding number of autoimmune diseases [5].
A major obstacle to three-loci-mismatched haploidentical HSCT is the HLA barrier, mainly in the GVH direction, and bidirectional alloreactivity (Figure 1). Therefore, various approaches to prevent GVHD are being investigated, including in vitro T-cell depletion of bone marrow or peripheral blood stem cells (PBSCs) or, more recently, in vivo T-cell depletion approaches using either granulocyte colony-stimulating factor (G-CSF)-mobilized bone marrow in combination with PBSCs and anti-thymocyte globulin or the administration of high-dose cyclophosphamide after transplantation of haploidentical bone marrow-derived progenitor cells [6].
GVHD is a multistage process incited by pre-transplantation conditioning regimens (radiation and/or chemotherapy) [9]. Increased tissue damage and generation of pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) trigger danger-recognising receptors, such as the Toll-like receptors (TLRs), and fully activate antigen presenting cells (APCs) (Figure 1), leading to the secretion of pro-inflammatory cytokines, as well as the activation and expansion of donor T cells. Trafficking of donor T cells towards target sites of GVHD is the second stage in GVHD pathogenesis, where chemokines and other inflammatory mediators released as a result of pre-transplantation conditioning regimens play a critical role. Tissue damage is mainly effected by cytotoxic T cells and FAS (CD95)-FAS ligand (CD95L) or perforin/granzyme-mediated pathways. Other cell types, such as natural killer (NK) cells, partake in GVHD initiation, as reviewed elsewhere [9].
Despite being a major clinical challenge, mild-to-moderate GVHD might have a therapeutic effect, commonly referred to as graft-versus-leukaemia (GVL), which mainly results from the recognition and killing of tumour cells by donor-derived alloreactive T cells. Different approaches have been developed in addition to pharmacological interventions to dampen GVH while preserving GVL responses, including the administration of genetically-modified T cells, dendritic cells (DCs), and NK cells, and the co-administration of mesenchymal stromal cells, as reviewed elsewhere [3,9,10].
This article focuses on the latest developments in graft engineering techniques to target T cells with the aim of reducing GVHD while preserving anti-tumour and anti-pathogen immune responses after haploidentical HSCT.
2. Depletion of T-Cell Subpopulations
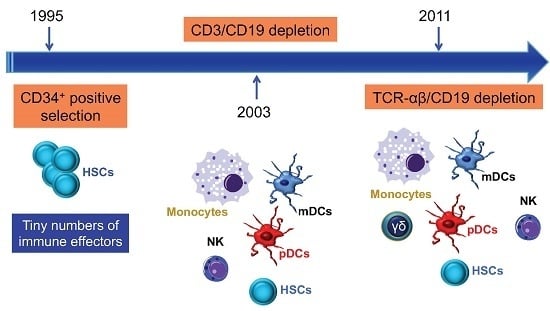
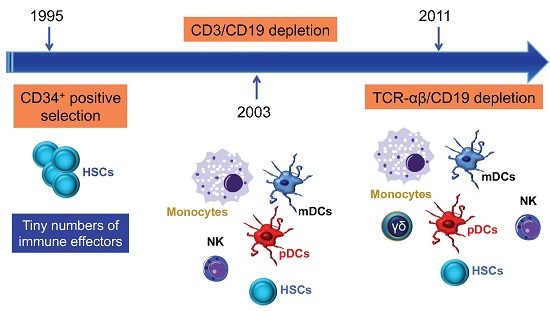
A major breakthrough in GVHD prevention was the depletion of donor T cells by physical or immunological techniques [11], including soybean lectin agglutination, rosette depletion, and monoclonal antibody-mediated methods. The development of preclinical models of transplantation of mega-doses of highly purified CD34+ progenitor cells described by Reisner et al. [12], and their subsequent application to patients by Aversa et al. [13], were major steps towards a broader clinical use of haploidentical HSCT. The introduction of the one-step, semi-automated MACS® device (Clini-MACS, Miltenyi Biotec, Bergisch Gladbach, Germany) brought further improvement, including the achievement of a median CD34-cell purity of 97% and an extensive depletion of T and B cells (Figure 2). Pioneering clinical trials with the Miltenyi semi-automated device were performed in children and showed a high engraftment rate with a low incidence of acute and chronic GVHD (reviewed in [6]).
2.1. Depletion of T-Cell Receptor (TCR)-αβ T Cells and CD19+ B Cells
In order to avoid extensive GVHD and to prevent post-transplantation Epstein-Barr virus (EBV)-induced lymphoproliferative disease (EBV-PTLD), CD3/CD19-depleted HLA-haploidentical HSCT has been evaluated within a prospective phase II trial in children with acute leukaemia and advanced myelodysplastic syndrome, who received a myeloablative conditioning regimen consisting of fludarabine or clofarabine (in patients with active disease only), thiotepa, melphalan, and serotherapy [15]. Using this graft manipulation procedure, the number of CD3+ T cells remained below the threshold value of 1 × 105 cells/kg of the recipient’s body weight, even in small children weighing less than 10 kg. Transplantation-related mortality (TRM) was 8% at 1 year and 20% at 5 years post-transplant. Primary engraftment was documented in 87% of children, with 1000/µL leukocytes and 20,000/µL platelets being reached 10 and 11 days after HSCT, respectively. Acute grade II and grade III-IV GVHD occurred in 26% and 6.5% of patients, respectively, whereas chronic GVHD was observed in 21% of evaluable patients. The cumulative incidence of cytomegalovirus (CMV) re-activation was 21% for all patients and 52% and 0% for recipient (R)+donor (D)+/R+D− and R−D+/R−D− subgroups, respectively, whereas the cumulative incidence of human adenovirus (HAdV)-DNA in stool was 53%. Notably, in most patients, an endogenous T-cell response to viral antigens was detected, with HAdV-reactive and CMV-reactive T cells being detected in vitro at a median frequency of 0.12% and 0.6%, respectively, after stimulation with hexon antigen/virus lysate and pp65. Pulmonary aspergillosis was diagnosed in 6 patients (proven in 1, probable in 5); 3 out of these 6 patients had pre-existing infiltrates prior to HSCT conditioning. However, no lethal fungal or viral infection occurred in any of the patients. The median time to reach >0.1 × 109/L CD3+CD4+ and CD3+CD8+ T cells was 61 and 107 days, respectively. By contrast, the recovery of CD56+ NK cells was prompt, with co-transfused NK cells being detectable in the first week after HSCT. Event-free survival (EFS) at 3 years for the whole group was 25% [15]. Remission status significantly influenced survival, with patients transplanted with active disease having a 3-year EFS of 15%. However, a subgroup analysis of patients who received a first haploidentical HSCT in the first, second, or third complete remission showed a favourable 3-year EFS of 46% [15]. The overall probability of relapse at 2 years was 38% in patients receiving the first HSCT in complete remission and 75% in those who were transplanted with the active disease.
A TCR-αβ depletion reagent has been available for the Clini-MACS system since 2009. To maintain additional anti-tumour and anti-infectious properties in the graft, a novel strategy aimed at depleting TCR-αβ+ T cells and B cells has been recently validated (Table 1) [14]. The authors performed 102 separations with a 4.7-log and 4.1-log depletion of TCR-αβ+ and CD19+ cells, respectively. Comparison with other techniques of T-cell depletion, including positive selection of CD34+ cells and CD3/CD19 depletion, revealed a comparable or better performance in terms of CD34 enrichment and CD3/CD19 depletion, with more constant results and lower coefficients of variation [14].
Comprehensive 10-colour flow cytometry panels have recently been developed for quality control purposes and to allow for the correct application of desired stem cell and T-cell dosages for clinical-scale TCR-αβ/CD19-depleted grafts [25].
Using the same approach (Figure 3), 23 children were treated with HLA-haploidentical HSCT for non-malignant disorders, without receiving any post-transplantation pharmacological GVHD prophylaxis [16]. The median number of CD34+ cells, TCR-αβ+ T cells, and B cells infused was 16.8 × 106/kg, 40.0 × 103/kg, and 40.0 × 103/kg, respectively. All patients but 4 engrafted. Three patients developed grade I–II acute GVHD of the skin, with no visceral acute or chronic GVHD being reported. The cumulative incidence of TRM was 9.3%. With a median follow-up of 18 months at the time of study publication, 21/23 children are alive and disease-free, with the 2-year probability of disease-free survival (DFS) being 91.1%. The recovery of γδ+ T cells was prompt, whereas αβ+ T cells progressively increased over time.
A retrospective study involving 34 adult leukaemia patients (AML and ALL) who received TCR-αβ-depleted haploidentical HSCT with CD34+ progenitors after a myeloablative conditioning regimen showed favourable GVHD-free survival rates [23]. Full engraftment and donor chimerism were established in 31 patients, and grade III–IV acute GVHD and chronic GVHD developed in four patients. One-year overall survival and disease-free survival were 54% and 42%, respectively. The only risk factor identified for overall survival was relapse, which was higher in ALL compared with AML patients. Neither EBV-PTLD nor CMV-related complications were observed in any patient with immune reconstitution.
B-cell reconstitution was not dissimilar to that observed after allogeneic HSCT [26], with >200 B cells/μL of blood being detected 4 months and 1 year after the infusion of TCR-αβ+/B-cell depleted grafts for primary immune deficiencies and acute leukaemia, respectively [16,19,20,21]. Further studies with larger cohorts of patients are needed to characterise long-term B-cell reconstitution in children and adults receiving extensively T-cell/B-cell depleted haploidentical grafts.
Overall, the studies published thus far point to the safety and efficacy of this graft manipulation approach for HLA-haploidentical HSCT, although refined treatment options to boost immunological reconstitution and to maintain long-term disease remission are needed.
2.2. Depletion of Naïve T Cells
Most T cells that cause GVHD reside within the naïve T-cell subset, unless the donor has developed a memory T-cell response through exposure to allogeneic cells after either pregnancy or blood product transfusion [27]. A single-arm first-in-human clinical trial enrolling 35 patients with high-risk leukaemia has shown that transplantation with >5.0 × 106 CD34+ cells/kg and a defined dose of naive CD45RA T-cell-depleted peripheral blood stem cell (PBSC) grafts after a total body irradiation (TBI)-containing myeloablative conditioning regimen translates into prompt engraftment [28] (Table 2). An average number of 3600 naïve T cells/kg was infused. Although the incidence of grade II-IV acute GVHD was 66%, GVHD was always responsive to corticosteroids. The estimated probability of chronic GVHD was 9% at 2 years compared with 50% rates in a contemporary cohort of patients receiving T cell-replete grafts. Rapid T-cell recovery and transfer of protective virus-specific immunity could be documented, and overall survival was 78% at 2 years [28].
A novel 2-step, good manufacturing practice (GMP)-compliant procedure to deplete naïve T cells, while preserving CD34+ HSCs and pathogen-specific memory T cells, has been recently developed [29]. CD34+ HSCs were initially selected from G-CSF-mobilized apheresis products, followed by depletion of CD45RA+ cells using a murine anti-CD45RA monoclonal antibodies directly conjugated to iron dextran beads. A theoretical advantage of CD45RA depletion, as compared with complete T-cell depletion (TCD), is that pathogen-specific memory T cells are retained in the graft and could transfer protective immunity to opportunistic pathogens, such as CMV and EBV. In addition to a 4.5–5.0-log depletion of naïve T cells, CD45RA-depleted products contained a lower number of regulatory T cells (Treg), B cells, and NK cells, all of which express CD45RA. Importantly, the frequency of multifunctional virus-specific CD4+ and CD8+ T cells was equivalent or even higher in CD45RA-depleted products compared with un-manipulated grafts. The cost of the 2-stage cell selection procedure was considerable and estimated to be approximately $20,000, potentially limiting a broad application of this methodology. However, if effective at preventing GVHD while sparing pathogen-specific immunity, this approach might compare favourably with other TCD methods, including the removal of TCR-αβ+ T cells and CD19+ B cells.
2.3. Depletion of Alloreactive T Cells
Photodynamic allodepletion aims at eliminating host-reactive donor T cells from allogeneic HSCT to prevent GVHD, while conserving donor immunity [30]. This clinical-scale, semi-closed process is based on three consecutive phases, i.e., coloration, extrusion, and light exposure, and targets activation-based changes in P-glycoprotein, which result in an altered efflux of the photosensitizer TH9402. Lymphocytes expanded with anti-CD3 and IL-2 served as APCs and were co-cultured with responder cells from HLA-matched or mismatched donors. In mismatched stimulator-responder pairs, alloreactivity was reduced by a median 474-fold compared with the un-manipulated responder cells. By contrast, third-party responses were maintained. In matched pairs, alloreactive helper T-lymphocyte precursors were reduced to <1:100,000, while third-party responses remained around or >1:10,000. Specific anti-viral and anti-bacterial immunity was preserved in photo-depleted products.
In a clinical trial designed to evaluate the efficacy of photo-depletion to prevent severe acute GVHD, 24 patients with haematological malignancies conditioned with fludarabine, cyclophosphamide, and TBI received a CD34-selected allograft from a MRD along with 5 × 106/kg photo-depleted donor T cells [31,32]. The photo-depleted product showed an inverted CD4+/CD8+ ratio, with the greatest depletion occurring in CD4+ naive and central memory T-cell subsets. By contrast, CD8+ naive and effector cells were relatively unaffected, reflecting the differential retention of TH9402 by T-cell subsets, which was greater in CD4+ and central memory cells and led to their preferential elimination during the photo-depletion procedure [32]. Engraftment was rapid, with 95% donor myeloid chimerism occurring by day 14, and 95% donor T-cell chimerism occurring by day 30 in most patients. Probabilities of acute GVHD were 38% for grades II–IV and 13% for grades III–IV, and 65% for chronic GVHD. The probability of haematological relapse was low (27%), considering the high-risk characteristics of the patient population. In multivariable analysis, low CD4+ central memory frequency in photo-depleted products was associated with chronic GVHD and worse overall survival (OS) [32]. An unexpected outcome of this study was the high rate of late viral, bacterial, and fungal infections that accompanied chronic or persisting acute GVHD, with 53% non-relapse mortality and median survival of only 568 days. In vitro proliferative responses to CMV were significantly reduced in the photo-depleted product, and reactivity to CMV remained much lower than in a control cohort of 35 patients given TCD HLA-matched transplants [32].
A cell-based therapeutic consisting of photo-depleted lymphocytes, ATIRTM (Allodepleted T-cell ImmunotheRapeutic), has been developed by Kiadis Pharma. An open-label, multi-centre phase 2 study (CR-AIR-007; NCT01794299) enrolled 23 patients with acute leukaemia who received haploidentical CD34+ cells followed by a fixed dose of 2 × 106/kg photo-depleted donor lymphocytes (ATIR101) at a median of 28 days after HSCT, without the use of post-transplant GVHD prophylaxis [33]. Ex vivo photo-depletion translated into the inactivation of host tissue-specific, proliferating T cells while maintaining anti-third party and anti-CD3/CD28 reactivity. No grade III/IV GVHD was observed after the infusion of allo-depleted T cells. Furthermore, 1-year OS was higher in patients receiving ATIR101 compared with a historic control group (61% in the HSCT + ATIR101 group vs. 20% in the control group). As of August 1st, 2016, two patients relapsed within the first year after HSCT. Severe infections occurred in 9/23 patients within the first 6 months after HSCT, which were viral infections in 7/23 subjects. GVHD-free, relapse-free survival (GRFS) for patients receiving HSCT + ATIR101 was estimated to be 57% at 1 year after HSCT, which compares favourably with the control group of patients receiving TCD-HSCT only (20%).
Overall, this TCD strategy is promising, although further pre-clinical development is warranted to avoid the undesired elimination of clinically useful T-cell subsets that contribute to post-transplantation immune recovery and control of infections.
In vivo depletion of alloreactive T cells with high-dose cyclophosphamide administered early post-transplantation (PTCy) allows the engraftment of haploidentical grafts from either bone marrow or peripheral blood without severe acute and chronic GVHD [36]. Outcomes for the first patients transplanted at Johns Hopkins were published in 2002 [37]. This strategy has been used in patients receiving tacrolimus and mycophenolate as GVHD prophylaxis and following reduced-intensity or non-myeloablative conditioning. Although this approach is technically less demanding and considerably more affordable compared with in vitro T-cell depletion, studies have shown ~50% relapse rates 1 year after transplantation [38], which could be the result of alloreactive T-cell depletion and reduced GVL responses. More recently, haploidentical bone marrow transplantation (BMT) with PTCy was shown to yield similar survivals to those observed in patients receiving HLA-matched BMT [39]. When patients in this retrospective analysis were risk-stratified using the Disease Risk Index (DRI), low-risk, intermediate-risk, and high/very high-risk patients had 3-year progression-free survival (PFS) estimates of 65%, 37%, and 22%, with corresponding 3-year OS estimates of 71%, 48%, and 35%, respectively [39]. Other investigators are attempting to further reduce relapse rates after haploidentical BMT with PTCy by using myeloablative busulfan and fludarabine conditioning [40]. However, non-relapse mortality at 100 days and 1 year were 9% and 16%, respectively, predominantly as a result of infections. Another encouraging report of 148 patients with a variety of haematological malignancies showed a 13% cumulative incidence of TRM after unmanipulated haploidentical BMT with PCTy [41,42]. The cumulative incidence of grades II–IV acute GVHD, grades III–IV acute GVHD, and chronic GVHD were 24%, 10%, and 12%, respectively.
3. Treatment of Donors to Prevent Graft-Versus-Host Disease (GVHD)
3.1. Granulocyte Colony-Stimulating Factor (G-CSF)
Experimental observations in the mid-1990s linked G-CSF to immune deviation in humans. G-CSF promotes the differentiation of type 1 Treg cells (Tr1) cells, endowed with the ability to release interleukin (IL)-10 and transforming growth factor (TGF)-β1, and to suppress T-cell proliferation in a cytokine-dependent manner [43]. Finally, G-CSF indirectly modulates DC function in humans, by inducing hepatocyte growth factor (HGF), IL-10, and interferon (IFN)-α, and mobilizes type 2 DCs (DC2) [44]. Overall, the available data imply that G-CSF-mobilized cell therapy products may be intrinsically less capable of inducing uncontrollable GVHD.
However, published evidence suggests that G-CSF might exert protective effects when given to haematopoietic stem cell (HSC) donors but not to HSCT recipients. In mice, the transplantation of G-CSF-mobilized splenocytes was associated with a significant reduction in tumour necrosis factor (TNF)-α and lipopolysaccharide (LPS) production and in GVHD score, translating into improved survival. In contrast, the administration of G-CSF to recipient animals exerted no effect on acute GVHD-related survival or in vivo TNF-α production in the absence of G-CSF pretreatment of the donor, indicating that G-CSF effects on the donor rather than on the recipient mice might account for the GVHD improvement [45].
Associations between immune cell subsets in G-CSF-mobilised grafts and clinical outcomes, including GVHD occurrence, were reported in patients receiving PBSC transplantation [46]. The frequencies of activated NK cells and Natural Killer T (NKT) cells were correlated with a significantly lower risk of acute GVHD. By contrast, late activated, HLA-DR-expressing T cells were associated with a significantly higher risk of acute and chronic GVHD. Intriguingly, the frequency of CD34-expressing monocytes in G-CSF-mobilised allografts inversely correlates with the incidence of acute GVHD, suggesting that in vitro-expanded G-CSF-mobilised monocytes might be used as GVHD therapeutics [47]. In the context of MUD HSCT, the intra-graft content of monocytic myeloid-derived suppressor cells (MDSCs), which are expanded as a result of G-CSF administration, was the only predictor of acute GVHD [48]. The cumulative incidence of acute GVHD at 180 days after transplantation for recipients receiving monocytic MDSC doses below and above the median was 63% and 22%, respectively. However, monocytic MDSCs had no apparent impact on relapse rates or TRM rates. There is also evidence that the administration of G-CSF-primed bone marrow grafts shares the advantages of G-CSF-mobilised peripheral blood grafts without being associated with increased risk of GVHD [49]. Direct comparisons of the immunological properties of G-CSF-mobilised blood and G-CSF-primed bone marrow samples indicate that G-CSF-primed bone marrows have reduced T-cell cytokine production, lower expression of CD28 costimulatory molecules on T cells, lower DC content, and diminished proliferation capacity [50].
Recently, a transplantation protocol (GIAC) involving sequential, in vivo modulation of T-cell functions in both recipients and donors was developed in China [51], the main elements of which stand for donor treatment with G-CSF (G), intensified immunologic suppression (I), anti-human thymocyte immunoglobulin for GVHD prevention (A), and the combination (C) of peripheral blood and bone marrow transplantation. This approach resulted in high rates of engraftment and similar incidence of GVHD as that observed in allogeneic HSCT from MRD, and was inspired by previous data from the same group showing that the combined use of G-CSF-primed BM and peripheral blood HSCs in proportions ranging from 2:1 to 1:2 maintained T-cell hyporesponsiveness and polarization towards a Th2 phenotype [52].
Taken together, these studies suggest that immune modulation by G-CSF might be used to affect graft composition and to prevent GVHD.
3.2. Low and Ultra-Low Dose IL-2
Treatment with IL-2 is a promising approach to expand Treg cells and NK cells in HSC donors. Ultra-low dose (ULD) IL-2 has been given to 21 healthy donors as an immune-modulating agent with the aim of preventing GVHD after HSCT [53]. Safety, dose level, and immune signatures were evaluated after the administration of 50,000 to 200,000 units/m2/day IL-2 for 5 consecutive days. The treatment was well tolerated and was associated with an increase of Treg cells with potent suppressive activity, as well as of CD56bright NK cells with enhanced IFN-γ release. IFN-γ-induced protein 10 (IP10) was increased in the serum. Gene expression profiling revealed a significant change in a restricted set of genes, including FOXP3 and IL-2RA.
Low-dose IL-2 has been tested in a controlled, open-label randomized trial that included 90 recipients of allogeneic HSCT [54]. Patients in the IL-2 arm received a subcutaneous injection of low-dose IL-2 (1 × 106 U/d) on day 60 after HSCT for 14 consecutive days, followed by a 14-day hiatus. Detection of the minimal residual disease occurred more frequently in IL-2-treated patients compared with the control arm (36% vs. 15%). The cumulative incidence of moderate-to-severe chronic GVHD was significantly lower in IL-2-treated patients compared with controls (33% vs. 57%), leading to significantly higher 3-year GVHD-free PFS rates (47% in IL-2 treated patients vs. 31% in controls). In line with previous findings in patients treated with IL-2 for steroid-refractory chronic GVHD [55], blood Treg cells, NK cells, and NK-cell cytotoxicity were increased in IL-2-treated subjects between 3 and 6 months after HSCT [54].
4. T-Cell Depleted (TCD) Haploidentical Haematopoietic Stem Cell Transplantation (HSCT) as a Platform for Adoptive Immunotherapy
Many groups are currently evaluating adoptive immunotherapy with transplant donor-derived T cells to treat and/or prevent leukaemia relapses and drug-resistant infectious complications [11,56]. Investigators at Memorial Sloan Kettering Cancer Centre, New York, have pioneered the use of in vitro expanded T cells specific for peptide epitopes of the Wilms Tumour 1 (WT-1) protein in patients with WT-1+ hematologic malignancies [11]. WT-1 is a suitable target for adoptive immunotherapy being differentially expressed in over 70% of AMLs and myelomas and being also expressed at high levels in advanced MDS. Importantly, in transplant recipients treated with unselected donor lymphocytes for the relapse of AML or myeloma, expansion of WT-1 specific T-cells closely correlates with the eradication of tumour cells and the achievement of complete remission [57]. WT-1 specific T cells generated from normal HSCT donors were administered at escalating doses (3.8 × 108 to 3.3 × 109/m2) to 11 patients with WT-1+ leukaemia, MDS, or myeloma in disease relapse post-transplantation [58]. At high doses, WT-1 specific T-cells expanded in vivo, preferentially localised to the bone marrow compartment and were detected in the blood for more than 7–14 months, with the induction of complete remissions.
4.1. Suicide Gene-Transduced T Cells to Provide Graft-Versus-Leukaemia (GVL) Responses without GVHD
Several groups are using suicide gene-transduced donor T cells to control GVHD, while promoting post-transplantation immune reconstitution. A recent phase I/II multi-centre clinical trial of transplantation with TCD haploidentical HSCs explored the potential benefit of infusing donor lymphocytes genetically engineered to express the human herpes simplex virus thymidine kinase type 1 (HSVtk) suicide gene, with the aim of boosting immune reconstitution and improving disease control in patients with high-risk leukaemia [59]. Gene-modified cells were infused starting at day 28 after HSCT, with an initial dose of 1 × 106 cells/kg. In the absence of GVHD and immune recovery, a second dose of 1 × 107 cells/kg, a third dose of 1 × 106 cells/kg plus subcutaneous recombinant human IL-2 (rhIL-2), and a fourth dose of 1 × 107 cells/kg plus rhIL-2 were administered at monthly intervals. Collectively, no acute or chronic events related to the gene-transfer procedure were observed. Engraftment was documented in 22 out of the 28 patients infused, and CD3+ T-cell counts >100/µL were achieved by day 75 after HSCT [59]. Importantly, CMV- and EBV-specific, IFN-γ-producing T cells were detected at the time of immune recovery. Anti-viral responses progressively normalized in patients who attained immune recovery. The infusion of HSVtk cells was correlated with the development of GVHD in 10 out of 22 immune-reconstituted patients. Treatment with ganciclovir translated into a significant reduction of circulating HSVtk cells, but not CD3+ T cells, reassuring about the lack of impact of ganciclovir on long-term immune recovery. Collectively, the overall survival in patients with de novo acute leukaemia transplanted in any complete remission was 49% at 3 years [59].
A different gene construct was used for the transduction of donor T cells by Di Stasi and co-workers [17]. Human caspase-9 (C9) was fused to a modified human FK506-binding protein whose dimerization, in the presence of a synthetic bioinert drug, was shown to trigger the activation of C9 and death of the cells expressing the construct [17]. ∆CD19 was used as a “selectable marker” to isolate transduced T cells that were then administered to 5 patients after haploidentical HSCT with purified CD34+ cells. Modified T cells displayed a CD3+∆CD19+ phenotype and were detectable in vivo within 3–7 days after the first infusion. Concomitantly with the expansion of gene-modified T cells, mild GVHD of the skin occurred in 4 of 10 patients [60]. Each patient with GVHD received one infusion of the dimerizing drug AP1903, which induced a more than 90% decline of blood transgenic cells within 30 min [17]. GVHD-associated abnormalities were resolved within 24 h after infusion and without additional immune suppressive therapy. Gene-modified T cells were detected in stable numbers for more than 1 year and mainly comprised polyclonal CD3+CD19+ T cells that also contained virus-reactive T cells. In line with this finding, no patient experienced reactivation of CMV, EBV, HAdV, BK virus, TCD Haploidentical HSCT As a Platform for Adoptive Immunotherapy, and Aspergillus after T-cell infusion and treatment with AP1903. The authors confirmed these findings in patients treated with iC9-DLI without any prior allodepletion step [61]. Safety switch-modified T cells persisted in vivo for more than 2 years and accelerated the recovery of endogenous T cells, including CD4+ T cells of thymic origin [60].
Although further studies are required to confirm efficacy, the infusion of suicide-gene transduced T cells is a very promising approach to enhance immune recovery without inducing GVHD [61].
4.2. Virus-Specific T Cells
Viral infections continue to account for substantial post-transplant morbidity and mortality after allogeneic HSCT, as reviewed elsewhere [62]. Immunotherapeutic strategies are increasingly being exploited to prevent and treat viral infections when anti-viral drugs are ineffective or cause excessive toxicity. Anecdotal reports suggest the safety and tolerability of infusing virus-specific T cells activated ex vivo using pools of overlapping peptides [56]. Virus-specific T cells stimulated with overlapping peptides derived from the immunodominant HAdV5 hexon protein (MACS GMP PeptivatorTM AdV5 hexon, Miltenyi Biotec) were recently used to treat disseminated HAdV infection after TCD haploidentical HSCT. The IFN-γ-secreting T cells are then labelled and magnetically enriched using the Cytokine Secretion System and the Clini-MACSTM device. The adoptive transfer of HAdV-specific T cells was safe and not associated with any adverse event, including alloreactivity against the recipient’s tissues. Recovering CD4+ and CD8+ T cells mostly displayed a CD45RO+ memory phenotype, released IFN-γ in response to HAdV-derived peptides, but lacked in vitro responses against other dominant HAdV antigens not used for T-cell activation, as well as responses to other viral pathogens, such as CMV and EBV [56].
The Memorial Sloan Kettering Cancer Research Centre and Baylor College of Medicine Groups have established consortia to foster the implementation of multi-centre clinical trials of banked third party T cells for EBV, CMV, and other life-threatening viral infections complicating HSCT [63].
5. Novel Strategies for Controlling GVHD
While TCD strategies remain a cornerstone for GVHD control, other avenues are being explored to reduce GVHD while preserving anti-viral and anti-tumour responses. Targeting epigenetic modifiers such as acetyl and methyl-transferases, micro-RNAs, inhibiting Notch signalling and mitochondrial ATP-ase, inhibiting protein kinase-C, and JAK/STAT signalling have all emerged as potential therapeutic targets for GVHD control. Earlier studies using histone deacetylase inhibitors (HDACs) such as suberoylanilide hydroxamic acid (SAHA/vorinostat) showed that these drugs can mitigate the effect of GVHD by impairing the function of host APCs [64]. Treatment of DCs with HDACs also led to the induction of the tryptophan catabolising enzyme indoleamine 2,3-dioxygenase-1 (IDO1), which is an inhibitor of DC and T-cell function [65]. Other studies in mouse models have shown that the inhibition of HDAC6 (a non-histone deacetylase) specifically abrogates CD8 T-cell function and significantly reduces GVHD-like manifestations [66]. Similarly, the inhibition of histone methylation using DZNep (3-deazaneplanocin A) resulted in the apoptosis of activated alloreactive T cells and diminished tissue damage and injury in the host [67,68].
Notch signalling is one of the key regulators of T-cell and DC development. Targeting Notch signalling to control murine GVHD has attracted considerable interest. The inhibition of Notch in T cells resulted in reduced pro-inflammatory cytokine production without compromising immune cell proliferation [69]. Gatza et al. also showed that targeting mitochondrial ATPase could reduce the frequency of alloreactive T cells in GVHD without affecting T-cell responses [70]. PKC-α is a key regulator of T-cell signalling through its interaction with several transcription factors, including NF-AT. Small-molecule inhibition of PKC-θ and PKC-α has been successfully pursued in murine models of GVHD [71]. Several other potential candidates, such as JAK-STAT inhibitors [72], miRNA inhibitors [73] and aurora kinase-A inhibitors [74] are currently being evaluated. However, the clinical efficacy of the above candidate drugs has yet to be substantiated in clinical trials.
6. Conclusions
Haploidentical HSCT could be offered to patients with an indication for allogeneic HSCT, but we do not have a MRD or a MUD available within a reasonable time frame. TCD haploidentical HSCs can secure consistent engraftment without an increase in relapse in patients transplanted for acute leukaemia. Unquestionably, T-cell depletion is an efficient tool for preventing acute and chronic GVHD after haploidentical HSCT. However, immunological reconstitution may be delayed, requiring cellular therapy approaches to boost the recovery of anti-leukaemia and pathogen-specific immunity. The continuous availability of haploidentical donors will allow us to address the issues of delayed immune reconstitution and post-transplantation leukaemia recurrence by adoptively transferring non-alloreactive T cells, either genetically modified (iC9 or HSVtk) or specifically selected. Innovative protocols for graft engineering, including the immunomagnetic depletion of GVHD-inducing CD45RA-expressing T cells, are currently being tested with varying degrees of success.
In vitro T-cell depletion protocols are laborious and expensive, compared with other approaches that are being successfully implemented in the clinic, including the administration of PTCy. Randomised clinical trials to assess the superiority of each approach have not yet been conducted.
In conclusion, the generation of “designer grafts” will hopefully pave the way to safer HSCT approaches in patients with malignancies or will serve as a platform for tolerance induction in patients’ with autoimmune diseases.
Acknowledgments
Sergio Rutella is the recipient of a National Priorities Research Program (NPRP) Grant from the Qatar National Research Fund (NPRP#8-2297-3-494), of research funds from the Roger Counter Foundation (Dorset, United Kingdom) and mainstream quality-related (QR) funds from the Higher Education Funding Council for England (HEFCE).
Author Contributions
Jayakumar Vadakekolathu and Sergio Rutella mined the literature and wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ballen, K.K.; Koreth, J.; Chen, Y.B.; Dey, B.R.; Spitzer, T.R. Selection of optimal alternative graft source: Mismatched unrelated donor, umbilical cord blood, or haploidentical transplant. Blood 2012, 119, 1972–1980. [Google Scholar] [CrossRef] [PubMed]
- Kanakry, C.G.; Fuchs, E.J.; Luznik, L. Modern approaches to HLA-haploidentical blood or marrow transplantation. Nat. Rev. Clin. Oncol. 2016, 13, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Shlomchik, W.D. Graft-versus-host disease. Nat. Rev. Immunol. 2007, 7, 340–352. [Google Scholar] [CrossRef] [PubMed]
- Im, A.; Hakim, F.T.; Pavletic, S.Z. Novel targets in the treatment of chronic graft-versus-host disease. Leukemia 2017, 31, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Snowden, J.A.; Pearce, R.M.; Lee, J.; Kirkland, K.; Gilleece, M.; Veys, P.; Clark, R.E.; Kazmi, M.; Abinun, M.; Jackson, G.H.; et al. Haematopoietic stem cell transplantation (HSCT) in severe autoimmune diseases: Analysis of UK outcomes from the British Society of Blood and Marrow Transplantation (BSBMT) data registry 1997–2009. Br. J. Haematol. 2012, 157, 742–746. [Google Scholar] [CrossRef] [PubMed]
- Handgretinger, R. New approaches to graft engineering for haploidentical bone marrow transplantation. Semin. Oncol. 2012, 39, 664–673. [Google Scholar] [CrossRef] [PubMed]
- Filippini, P.; Rutella, S. Recent advances on cellular therapies and immune modulators for graft-versus-host disease. Expert Rev. Clin. Immunol. 2014, 10, 1357–1374. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.C.; Ferrara, J.L.; Levine, J.E. Advances in predicting acute GVHD. Br. J. Haematol. 2013, 160, 288–302. [Google Scholar] [CrossRef] [PubMed]
- Blazar, B.R.; Murphy, W.J.; Abedi, M. Advances in graft-versus-host disease biology and therapy. Nat. Rev. Immunol. 2012, 12, 443–458. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, M.E.; Fibbe, W.E. Mesenchymal stromal cells: Sensors and switchers of inflammation. Cell Stem Cell 2013, 13, 392–402. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, R.J.; Koehne, G.; Hasan, A.N.; Doubrovina, E.; Prockop, S. T-cell depleted allogeneic hematopoietic cell transplants as a platform for adoptive therapy with leukemia selective or virus-specific T-cells. Bone Marrow Transpl. 2015, 50, S43–S50. [Google Scholar] [CrossRef] [PubMed]
- Or-Geva, N.; Reisner, Y. The evolution of T-cell depletion in haploidentical stem-cell transplantation. Br. J. Haematol. 2016, 172, 667–684. [Google Scholar] [CrossRef] [PubMed]
- Aversa, F.; Martelli, M.F.; Velardi, A. Haploidentical hematopoietic stem cell transplantation with a megadose T-cell-depleted graft: Harnessing natural and adaptive immunity. Semin. Oncol. 2012, 39, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Schumm, M.; Lang, P.; Bethge, W.; Faul, C.; Feuchtinger, T.; Pfeiffer, M.; Vogel, W.; Huppert, V.; Handgretinger, R. Depletion of T-cell receptor ab and CD19 positive cells from apheresis products with the CliniMACS device. Cytotherapy 2013, 15, 1253–1258. [Google Scholar] [CrossRef] [PubMed]
- Lang, P.; Teltschik, H.M.; Feuchtinger, T.; Muller, I.; Pfeiffer, M.; Schumm, M.; Ebinger, M.; Schwarze, C.P.; Gruhn, B.; Schrauder, A.; et al. Transplantation of CD3/CD19 depleted allografts from haploidentical family donors in paediatric leukaemia. Br. J. Haematol. 2014, 165, 688–698. [Google Scholar] [CrossRef] [PubMed]
- Bertaina, A.; Merli, P.; Rutella, S.; Pagliara, D.; Bernardo, M.E.; Masetti, R.; Pende, D.; Falco, M.; Handgretinger, R.; Moretta, F.; et al. HLA-haploidentical stem cell transplantation after removal of αβ+ T and B-cells in children with non-malignant disorders. Blood 2014, 124, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Di Stasi, A.; Tey, S.K.; Dotti, G.; Fujita, Y.; Kennedy-Nasser, A.; Martinez, C.; Straathof, K.; Liu, E.; Durett, A.G.; Grilley, B.; et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N. Engl. J. Med. 2011, 365, 1673–1683. [Google Scholar] [CrossRef] [PubMed]
- Feucht, J.; Opherk, K.; Lang, P.; Kayser, S.; Hartl, L.; Bethge, W.; Matthes-Martin, S.; Bader, P.; Albert, M.H.; Maecker-Kolhoff, B.; et al. Adoptive T-cell therapy with hexon-specific Th1 cells as a treatment of refractory adenovirus infection after HSCT. Blood 2015, 125, 1986–1994. [Google Scholar] [CrossRef] [PubMed]
- Maschan, M.; Shelikhova, L.; Ilushina, M.; Kurnikova, E.; Boyakova, E.; Balashov, D.; Persiantseva, M.; Skvortsova, Y.; Laberko, A.; Muzalevskii, Y.; et al. TCR-αβ and CD19 depletion and treosulfan-based conditioning regimen in unrelated and haploidentical transplantation in children with acute myeloid leukemia. Bone Marrow Transpl. 2016, 51, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Balashov, D.; Shcherbina, A.; Maschan, M.; Trakhtman, P.; Skvortsova, Y.; Shelikhova, L.; Laberko, A.; Livshits, A.; Novichkova, G.; Maschan, A.; et al. Single-center experience of unrelated and haploidentical stem cell transplantation with TCR-αβ and CD19 depletion in children with primary immunodeficiency syndromes. Biol. Blood Marrow Transpl. 2015, 21, 1955–1962. [Google Scholar] [CrossRef] [PubMed]
- Lang, P.; Feuchtinger, T.; Teltschik, H.M.; Schwinger, W.; Schlegel, P.; Pfeiffer, M.; Schumm, M.; Lang, A.M.; Lang, B.; Schwarze, C.P.; et al. Improved immune recovery after transplantation of TCR-αβ/CD19-depleted allografts from haploidentical donors in pediatric patients. Bone Marrow Transpl. 2015, 50, S6–S10. [Google Scholar] [CrossRef] [PubMed]
- Rutella, S.; Filippini, P.; Bertaina, V.; Li Pira, G.; Altomare, L.; Ceccarelli, S.; Brescia, L.P.; Lucarelli, B.; Girolami, E.; Conflitti, G.; et al. Mobilization of healthy donors with plerixafor affects the cellular composition of T-cell receptor (TCR)-αβ/CD19-depleted haploidentical stem cell grafts. J. Transl. Med. 2014, 12, 240. [Google Scholar] [CrossRef] [PubMed]
- Kaynar, L.; Demir, K.; Turak, E.E.; Ozturk, C.P.; Zararsiz, G.; Gonen, Z.B.; Gokahmetoglu, S.; Sivgin, S.; Eser, B.; Koker, Y.; et al. TCR-αβ-depleted haploidentical transplantation results in adult acute leukemia patients. Hematology 2017, 22, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, F.; Merli, P.; Pagliara, D.; Li Pira, G.; Falco, M.; Pende, D.; Rondelli, R.; Lucarelli, B.; Brescia, L.P.; Masetti, R.; et al. Outcome of children with acute leukemia given HLA-haploidentical HSCT after αβ T-cell and B-cell depletion. Blood 2017. [Google Scholar] [CrossRef] [PubMed]
- Bremm, M.; Cappel, C.; Erben, S.; Jarisch, A.; Schumm, M.; Arendt, A.; Bonig, H.; Klingebiel, T.; Koehl, U.; Bader, P.; et al. Generation and flow cytometric quality control of clinical-scale TCR-αβ/CD19-depleted grafts. Cytom. B Clin. Cytom. 2017, 92, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Ogonek, J.; Kralj Juric, M.; Ghimire, S.; Varanasi, P.R.; Holler, E.; Greinix, H.; Weissinger, E. Immune reconstitution after allogeneic hematopoietic stem cell transplantation. Front. Immunol. 2016, 7, 507. [Google Scholar] [CrossRef] [PubMed]
- Anderson, B.E.; McNiff, J.; Yan, J.; Doyle, H.; Mamula, M.; Shlomchik, M.J.; Shlomchik, W.D. Memory CD4+ T cells do not induce graft-versus-host disease. J. Clin. Investig. 2003, 112, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Bleakley, M.; Heimfeld, S.; Loeb, K.R.; Jones, L.A.; Chaney, C.; Seropian, S.; Gooley, T.A.; Sommermeyer, F.; Riddell, S.R.; Shlomchik, W.D. Outcomes of acute leukemia patients transplanted with naive T cell-depleted stem cell grafts. J. Clin. Investig. 2015, 125, 2677–2689. [Google Scholar] [CrossRef] [PubMed]
- Bleakley, M.; Heimfeld, S.; Jones, L.A.; Turtle, C.; Krause, D.; Riddell, S.R.; Shlomchik, W. Engineering human peripheral blood stem cell grafts that are depleted of naive T cells and retain functional pathogen-specific memory T cells. Biol. Blood Marrow Transpl. 2014, 20, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Mielke, S.; Nunes, R.; Rezvani, K.; Fellowes, V.S.; Venne, A.; Solomon, S.R.; Fan, Y.; Gostick, E.; Price, D.A.; Scotto, C.; et al. A clinical-scale selective allodepletion approach for the treatment of HLA-mismatched and matched donor-recipient pairs using expanded T lymphocytes as antigen-presenting cells and a TH9402-based photodepletion technique. Blood 2008, 111, 4392–4402. [Google Scholar] [CrossRef] [PubMed]
- Mielke, S.; McIver, Z.A.; Shenoy, A.; Fellowes, V.; Khuu, H.; Stroncek, D.F.; Leitman, S.F.; Childs, R.; Battiwalla, M.; Koklanaris, E.; et al. Selectively T cell-depleted allografts from HLA-matched sibling donors followed by low-dose posttransplantation immunosuppression to improve transplantation outcome in patients with hematologic malignancies. Biol. Blood Marrow Transpl. 2011, 17, 1855–1861. [Google Scholar] [CrossRef] [PubMed]
- McIver, Z.A.; Melenhorst, J.J.; Grim, A.; Naguib, N.; Weber, G.; Fellowes, V.; Khuu, H.; Stroncek, D.S.; Leitman, S.F.; Battiwalla, M.; et al. Immune reconstitution in recipients of photodepleted HLA-identical sibling donor stem cell transplantations: T cell subset frequencies predict outcome. Biol. Blood Marrow Transpl. 2011, 17, 1846–1854. [Google Scholar] [CrossRef] [PubMed]
- Roy, D.C.; Lachance, S.; Roy, J.; Walker, I.; Maerten, J.; Delisle, J.S.; Foley, S.R.; Lewalle, P.; Olavarria, E.; Selleslag, D.; et al. Donor lymphocytes depleted of alloreactive T cells (ATIR101) improve event-free survival (GRFS) and overall survival in a T-cell depleted haploidentical HSCT: Phase 2 trial in patients with AML and ALL. Blood 2016, 128, 1226. [Google Scholar]
- Shook, D.R.; Triplett, B.M.; Eldridge, P.W.; Kang, G.; Srinivasan, A.; Leung, W. Haploidentical stem cell transplantation augmented by CD45RA negative lymphocytes provides rapid engraftment and excellent tolerability. Pediatr. Blood Cancer 2015, 62, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Triplett, B.M.; Shook, D.R.; Eldridge, P.; Li, Y.; Kang, G.; Dallas, M.; Hartford, C.; Srinivasan, A.; Chan, W.K.; Suwannasaen, D.; et al. Rapid memory T-cell reconstitution recapitulating CD45RA-depleted haploidentical transplant graft content in patients with hematologic malignancies. Bone Marrow Transpl. 2015, 50, 968–977. [Google Scholar] [CrossRef] [PubMed]
- Robinson, T.M.; O’Donnell, P.V.; Fuchs, E.J.; Luznik, L. Haploidentical bone marrow and stem cell transplantation: Experience with post-transplantation cyclophosphamide. Semin. Hematol. 2016, 53, 90–97. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, P.V.; Luznik, L.; Jones, R.J.; Vogelsang, G.B.; Leffell, M.S.; Phelps, M.; Rhubart, P.; Cowan, K.; Piantados, S.; Fuchs, E.J.; et al. Nonmyeloablative bone marrow transplantation from partially HLA-mismatched related donors using posttransplantation cyclophosphamide. Biol. Blood Marrow Transpl. 2002, 8, 377–386. [Google Scholar] [CrossRef]
- Luznik, L.; O’Donnell, P.V.; Symons, H.J.; Chen, A.R.; Leffell, M.S.; Zahurak, M.; Gooley, T.A.; Piantadosi, S.; Kaup, M.; Ambinder, R.F.; et al. HLA-haploidentical bone marrow transplantation for hematologic malignancies using nonmyeloablative conditioning and high-dose, posttransplantation cyclophosphamide. Biol. Blood Marrow Transpl. 2008, 14, 641–650. [Google Scholar] [CrossRef] [PubMed]
- McCurdy, S.R.; Kanakry, J.A.; Showel, M.M.; Tsai, H.L.; Bolanos-Meade, J.; Rosner, G.L.; Kanakry, C.G.; Perica, K.; Symons, H.J.; Brodsky, R.A.; et al. Risk-stratified outcomes of nonmyeloablative HLA-haploidentical BMT with high-dose posttransplantation cyclophosphamide. Blood 2015, 125, 3024–3031. [Google Scholar] [CrossRef] [PubMed]
- Kanakry, C.G.; O’Donnell, P.V.; Furlong, T.; de Lima, M.J.; Wei, W.; Medeot, M.; Mielcarek, M.; Champlin, R.E.; Jones, R.J.; Thall, P.F.; et al. Multi-institutional study of post-transplantation cyclophosphamide as single-agent graft-versus-host disease prophylaxis after allogeneic bone marrow transplantation using myeloablative busulfan and fludarabine conditioning. J. Clin. Oncol. 2014, 32, 3497–3505. [Google Scholar] [CrossRef] [PubMed]
- Raiola, A.M.; Dominietto, A.; Ghiso, A.; di Grazia, C.; Lamparelli, T.; Gualandi, F.; Bregante, S.; van Lint, M.T.; Geroldi, S.; Luchetti, S.; et al. Unmanipulated haploidentical bone marrow transplantation and posttransplantation cyclophosphamide for hematologic malignancies after myeloablative conditioning. Biol. Blood Marrow Transpl. 2013, 19, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Bacigalupo, A.; Dominietto, A.; Ghiso, A.; di Grazia, C.; Lamparelli, T.; Gualandi, F.; Bregante, S.; van Lint, M.T.; Geroldi, S.; Luchetti, S.; et al. Unmanipulated haploidentical bone marrow transplantation and post-transplant cyclophosphamide for hematologic malignanices following a myeloablative conditioning: An update. Bone Marrow Transpl. 2015, 50, S37–S39. [Google Scholar] [CrossRef] [PubMed]
- Rutella, S.; Pierelli, L.; Bonanno, G.; Sica, S.; Ameglio, F.; Capoluongo, E.; Mariotti, A.; Scambia, G.; d’Onofrio, G.; Leone, G.; et al. Role for granulocyte colony-stimulating factor in the generation of human T regulatory type 1 cells. Blood 2002, 100, 2562–2571. [Google Scholar] [CrossRef] [PubMed]
- Rutella, S.; Bonanno, G.; Pierelli, L.; Mariotti, A.; Capoluongo, E.; Contemi, A.M.; Ameglio, F.; Curti, A.; de Ritis, D.G.; Voso, M.T.; et al. Granulocyte colony-stimulating factor promotes the generation of regulatory DC through induction of IL-10 and IFN-α. Eur. J. Immunol. 2004, 34, 1291–1302. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.; Hill, G.R.; Pan, L.; Gerbitz, A.; Teshima, T.; Brinson, Y.; Ferrara, J.L. G-CSF modulates cytokine profile of dendritic cells and decreases acute graft-versus-host disease through effects on the donor rather than the recipient. Transplantation 2000, 69, 691–693. [Google Scholar] [CrossRef] [PubMed]
- Vasu, S.; Geyer, S.; Bingman, A.; Auletta, J.J.; Jaglowski, S.; Elder, P.; Donnell, L.O.; Bradbury, H.; Kitzler, R.; Andritsos, L.; et al. Granulocyte colony-stimulating factor-mobilized allografts contain activated immune cell subsets associated with risk of acute and chronic graft-versus-host disease. Biol. Blood Marrow Transpl. 2016, 22, 658–668. [Google Scholar] [CrossRef] [PubMed]
- D’Aveni, M.; Rossignol, J.; Coman, T.; Sivakumaran, S.; Henderson, S.; Manzo, T.; Santos e Sousa, P.; Bruneau, J.; Fouquet, G.; Zavala, F.; et al. G-CSF mobilizes CD34+ regulatory monocytes that inhibit graft-versus-host disease. Sci. Transl. Med. 2015, 7, 281ra242. [Google Scholar] [CrossRef] [PubMed]
- Vendramin, A.; Gimondi, S.; Bermema, A.; Longoni, P.; Rizzitano, S.; Corradini, P.; Carniti, C. Graft monocytic myeloid-derived suppressor cell content predicts the risk of acute graft-versus-host disease after allogeneic transplantation of granulocyte colony-stimulating factor-mobilized peripheral blood stem cells. Biol. Blood Marrow Transpl. 2014, 20, 2049–2055. [Google Scholar] [CrossRef] [PubMed]
- Pessach, I.; Resnick, I.; Shimoni, A.; Nagler, A. G-CSF-primed BM for allogeneic SCT: Revisited. Bone Marrow Transpl. 2015, 50, 892–898. [Google Scholar] [CrossRef] [PubMed]
- Jun, H.X.; Jun, C.Y.; Yu, Z.X. A direct comparison of immunological characteristics of granulocyte colony-stimulating factor (G-CSF)-primed bone marrow grafts and G-CSF-mobilized peripheral blood grafts. Haematologica 2005, 90, 715–716. [Google Scholar] [PubMed]
- Huang, X.J.; Liu, D.H.; Liu, K.Y.; Xu, L.P.; Chen, H.; Han, W.; Chen, Y.H.; Zhang, X.H.; Lu, D.P. Treatment of acute leukemia with unmanipulated HLA-mismatched/haploidentical blood and bone marrow transplantation. Biol. Blood Marrow Transpl. 2009, 15, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.J.; Chang, Y.J.; Zhao, X.Y. Maintaining hyporesponsiveness and polarization potential of T cells after in vitro mixture of G-CSF mobilized peripheral blood grafts and G-CSF primed bone marrow grafts in different proportions. Transpl. Immunol. 2007, 17, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Bollard, C.M.; Carlsten, M.; Melenhorst, J.J.; Biancotto, A.; Wang, E.; Chen, J.; Kotliarov, Y.; Cheung, F.; Xie, Z.; et al. Ultra-low dose interleukin-2 promotes immune-modulating function of regulatory T cells and natural killer cells in healthy volunteers. Mol. Ther. 2014, 22, 1388–1395. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.Y.; Zhao, X.S.; Wang, Y.T.; Chen, Y.H.; Xu, L.P.; Zhang, X.H.; Han, W.; Chen, H.; Wang, Y.; Yan, C.H.; et al. Prophylactic use of low-dose interleukin-2 and the clinical outcomes of hematopoietic stem cell transplantation: A randomized study. Oncoimmunology 2016, 5, e1250992. [Google Scholar] [CrossRef] [PubMed]
- Koreth, J.; Matsuoka, K.; Kim, H.T.; McDonough, S.M.; Bindra, B.; Alyea, E.P., 3rd; Armand, P.; Cutler, C.; Ho, V.T.; Treister, N.S.; et al. Interleukin-2 and regulatory T cells in graft-versus-host disease. N. Engl. J. Med. 2011, 365, 2055–2066. [Google Scholar] [CrossRef] [PubMed]
- Di Nardo, M.; Li Pira, G.; Amodeo, A.; Cecchetti, C.; Giorda, E.; Ceccarelli, S.; Brescia, L.P.; Pirozzi, N.; Rutella, S.; Locatelli, F.; et al. Adoptive immunotherapy with antigen-specific T cells during extracorporeal membrane oxygenation (ECMO) for adenovirus-related respiratory failure in a child given haploidentical stem cell transplantation. Pediatr. Blood Cancer 2014, 61, 376–379. [Google Scholar] [CrossRef] [PubMed]
- Tyler, E.M.; Jungbluth, A.A.; O’Reilly, R.J.; Koehne, G. WT1-specific T-cell responses in high-risk multiple myeloma patients undergoing allogeneic T cell-depleted hematopoietic stem cell transplantation and donor lymphocyte infusions. Blood 2013, 121, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Chapuis, A.G.; Ragnarsson, G.B.; Nguyen, H.N.; Chaney, C.N.; Pufnock, J.S.; Schmitt, T.M.; Duerkopp, N.; Roberts, I.M.; Pogosov, G.L.; Ho, W.Y.; et al. Transferred WT1-reactive CD8+ T cells can mediate antileukemic activity and persist in post-transplant patients. Sci. Transl. Med. 2013, 5, 174ra127. [Google Scholar] [CrossRef] [PubMed]
- Ciceri, F.; Bonini, C.; Stanghellini, M.T.; Bondanza, A.; Traversari, C.; Salomoni, M.; Turchetto, L.; Colombi, S.; Bernardi, M.; Peccatori, J.; et al. Infusion of suicide-gene-engineered donor lymphocytes after family haploidentical haemopoietic stem-cell transplantation for leukaemia (the TK007 trial): A non-randomised phase I-II study. Lancet Oncol. 2009, 10, 489–500. [Google Scholar] [CrossRef]
- Zhou, X.; di Stasi, A.; Tey, S.K.; Krance, R.A.; Martinez, C.; Leung, K.S.; Durett, A.G.; Wu, M.F.; Liu, H.; Leen, A.M.; et al. Long-term outcome after haploidentical stem cell transplant and infusion of T cells expressing the inducible caspase 9 safety transgene. Blood 2014, 123, 3895–3905. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Dotti, G.; Krance, R.A.; Martinez, C.A.; Naik, S.; Kamble, R.T.; Durett, A.G.; Dakhova, O.; Savoldo, B.; di Stasi, A.; et al. Inducible caspase-9 suicide gene controls adverse effects from alloreplete T cells after haploidentical stem cell transplantation. Blood 2015, 125, 4103–4113. [Google Scholar] [CrossRef] [PubMed]
- Tzannou, I.; Leen, A.M. Preventing stem cell transplantation-associated viral infections using T-cell therapy. Immunotherapy 2015, 7, 793–810. [Google Scholar] [CrossRef] [PubMed]
- Leen, A.M.; Bollard, C.M.; Mendizabal, A.M.; Shpall, E.J.; Szabolcs, P.; Antin, J.H.; Kapoor, N.; Pai, S.Y.; Rowley, S.D.; Kebriaei, P.; et al. Multicenter study of banked third-party virus-specific T cells to treat severe viral infections after hematopoietic stem cell transplantation. Blood 2013, 121, 5113–5123. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.; Maeda, Y.; Hotary, K.; Liu, C.; Reznikov, L.L.; Dinarello, C.A.; Ferrara, J.L. Histone deacetylase inhibitor suberoylanilide hydroxamic acid reduces acute graft-versus-host disease and preserves graft-versus-leukemia effect. Proc. Natl. Acad. Sci. USA 2004, 101, 3921–3926. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.; Sun, Y.; Toubai, T.; Duran-Struuck, R.; Clouthier, S.G.; Weisiger, E.; Maeda, Y.; Tawara, I.; Krijanovski, O.; Gatza, E.; et al. Histone deacetylase inhibition modulates indoleamine 2,3-dioxygenase-dependent DC functions and regulates experimental graft-versus-host disease in mice. J. Clin. Investig. 2008, 118, 2562–2573. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, G.; Okiyama, N.; Villarroel, V.A.; Katz, S.I. Histone deacetylase 6 inhibition impairs effector CD8 T-cell functions during skin inflammation. J. Allergy Clin. Immunol. 2015, 135, 1228–1239. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Wang, J.; Kato, K.; Xie, F.; Varambally, S.; Mineishi, S.; Kuick, R.; Mochizuki, K.; Liu, Y.; Nieves, E.; et al. Inhibition of histone methylation arrests ongoing graft-versus-host disease in mice by selectively inducing apoptosis of alloreactive effector T cells. Blood 2012, 119, 1274–1282. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhang, Y.; Xu, M.; Rong, R.; Wang, J.; Zhu, T. Inhibition of histone methyltransferase EZH2 ameliorates early acute renal allograft rejection in rats. BMC Immunol. 2016, 17, 41. [Google Scholar] [CrossRef] [PubMed]
- Sandy, A.R.; Chung, J.; Toubai, T.; Shan, G.T.; Tran, I.T.; Friedman, A.; Blackwell, T.S.; Reddy, P.; King, P.D.; Maillard, I. T cell-specific notch inhibition blocks graft-versus-host disease by inducing a hyporesponsive program in alloreactive CD4+ and CD8+ T cells. J. Immunol. 2013, 190, 5818–5828. [Google Scholar] [CrossRef] [PubMed]
- Gatza, E.; Wahl, D.R.; Opipari, A.W.; Sundberg, T.B.; Reddy, P.; Liu, C.; Glick, G.D.; Ferrara, J.L. Manipulating the bioenergetics of alloreactive T cells causes their selective apoptosis and arrests graft-versus-host disease. Sci. Transl. Med. 2011, 3, 67ra68. [Google Scholar] [CrossRef] [PubMed]
- Haarberg, K.M.; Li, J.; Heinrichs, J.; Wang, D.; Liu, C.; Bronk, C.C.; Kaosaard, K.; Owyang, A.M.; Holland, S.; Masuda, E.; et al. Pharmacologic inhibition of PKCα and PKCθ prevents GVHD while preserving GVL activity in mice. Blood 2013, 122, 2500–2511. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Cooper, M.L.; Alahmari, B.; Ritchey, J.; Collins, L.; Holt, M.; di Persio, J.F. Pharmacologic blockade of JAK1/JAK2 reduces GvHD and preserves the graft-versus-leukemia effect. PLoS ONE 2014, 9, e109799. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Oravecz-Wilson, K.; Mathewson, N.; Wang, Y.; McEachin, R.; Liu, C.; Toubai, T.; Wu, J.; Rossi, C.; Braun, T.; et al. Mature T cell responses are controlled by microRNA-142. J. Clin. Investig. 2015, 125, 2825–2840. [Google Scholar] [CrossRef] [PubMed]
- Furlan, S.N.; Watkins, B.; Tkachev, V.; Flynn, R.; Cooley, S.; Ramakrishnan, S.; Singh, K.; Giver, C.; Hamby, K.; Stempora, L.; et al. Transcriptome analysis of GVHD reveals aurora kinase A as a targetable pathway for disease prevention. Sci. Transl. Med. 2015, 7, 315ra191. [Google Scholar] [CrossRef] [PubMed]
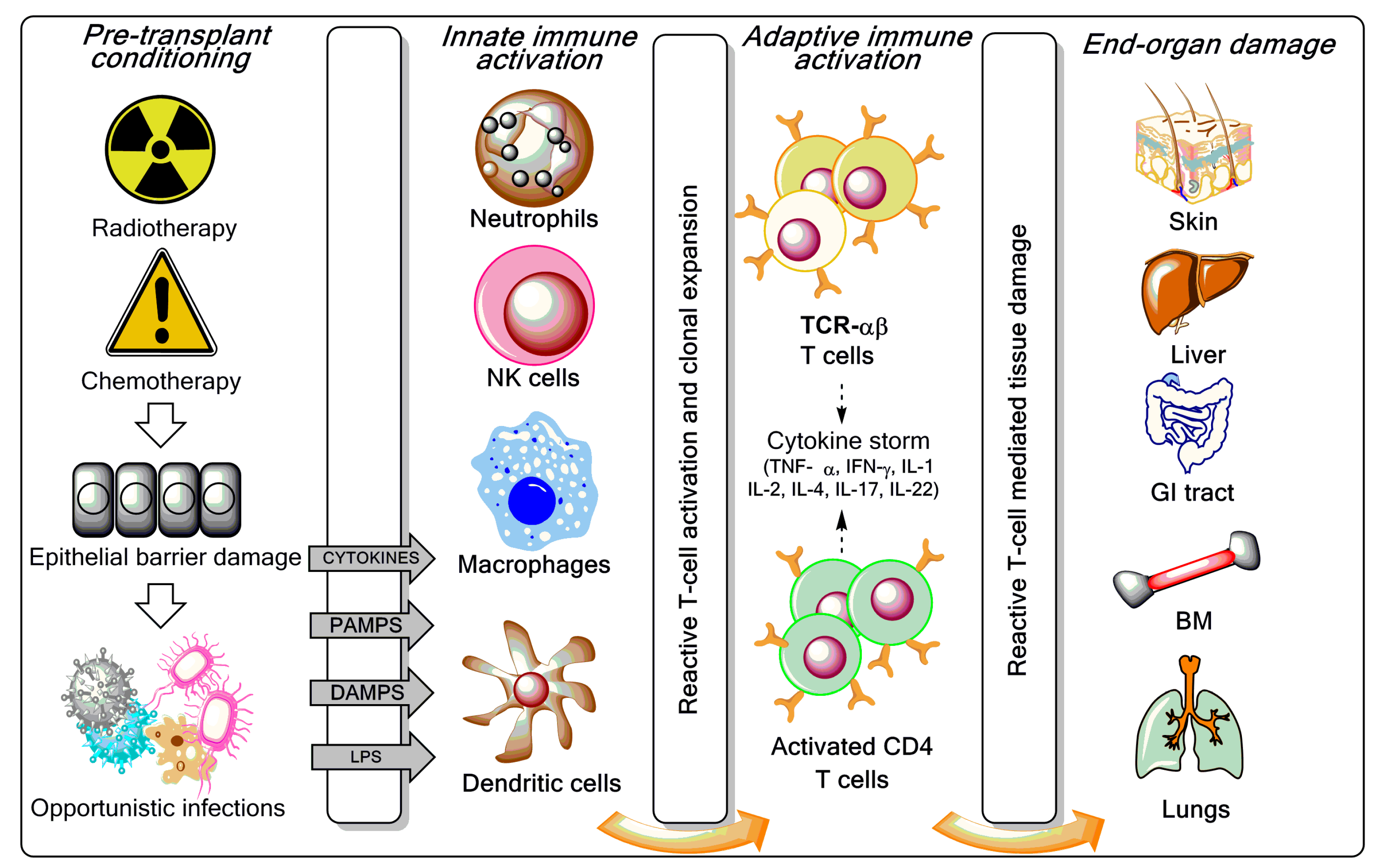
Figure 1.
Overview of graft-versus-host disease (GVHD) pathogenesis. As reviewed elsewhere [3,7,8], the pre-transplant conditioning regimen (chemotherapy with or without radiotherapy) contributes to GVHD induction via tissue destruction, bacterial translocation across gut mucosal cells, and release of pro-inflammatory cytokines such as tumour necrosis factor (TNF)-α, interleukin (IL)-1, and IL-7. Lipopolysaccharide (LPS) and other components of the bacterial cell wall leaking through the damaged intestinal mucosa stimulate mononuclear cells, amplify the production of inflammatory cytokines, and contribute to apoptosis. Innate immune cells partake in tissue damage and cytokine production, a phenomenon referred to as “cytokine storm”. Both host and donor antigen-presenting cells (APCs) initiate GVH responses through the release of IL-12 and IL-23. Activated T cells, natural killer (NK) cells, and macrophages mediate end-organ damage through cytokine production and direct cytotoxic effects on target tissues (release of cytolytic granules and expression of the CD95 ligand). Acute GVHD develops in parenchymal targets containing highly proliferating cells (bone marrow, skin, liver, gut, and lungs). CD4+ T cells are also activated by macrophages and dendritic cells (DCs) to produce pro-inflammatory mediators. CTL = cytotoxic T lymphocyte; IFN = interferon; PAMP = pathogen-associated molecular pattern; DAMP = damage-associated molecular pattern; TCR = T-cell receptor; BM = bone marrow; GI = gastrointestinal.
Figure 1.
Overview of graft-versus-host disease (GVHD) pathogenesis. As reviewed elsewhere [3,7,8], the pre-transplant conditioning regimen (chemotherapy with or without radiotherapy) contributes to GVHD induction via tissue destruction, bacterial translocation across gut mucosal cells, and release of pro-inflammatory cytokines such as tumour necrosis factor (TNF)-α, interleukin (IL)-1, and IL-7. Lipopolysaccharide (LPS) and other components of the bacterial cell wall leaking through the damaged intestinal mucosa stimulate mononuclear cells, amplify the production of inflammatory cytokines, and contribute to apoptosis. Innate immune cells partake in tissue damage and cytokine production, a phenomenon referred to as “cytokine storm”. Both host and donor antigen-presenting cells (APCs) initiate GVH responses through the release of IL-12 and IL-23. Activated T cells, natural killer (NK) cells, and macrophages mediate end-organ damage through cytokine production and direct cytotoxic effects on target tissues (release of cytolytic granules and expression of the CD95 ligand). Acute GVHD develops in parenchymal targets containing highly proliferating cells (bone marrow, skin, liver, gut, and lungs). CD4+ T cells are also activated by macrophages and dendritic cells (DCs) to produce pro-inflammatory mediators. CTL = cytotoxic T lymphocyte; IFN = interferon; PAMP = pathogen-associated molecular pattern; DAMP = damage-associated molecular pattern; TCR = T-cell receptor; BM = bone marrow; GI = gastrointestinal.
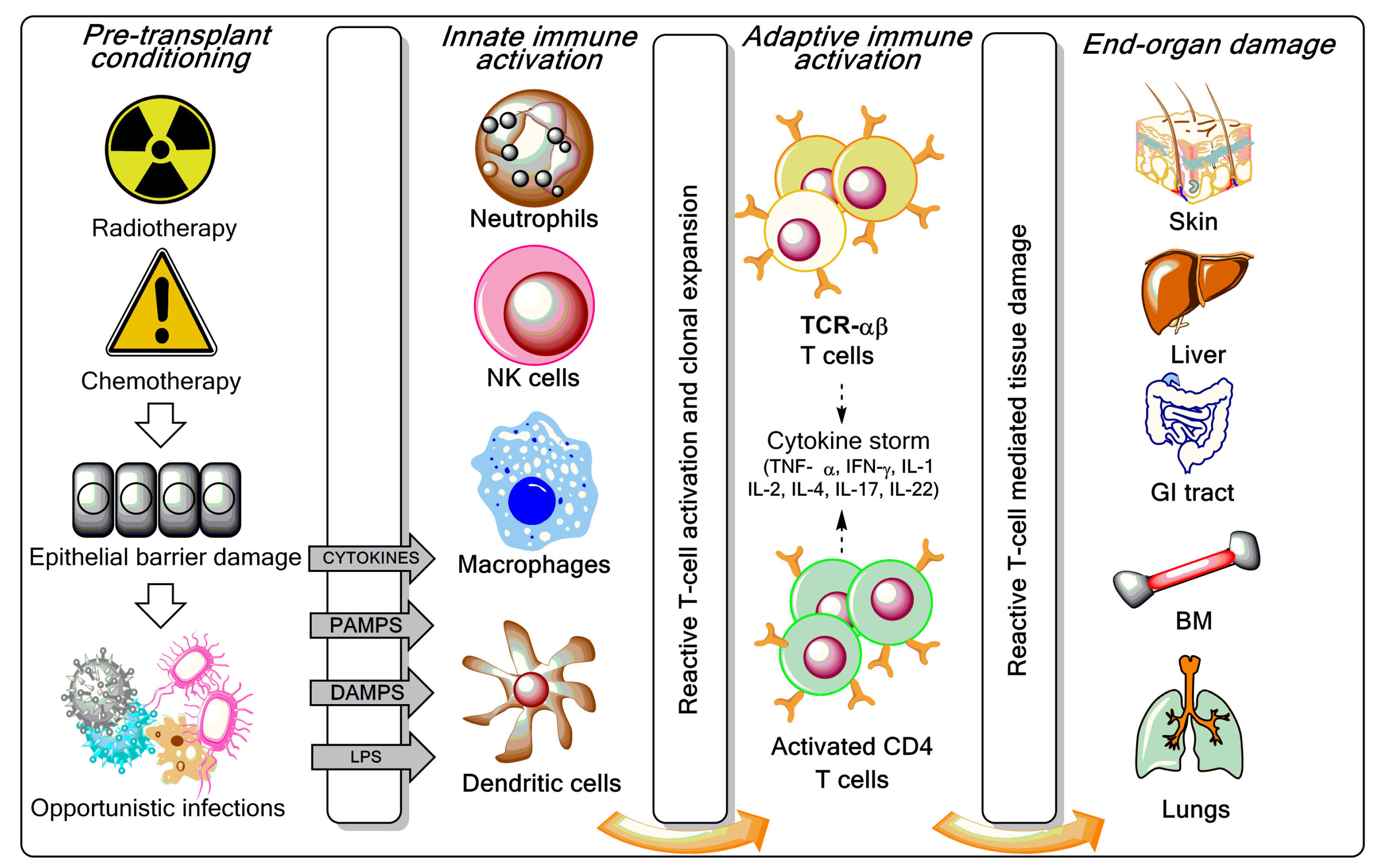
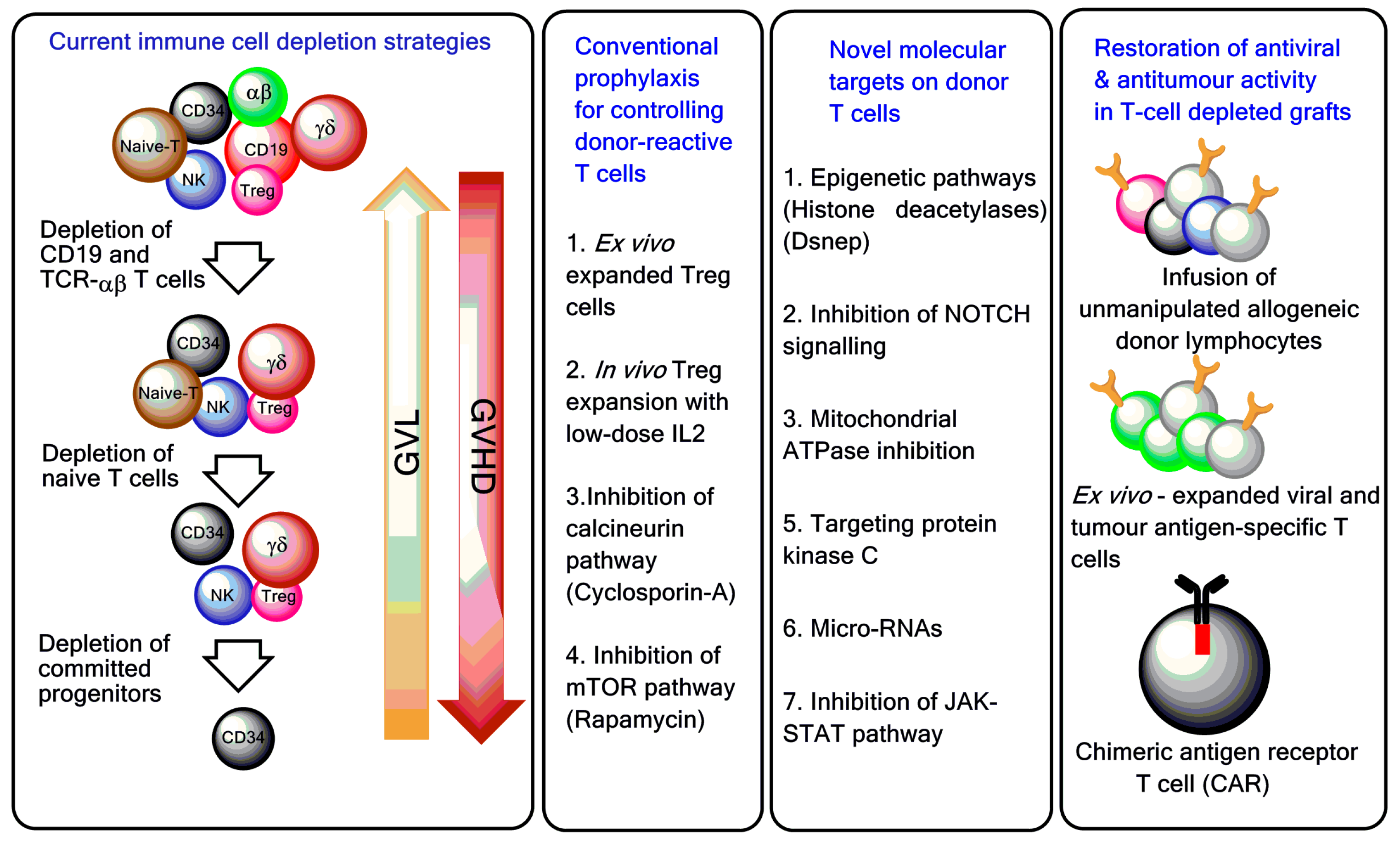
Figure 2.
T-cell manipulation strategies to control GVHD and improve post-haematopoietic stem cell transplantation (HSCT) immune reconstitution. Ex vivo T-cell depletion techniques have evolved and currently include CD3/CD19 depletion, TCR-αβ/CD19 depletion, and infusion of CD45RA-depleted grafts [14,15,16]. To boost immune reconstitution after T-cell depleted haploidentical HSCT, genetically modified T cells and pathogen-specific T cells are increasingly used in the clinic [17,18]. Treg = regulatory T cell; GVL = graft-versus-leukaemia; mTOR = mechanistic target of rapamycin; NOTCH = neurogenic locus notch homolog protein; JAK-STAT = Janus kinase/signal transducer and activator of transcription.
Figure 2.
T-cell manipulation strategies to control GVHD and improve post-haematopoietic stem cell transplantation (HSCT) immune reconstitution. Ex vivo T-cell depletion techniques have evolved and currently include CD3/CD19 depletion, TCR-αβ/CD19 depletion, and infusion of CD45RA-depleted grafts [14,15,16]. To boost immune reconstitution after T-cell depleted haploidentical HSCT, genetically modified T cells and pathogen-specific T cells are increasingly used in the clinic [17,18]. Treg = regulatory T cell; GVL = graft-versus-leukaemia; mTOR = mechanistic target of rapamycin; NOTCH = neurogenic locus notch homolog protein; JAK-STAT = Janus kinase/signal transducer and activator of transcription.
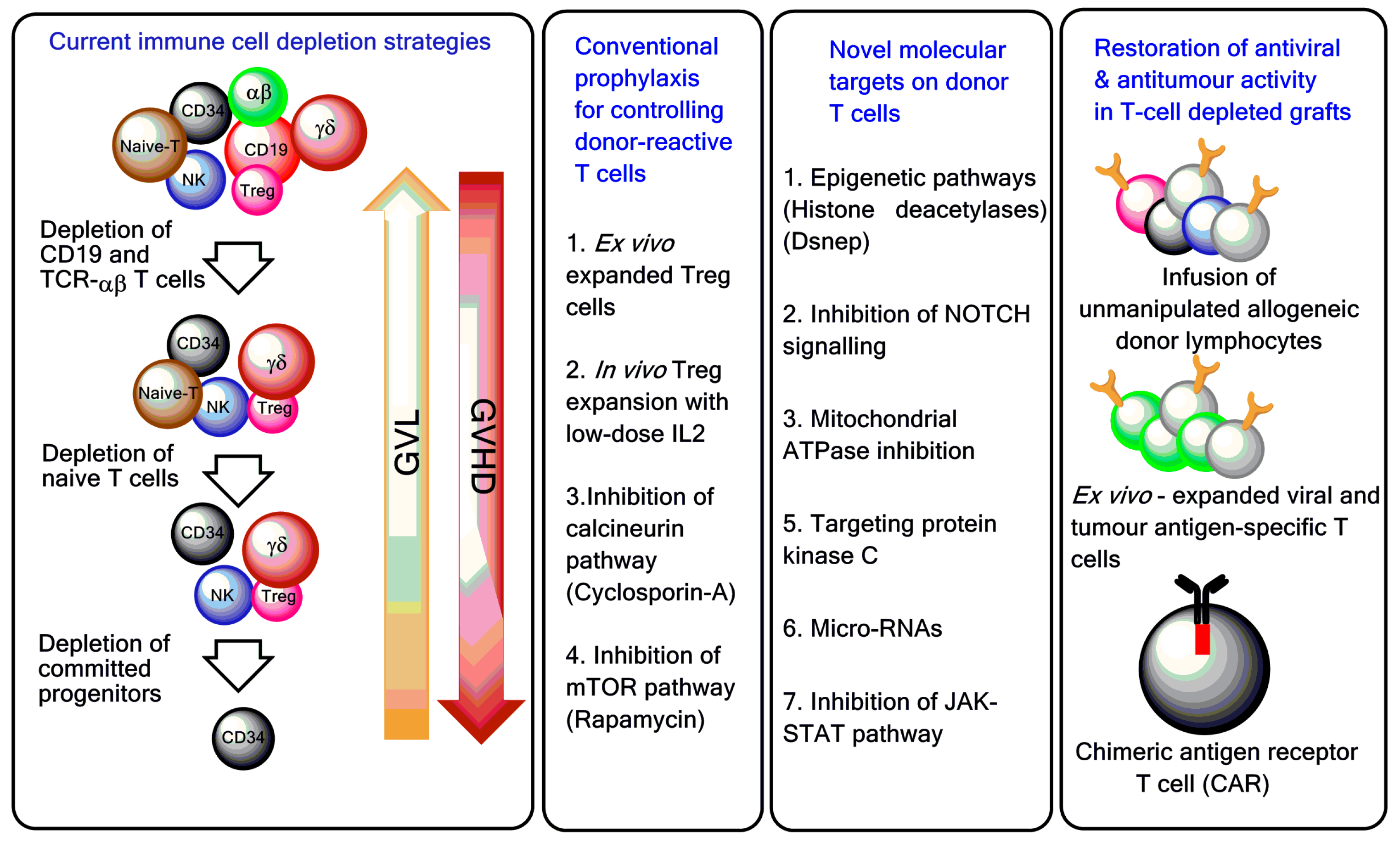
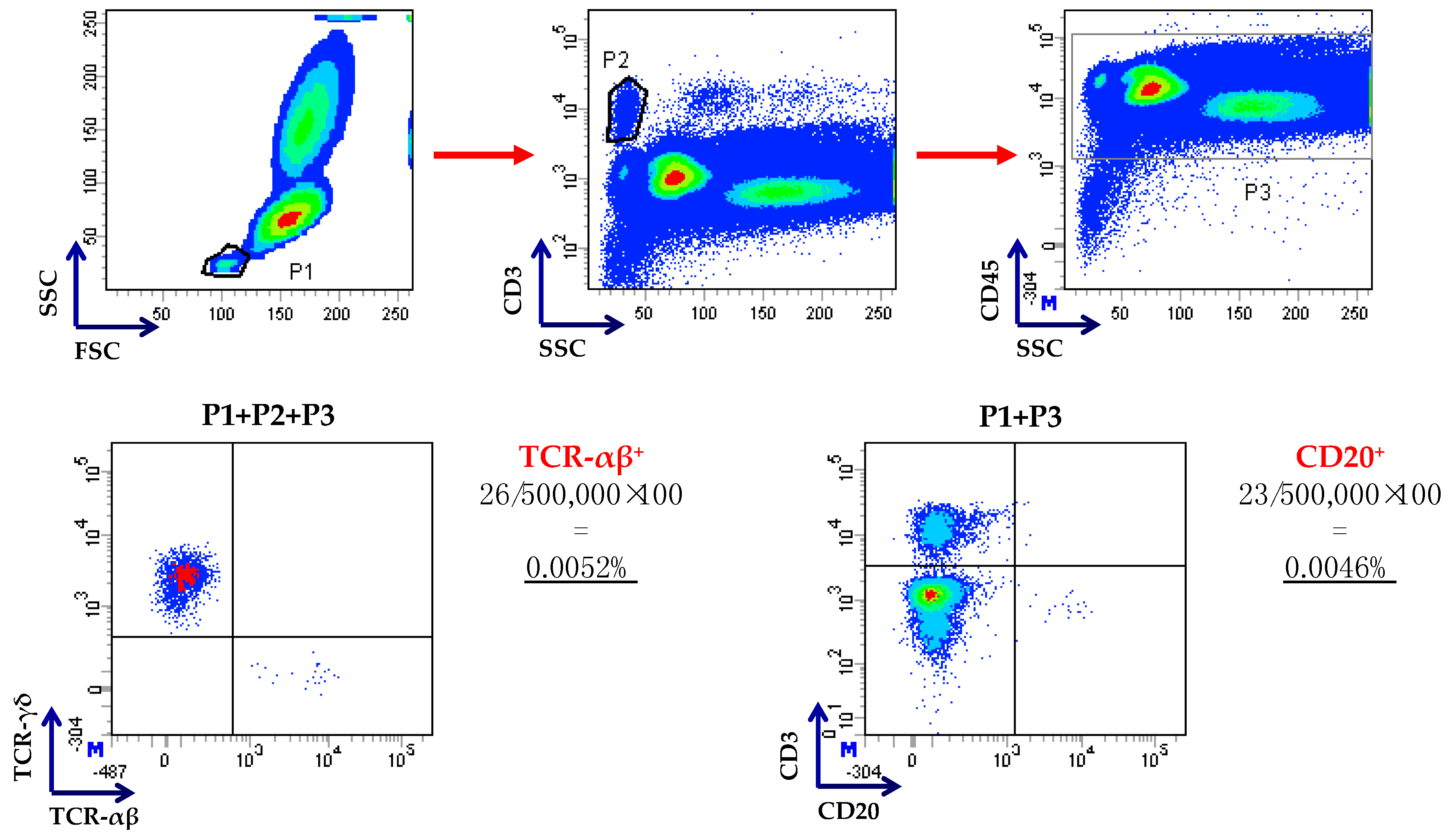
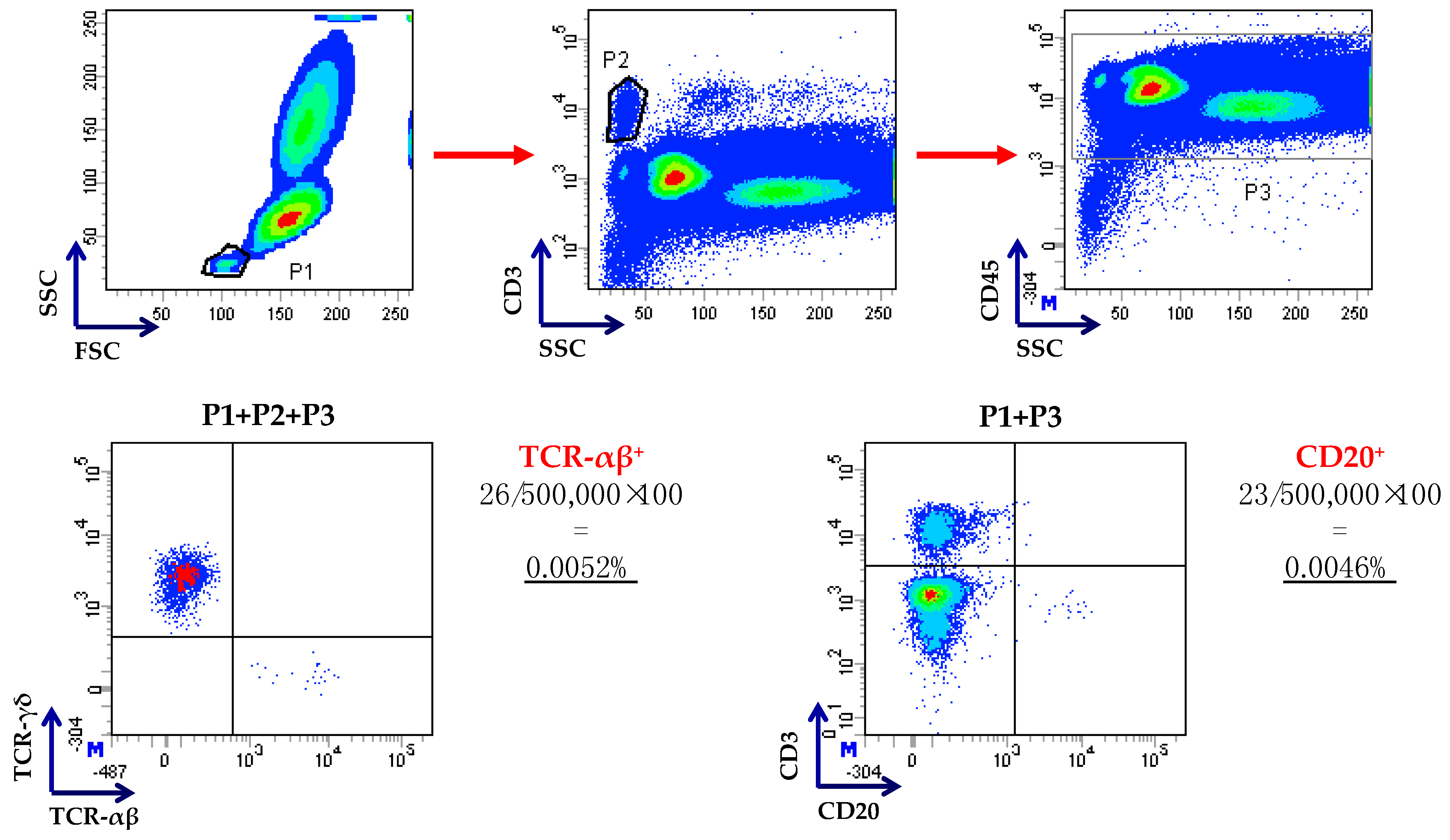
Figure 3.
Flow cytometry-based enumeration of residual T and B cells after TCR-αβ/CD19 depletion. Cells were gated on low side scatter/low forward scatter events (P1), followed by gating on CD3+ T cells (P2) and on CD45+ leukocytes (P3), as already published [22]. Residual TCR-αβ+ and CD20+ B cells were enumerated as shown in this representative procedure. Anti-CD20 monoclonal antibodies were used because of strong internalization of CD19 after the incubation of cells with CD19 reagent or steric hindrance by the beads, as suggested in [14].
Figure 3.
Flow cytometry-based enumeration of residual T and B cells after TCR-αβ/CD19 depletion. Cells were gated on low side scatter/low forward scatter events (P1), followed by gating on CD3+ T cells (P2) and on CD45+ leukocytes (P3), as already published [22]. Residual TCR-αβ+ and CD20+ B cells were enumerated as shown in this representative procedure. Anti-CD20 monoclonal antibodies were used because of strong internalization of CD19 after the incubation of cells with CD19 reagent or steric hindrance by the beads, as suggested in [14].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Clinical trials with TCR-αβ/CD19-depleted haematopoietic stem cells (HSCs).
| Patients | Disease | Graft-versus-Host Disease (GVHD) Prophylaxis | Acute/Chronic GVHD | TRM | EFS(DFS)/OS | Reference |
|---|---|---|---|---|---|---|
| 28 | HR-AML | FK506, MTX | 39%/30% | 10% | 60%/67% (2 years) | [19] |
| 37 | PID | FK506, MTX; FK506, MMF; CYA, MTX | 22% | 3.3% (27% GF) | 96.7% (15 months) | [20] |
| 41 | AL | MMF | 10%/9% | N.A. | 21/41 patients alive after 1.6 years | [21] |
| 23 | Non-malignant | None | 13%/0% | 9.3% | 91% (2 years) | [16,22] |
| 34 | HR-AL | N.A. | 5.9%/6.1% | 14.7% | 42%/54% (1 year) | [23] |
| 80 | AL | None | 30%; no extensive chronic GVHD | 5% | 71%/72% (5 years) | [24] |
Legend: HR-AML = high-risk acute myeloid leukaemia; CYA = cyclosporine-A; MTX = methotrexate; DFS = disease-free survival; EFS = event-free survival; OS = overall survival; GF = graft failure; PID = primary immune deficiencies; AL = acute leukaemia; HR-AL = high-risk acute leukaemia; MMF = mycophenolate mofetil; N.A. = not available; TRM = transplantation-related mortality.
Table 2.
Clinical trials with CD45RA T-cell depletion.
| Patients | Disease | Graft-Versus-Host Disease (GVHD) Prophylaxis | Acute/Chronic GVHD | TRM | EFS(DFS)/OS | Reference |
|---|---|---|---|---|---|---|
| 35 | High-risk leukaemia | Tacrolimus | 66%; 9% | 9% | 70%/78% (2 years) | [29] |
| 8 | Solid tumours | Sirolimus | No acute GVHD or GF | 1 patient died of sinusoidal obstruction syndrome | N.A. (median follow-up was 184 days) | [34] |
| 17 | Haematological malignancies | Sirolimus and MMF | 17.6% grades III–IV acute GVHD/6 patients with signs of oral or skin chronic GVHD | 11.7% | 76.5% of patients alive at a median of 223 days after haematopoietic stem cell transplantation (HSCT) | [35] |
DFS = disease-free survival; EFS = event-free survival; OS = overall survival; TRM = transplantation-related mortality; GF = graft failure; MMF = mycophenolate mofetil; N.A. = not available.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Vadakekolathu, J.; Rutella, S. T-Cell Manipulation Strategies to Prevent Graft-Versus-Host Disease in Haploidentical Stem Cell Transplantation. Biomedicines 2017, 5, 33. https://doi.org/10.3390/biomedicines5020033
AMA Style
Vadakekolathu J, Rutella S. T-Cell Manipulation Strategies to Prevent Graft-Versus-Host Disease in Haploidentical Stem Cell Transplantation. Biomedicines. 2017; 5(2):33. https://doi.org/10.3390/biomedicines5020033
Chicago/Turabian StyleVadakekolathu, Jayakumar, and Sergio Rutella. 2017. "T-Cell Manipulation Strategies to Prevent Graft-Versus-Host Disease in Haploidentical Stem Cell Transplantation" Biomedicines 5, no. 2: 33. https://doi.org/10.3390/biomedicines5020033
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.