Updates in the Development of ImmunoRNases for the Selective Killing of Tumor Cells


Abstract
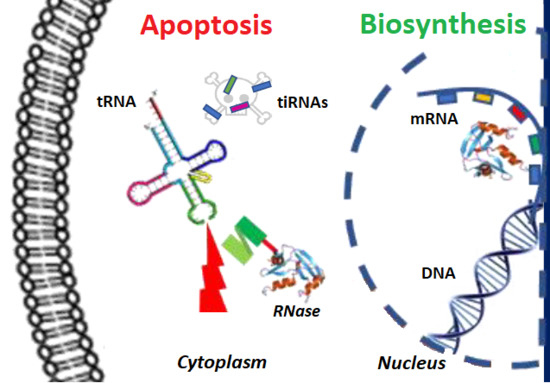
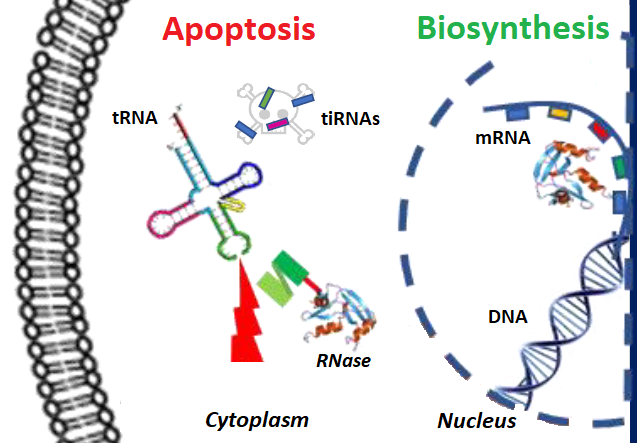
:
1. Introduction to Targeted Therapy and Immunotoxins
2. RNases Studied for Cytotoxicity and Anti-Cancer Activity
2.1. Anti-Tumor Activities of RNases
2.2. Amphibian Ranpirnase: An RNase of Clinical Significance
2.3. Targeted RNases as Anti-Cancer Agents
2.4. Humanized Immunofusions: More of the Good, Less of the Bad
2.5. Human RNases with Therapeutic Potential
2.5.1. Human Pancreatic RNase or RNase 1
2.5.2. Eosinophil-Derived Neurotoxin or RNase 2
2.5.3. Angiogenin or RNase 5
2.6. RNase Inhibitors Limit ImmunoRNase Potency
3. Angiogenin Mutants and Inhibition by RI/RNH1
RNase Mutants/Variant Designed to Be Resistant to RI
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Blythman, H.E.; Casellas, P.; Gros, O.; Gros, P.; Jansen, F.K.; Paolucci, F.; Pau, B.; Vidal, H. Immunotoxins: Hybrid molecules of monoclonal antibodies and a toxin subunit specifically kill tumour cells. Nature 1981, 290, 145–146. [Google Scholar] [CrossRef] [PubMed]
- De Lorenzo, C.; Arciello, A.; Cozzolino, R.; Palmer, D.B.; Laccetti, P.; Piccoli, R.; D’Alessio, G. A fully human antitumor immunoRNase selective for ErbB-2-positive carcinomas. Cancer Res. 2004, 64, 4870–4874. [Google Scholar] [CrossRef] [PubMed]
- Gorczyca, W.; Gong, J.; Ardelt, B.; Traganos, F.; Darzynkiewicz, Z. The cell cycle related differences in susceptibility of HL-60 cells to apoptosis induced by various antitumor agents. Cancer Res. 1993, 53, 3186–3192. [Google Scholar] [PubMed]
- Igney, F.H.; Krammer, P.H. Immune escape of tumors: Apoptosis resistance and tumor counterattack. J. Leukocyte Biol. 2002, 71, 907–920. [Google Scholar] [PubMed]
- Shah, M.A.; Schwartz, G.K. Cell cycle-mediated drug resistance. Clin. Cancer Res. 2001, 7, 2168–2181. [Google Scholar] [PubMed]
- Rybak, S.M.; Saxena, S.; Ackerman, E.; Youle, R. Cytotoxic potential of ribonuclease and ribonuclease hybrid proteins. J. Biol. Chem. 1991, 266, 21202–21207. [Google Scholar] [PubMed]
- Benito, A.; Ribó, M.; Vilanova, M. On the track of antitumour ribonucleases. Mol. BioSyst. 2005, 1, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, P.; Emara, M.M.; Villen, J.; Gygi, S.P.; Anderson, P. Angiogenin-induced tRNA fragments inhibit translation initiation. Mol. Cell 2011, 43, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Deonarain, M.; Epenetos, A. Targeting enzymes for cancer therapy: Old enzymes in new roles. Br. J. Cancer 1994, 70, 786–794. [Google Scholar] [CrossRef] [PubMed]
- Krauss, J.; Arndt, M.A.; Dubel, S.; Rybak, S.M. Antibody-targeted RNase fusion proteins (immunoRNases) for cancer therapy. Curr. Pharm. Biotechnol. 2008, 9, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Schirrmann, T.; Krauss, J.; Arndt, M.A.; Rybak, S.M.; Dübel, S. Targeted therapeutic RNases (ImmunoRNases). Expert Opin. Biol. Ther. 2009, 9, 79–95. [Google Scholar] [CrossRef] [PubMed]
- Newton, D.; Ilercil, O.; Laske, D.; Oldfield, E.; Rybak, S.; Youle, R. Cytotoxic ribonuclease chimeras. Targeted tumoricidal activity in vitro and in vivo. J. Biol. Chem. 1992, 267, 19572–19578. [Google Scholar] [PubMed]
- Deonarain, M.; Epenetos, A. Design, characterization and anti-tumour cytotoxicity of a panel of recombinant, mammalian ribonuclease-based immunotoxins. Br. J. Cancer 1998, 77, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Edelweiss, E.; Balandin, T.G.; Ivanova, J.L.; Lutsenko, G.V.; Leonova, O.G.; Popenko, V.I.; Sapozhnikov, A.M.; Deyev, S.M. Barnase as a new therapeutic agent triggering apoptosis in human cancer cells. PLoS ONE 2008, 3, e2434. [Google Scholar] [CrossRef] [PubMed]
- Ardelt, W.; Shogen, K.; Darzynkiewicz, Z. Onconase and amphinase, the antitumor ribonucleases from Rana pipiens oocytes. Curr. Pharm. Biotechnol. 2008, 9, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Yuki, S.; Kondo, Y.; Kato, F.; Kato, M.; Matsuo, N. Noncytotoxic ribonuclease, RNase T1, induces tumor cell death via hemagglutinating virus of Japan envelope vector. Eur. J. Biochem. 2004, 271, 3567–3572. [Google Scholar] [CrossRef] [PubMed]
- Newton, D.L.; Nicholls, P.J.; Rybak, S.M.; Youle, R.J. Expression and characterization of recombinant human eosinophil-derived neurotoxin and eosinophil-derived neurotoxin-anti-transferrin receptor sFv. J. Biol. Chem. 1994, 269, 26739–26745. [Google Scholar] [PubMed]
- Rybak, S.M.; Hoogenboom, H.R.; Meade, H.M.; Raus, J.; Schwartz, D.; Youle, R.J. Humanization of immunotoxins. Proc. Natl. Acad. Sci. USA 1992, 89, 3165–3169. [Google Scholar] [CrossRef] [PubMed]
- Ardelt, W.; Mikulski, S.M.; Shogen, K. Amino acid sequence of an anti-tumor protein from Rana pipiens oocytes and early embryos. Homology to pancreatic ribonucleases. J. Biol. Chem. 1991, 266, 245–251. [Google Scholar] [PubMed]
- Lee, I. Ranpirnase (Onconase®), a cytotoxic amphibian ribonuclease, manipulates tumour physiological parameters as a selective killer and a potential enhancer for chemotherapy and radiation in cancer therapy. Expert Opin. Biol. Ther. 2008, 8, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.J.; Chao, T.-Y.; Lavis, L.D.; Raines, R.T. Cytotoxic ribonucleases: The dichotomy of Coulombic forces. Biochemistry 2007, 46, 10308–10316. [Google Scholar] [CrossRef] [PubMed]
- Iordanov, M.S.; Ryabinina, O.P.; Wong, J.; Dinh, T.-H.; Newton, D.L.; Rybak, S.M.; Magun, B.E. Molecular determinants of apoptosis induced by the cytotoxic ribonuclease onconase: Evidence for cytotoxic mechanisms different from inhibition of protein synthesis. Cancer Res. 2000, 60, 1983–1994. [Google Scholar] [PubMed]
- Zhao, H.; Ardelt, B.; Ardelt, W.; Shogen, K.; Darzynkiewicz, Z. The cytotoxic ribonuclease onconase targets RNA interference (siRNA). Cell Cycle 2008, 7, 3258–3261. [Google Scholar] [CrossRef] [PubMed]
- Suhasini, A.N.; Sirdeshmukh, R. Onconase action on tRNA Lys3, the primer for HIV-1 reverse transcription. Biochem. Biophys. Res. Commun. 2007, 363, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Altomare, D.A.; Rybak, S.M.; Pei, J.; Maizel, J.V.; Cheung, M.; Testa, J.R.; Shogen, K. Onconase responsive genes in human mesothelioma cells: Implications for an RNA damaging therapeutic agent. BMC Cancer 2010, 10. [Google Scholar] [CrossRef] [PubMed]
- Mikulski, S.M.; Grossman, A.M.; Carter, P.W.; Shogen, K.; Costanzi, J.J. Phase-I human clinical-trial of onconase (r)(p-30 protein) administered intravenously on a weekly schedule in cancer-patients with solid tumors. Int. J. Oncol. 1993, 3, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, J.N.; Sørensen, J.B. Review on clinical trials of targeted treatments in malignant mesothelioma. Cancer Chemother. Pharmacol. 2011, 68, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kiesgen, S.; Arndt, M.A.; Körber, C.; Arnold, U.; Weber, T.; Halama, N.; Keller, A.; Bötticher, B.; Schlegelmilch, A.; Liebers, N. An EGF receptor targeting Ranpirnase-diabody fusion protein mediates potent antitumour activity in vitro and in vivo. Cancer Lett. 2015, 357, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Rybak, S.M.; Newton, D.L. Natural and engineered cytotoxic ribonucleases: Therapeutic potential. Exp. Cell Res. 1999, 253, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Arnold, U.; Ulbrich-Hofmann, R. Natural and engineered ribonucleases as potential cancer therapeutics. Biotechnol. Lett. 2006, 28, 1615–1622. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, C.D.; D’Alessio, G. From immunotoxins to immunoRNases. Curr. Pharm. Biotechnol. 2008, 9, 210–214. [Google Scholar] [CrossRef] [PubMed]
- Rybak, S.; Arndt, M.; Schirrmann, T.; Dubel, S.; Krauss, J. Ribonucleases and immunoRNases as anticancer drugs. Curr. Pharm. Des. 2009, 15, 2665–2675. [Google Scholar] [CrossRef] [PubMed]
- Mathew, M.; Verma, R.S. Humanized immunotoxins: A new generation of immunotoxins for targeted cancer therapy. Cancer Sci. 2009, 100, 1359–1365. [Google Scholar] [CrossRef] [PubMed]
- Shlyakhovenko, V. Ribonucleases in tumor growth. Exp. Oncol. 2009, 31, 127–133. [Google Scholar] [PubMed]
- Jinno, H.; Ueda, M.; Ozawa, S.; Kikuchi, K.; Ikeda, T.; Enomoto, K.; Kitajima, M. Epidermal growth factor receptor-dependent cytotoxic effect by an EGF—Ribonuclease conjugate on human cancer cell lines-A trial for less immunogenic chimeric toxin. Cancer Chemother. Pharmacol. 1996, 38, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Krauss, J.; Arndt, M.A.; Vu, B.K.; Newton, D.L.; Seeber, S.; Rybak, S.M. Efficient killing of CD22+ tumor cells by a humanized diabody–RNase fusion protein. Biochem. Biophys. Res. Commun. 2005, 331, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Balandin, T.G.; Edelweiss, E.; Andronova, N.V.; Treshalina, E.M.; Sapozhnikov, A.M.; Deyev, S.M. Antitumor activity and toxicity of anti-HER2 immunoRNase scFv 4D5-dibarnase in mice bearing human breast cancer xenografts. Investig. New Drugs 2011, 29, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Tomé-Amat, J.; Menéndez-Méndez, A.; García-Ortega, L.; Batt, C.A.; Oñaderra, M.; Martínez-del-Pozo, Á.; Gavilanes, J.G.; Lacadena, J. Production and characterization of scFvA33T1, an immunoRNase targeting colon cancer cells. FEBS J. 2012, 279, 3022–3032. [Google Scholar] [CrossRef] [PubMed]
- Buchner, J.; Pastan, I.; Brinkmann, U. A method for increasing the yield of properly folded recombinant fusion proteins: Single-chain immunotoxins from renaturation of bacterial inclusion bodies. Anal. Biochem. 1992, 205, 263–270. [Google Scholar] [CrossRef]
- Zewe, M.; Rybak, S.M.; Dübel, S.; Coy, J.F.; Welschof, M.; Newton, D.L.; Little, M. Cloning and cytotoxicity of a human pancreatic RNase immunofusion. Immunotechnology 1997, 3, 127–136. [Google Scholar] [CrossRef]
- Newton, D.L.; Pollock, D.; DiTullio, P.; Echelard, Y.; Harvey, M.; Wilburn, B.; Williams, J.; Hoogenboom, H.R.; Raus, J.C.; Meade, H.M. Antitransferrin receptor antibody-RNase fusion protein expressed in the mammary gland of transgenic mice. J. Immunol. Methods 1999, 231, 159–167. [Google Scholar] [CrossRef]
- Yoon, J.M.; Han, S.H.; Kown, O.B.; Kim, S.H.; Park, M.H.; Kim, B.K. Cloning and cytotoxicity of fusion proteins of EGF and angiogenin. Life Sci. 1999, 64, 1435–1445. [Google Scholar] [CrossRef]
- Futami, J.; Seno, M.; Ueda, M.; Tada, H.; Yamada, H. Inhibition of cell growth by a fused protein of human ribonuclease 1 and human basic fibroblast growth factor. Protein Eng. 1999, 12, 1013–1019. [Google Scholar] [CrossRef] [PubMed]
- Psarras, K.; Ueda, M.; Tanabe, M.; Kitajima, M.; Aiso, S.; Komatsu, S.; Seno, M. Targeting activated lymphocytes with an entirely human immunotoxin analogue: Human pancreatic RNase1-human IL-2 fusion. Cytokine 2000, 12, 786–790. [Google Scholar] [CrossRef] [PubMed]
- Arndt, M.A.; Krauss, J.; Vu, B.K.; Newton, D.L.; Rybak, S.M. A dimeric angiogenin immunofusion protein mediates selective toxicity toward CD22+ tumor cells. J. Immunother. 2005, 28, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Menzel, C.; Schirrmann, T.; Konthur, Z.; Jostock, T.; Dübel, S. Human antibody RNase fusion protein targeting CD30+ lymphomas. Blood 2008, 111, 3830–3837. [Google Scholar] [CrossRef] [PubMed]
- Cremer, C.; Braun, H.; Mladenov, R.; Schenke, L.; Cong, X.; Jost, E.; Brummendorf, T.H.; Fischer, R.; Carloni, P.; Barth, S.; et al. Novel angiogenin mutants with increased cytotoxicity enhance the depletion of pro-inflammatory macrophages and leukemia cells ex vivo. Cancer Immunol. Immunother. 2015, 64, 1575–1586. [Google Scholar] [CrossRef] [PubMed]
- Cremer, C.; Vierbuchen, T.; Hein, L.; Fischer, R.; Barth, S.; Nachreiner, T. Angiogenin mutants as novel effector molecules for the generation of fusion proteins with increased cytotoxic potential. J. Immunother. 2015, 38, 85–95. [Google Scholar] [CrossRef] [PubMed]
- D’Avino, C.; Palmieri, D.; Braddom, A.; Zanesi, N.; James, C.; Cole, S.; Salvatore, F.; Croce, C.M.; de Lorenzo, C. A novel fully human anti-NCL immunoRNase for triple-negative breast cancer therapy. Oncotarget 2016, 7, 87016–87030. [Google Scholar] [CrossRef] [PubMed]
- Newton, D.L.; Hansen, H.J.; Liu, H.; Ruby, D.; Iordanov, M.S.; Magun, B.E.; Goldenberg, D.M.; Rybak, S.M. Specifically targeting the CD22 receptor of human B-cell lymphomas with RNA damaging agents. Crit. Rev. Oncol. Hematol. 2001, 39, 79–86. [Google Scholar] [CrossRef]
- Weber, T.; Mavratzas, A.; Kiesgen, S.; Haase, S.; Bötticher, B.; Exner, E.; Mier, W.; Grosse-Hovest, L.; Jäger, D.; Arndt, M.A. A humanized anti-CD22-onconase antibody-drug conjugate mediates highly potent destruction of targeted tumor cells. J. Immunol. Res. 2015, 2015, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Borrebaeck, C.A.; Malmborg, A.-C.; Ohlin, M. Does endogenous glycosylation prevent the use of mouse monoclonal antibodies as cancer therapeutics? Immunol. Today 1993, 14, 477–479. [Google Scholar] [CrossRef]
- Hristodorov, D.; Nordlohne, J.; Mladenov, R.; Huhn, M.; Fischer, R.; Thepen, T.; Barth, S. Human microtubule-associated protein tau mediates targeted killing of CD30(+) lymphoma cells in vitro and inhibits tumour growth in vivo. Br. J. Haematol. 2014, 164, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Brehm, H.; Hristodorov, D.; Pardo, A.; Mladenov, R.; Niesen, J.; Fischer, R.; Tur, M.K.; Barth, S. Targeted killing of rhabdomyosarcoma cells by a MAP-based human cytolytic fusion protein. Cancer Lett. 2015, 365, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Brehm, H.; Niesen, J.; Mladenov, R.; Stein, C.; Pardo, A.; Fey, G.; Helfrich, W.; Fischer, R.; Gattenlohner, S.; Barth, S. A CSPG4-specific immunotoxin kills rhabdomyosarcoma cells and binds to primary tumor tissues. Cancer Lett. 2014, 352, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Amoury, M.; Mladenov, R.; Nachreiner, T.; Pham, A.T.; Hristodorov, D.; di Fiore, S.; Helfrich, W.; Pardo, A.; Fey, G.; Schwenkert, M.; et al. A novel approach for targeted elimination of CSPG4-positive triple-negative breast cancer cells using a MAP tau-based fusion protein. Int. J. Cancer 2016, 139, 916–927. [Google Scholar] [CrossRef] [PubMed]
- Lord, S.J.; Rajotte, R.V.; Korbutt, G.S.; Bleackley, R.C. Granzyme B: A natural born killer. Immunol. Rev. 2003, 193, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Stahnke, B.; Thepen, T.; Stocker, M.; Rosinke, R.; Jost, E.; Fischer, R.; Tur, M.K.; Barth, S. Granzyme B-H22(scFv), a human immunotoxin targeting CD64 in acute myeloid leukemia of monocytic subtypes. Mol. Cancer Ther. 2008, 7, 2924–2932. [Google Scholar] [CrossRef] [PubMed]
- Weickmann, J.; Glitz, D. Human ribonucleases. Quantitation of pancreatic-like enzymes in serum, urine, and organ preparations. J. Biol. Chem. 1982, 257, 8705–8710. [Google Scholar] [PubMed]
- Beintema, J.J.; Wietzes, P.; Weickmann, J.L.; Glitz, D.G. The amino acid sequence of human pancreatic ribonuclease. Anal. Biochem. 1984, 136, 48–64. [Google Scholar] [CrossRef]
- Weickmann, J.L.; Elson, M.; Glitz, D.G. Purification and characterization of human pancreatic ribonuclease. Biochemistry 1981, 20, 1272–1278. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, S.; Naddeo, M.; Russo, A.; D’Alessio, G. Degradation of double-stranded RNA by human pancreatic ribonuclease: Crucial role of noncatalytic basic amino acid residues. Biochemistry 2003, 42, 10182–10190. [Google Scholar] [CrossRef] [PubMed]
- Ribó, M.; Benito, A.; Canals, A.; Nogués, M.V.; Cuchillo, C.M.; Vilanova, M. Purification of engineered human pancreatic ribonuclease. Methods Enzymol. 2001, 341, 221–234. [Google Scholar] [PubMed]
- Potenza, N.; Salvatore, V.; Migliozzi, A.; Martone, V.; Nobile, V.; Russo, A. Hybridase activity of human ribonuclease-1 revealed by a real-time fluorometric assay. Nucleic Acids Res. 2006, 34, 2906–2913. [Google Scholar] [CrossRef] [PubMed]
- Durack, D.T.; SAckerman, J.; Loegering, D.A.; Gleich, G.J. Purification of human eosinophil-derived neurotoxin. Proc. Natl. Acad. Sci. USA 1981, 78, 5165–5169. [Google Scholar] [CrossRef] [PubMed]
- Gleich, G.J.; Loegering, D.A.; Bell, M.P.; Checkel, J.L.; Ackerman, S.J.; McKean, D.J. Biochemical and functional similarities between human eosinophil-derived neurotoxin and eosinophil cationic protein: Homology with ribonuclease. Proc. Natl. Acad. Sci. USA 1986, 83, 3146–3150. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, H. Eosinophil-derived neurotoxin/RNase 2: Connecting the past, the present and the future. Curr. Pharm. Biotechnol. 2008, 9, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Slifman, N.; Loegering, D.; McKean, D.; Gleich, G. Ribonuclease activity associated with human eosinophil-derived neurotoxin and eosinophil cationic protein. J. Immunol. 1986, 137, 2913–2917. [Google Scholar] [PubMed]
- Domachowske, J.B.; Dyer, K.D.; Bonville, C.A.; Rosenberg, H.F. Recombinant human eosinophil-derived neurotoxin/RNase 2 functions as an effective antiviral agent against respiratory syncytial virus. J. Infect. Dis. 1998, 177, 1458–1464. [Google Scholar] [CrossRef] [PubMed]
- Daniels, T.R.; Delgado, T.; Helguera, G.; Penichet, M.L. The transferrin receptor part II: Targeted delivery of therapeutic agents into cancer cells. Clin. Immunol. 2006, 121, 159–176. [Google Scholar] [CrossRef] [PubMed]
- Vallee, B.; Riordan, J.; Lobb, R.; Higachi, N.; Fett, J.; Crossley, G.; Bühler, R.; Budzik, G.; Breddam, K.; Bethune, J. Tumor-derived angiogenesis factors from rat Walker 256 carcinoma: An experimental investigation and review. Cell. Mol. Life Sci. 1985, 41, 1–15. [Google Scholar] [CrossRef]
- Yang, X.-L.; Schimmel, P. Functional expansion of the tRNA world under stress. Mol. Cell 2011, 43, 500–502. [Google Scholar] [CrossRef] [PubMed]
- Strydom, D.J.; Fett, J.W.; Lobb, R.R.; Alderman, E.M.; Bethune, J.L.; Riordan, J.F.; Vallee, B.L. Amino acid sequence of human tumor derived angiogenin. Biochemistry 1985, 24, 5486–5494. [Google Scholar] [CrossRef] [PubMed]
- Saxena, S.; Rybak, S.; Davey, R.; Youle, R.; Ackerman, E. Angiogenin is a cytotoxic, tRNA-specific ribonuclease in the RNase A superfamily. J. Biol. Chem. 1992, 267, 21982–21986. [Google Scholar] [PubMed]
- Wiedłocha, A. Following angiogenin during angiogenesis: A journey from the cell surface to the nucleolus. Arch. Immunol. Ther. Exp. 1999, 47, 299–305. [Google Scholar]
- Hu, G.; Xu, C.; Riordan, J.F. Human angiogenin is rapidly translocated to the nucleus of human umbilical vein endothelial cells and binds to DNA. J. Cell. Biochem. 2000, 76, 452–462. [Google Scholar] [CrossRef]
- Tsuji, T.; Sun, Y.; Kishimoto, K.; Olson, K.A.; Liu, S.; Hirukawa, S.; Hu, G. Angiogenin is translocated to the nucleus of HeLa cells and is involved in ribosomal RNA transcription and cell proliferation. Cancer Rese. 2005, 65, 1352–1360. [Google Scholar] [CrossRef] [PubMed]
- Olson, K.A.; Fett, J.W.; French, T.C.; Key, M.E.; Vallee, B.L. Angiogenin antagonists prevent tumor growth in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 442–446. [Google Scholar] [CrossRef] [PubMed]
- St Clair, D.K.; Rybak, S.M.; Riordan, J.F.; Vallee, B.L. Angiogenin abolishes cell-free protein synthesis by specific ribonucleolytic inactivation of ribosomes. Proc. Natl. Acad. Sci. USA 1987, 84, 8330–8334. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.R.; Collins, K. Starvation-induced cleavage of the tRNA anticodon loop in Tetrahymena thermophila. J. Biol. Chem. 2005, 280, 42744–42749. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, L.-C.; Lin, S.-I.; Kuo, H.-F.; Chiou, T.-J. Abundance of tRNA-derived small RNAs in phosphate-starved Arabidopsis roots. Plant Signal. Behav. 2010, 5, 537–539. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Feng, J.; Liu, Q.; Sun, F.; Tie, Y.; Zhu, J.; Xing, R.; Sun, Z.; Zheng, X. Stress induces tRNA cleavage by angiogenin in mammalian cells. FEBS Lett. 2009, 583, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Gresch, G.; Schenke, L.; Mladenov, R.; Zwirner, S.; Cremer, C.; Niesen, J.; Grieger, E.; Brümmendorf, T.; Jost, E.; Fischer, R. Elimination of different leukaemia subtypes using novel CD89-specific human cytolytic fusion proteins. Br. J. Haematol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Schirrmann, T.; Frenzel, A.; Linden, L.; Stelte-Ludwig, B.; Willuda, J.; Harrenga, A.; Dübel, S.; Müller-Tiemann, B.; Trautwein, M. Evaluation of human pancreatic RNase as effector molecule in a therapeutic antibody platform. MAbs 2014, 6, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Dickson, K.A.; Haigis, M.C.; Raines, R.T. Ribonuclease inhibitor: Structure and function. Prog. Nucleic Acid Res. Mol. Biol. 2005, 80, 349–374. [Google Scholar] [PubMed]
- Haigis, M.C.; Kurten, E.L.; Raines, R.T. Ribonuclease inhibitor as an intracellular sentry. Nucleic Acids Res. 2003, 31, 1024–1032. [Google Scholar] [CrossRef] [PubMed]
- Castro, J.; Ribó, M.; Benito, A.; Vilanova, M. Approaches to Endow Ribonucleases with Antitumor Activity: Lessons Learned from the Native Cytotoxic Ribonucleases, in Anti-cancer Drugs-Nature, Synthesis and Cell. InTech 2016. [Google Scholar] [CrossRef]
- Boix, E.; Wu, Y.; Vasandani, V.M.; Saxena, S.K.; Ardelt, W.; Ladner, J.; Youle, R.J. Role of the N terminus in RNase A homologues: Differences in catalytic activity, ribonuclease inhibitor interaction and cytotoxicity. J. Mol. Biol. 1996, 257, 992–1007. [Google Scholar] [CrossRef] [PubMed]
- Murthy, B.S.; de Lorenzo, C.; Piccoli, R.; D’Alessio, G.; Sirdeshmukh, R. Effects of protein RNase inhibitor and substrate on the quaternary structures of bovine seminal RNase. Biochemistry 1996, 35, 3880–3885. [Google Scholar] [CrossRef] [PubMed]
- Leland, P.A.; Schultz, L.W.; Kim, B.-M.; Raines, R.T. Ribonuclease A variants with potent cytotoxic activity. Proc. Natl. Acad. Sci. USA 1998, 95, 10407–10412. [Google Scholar] [CrossRef] [PubMed]
- Strong, L.E.; Kink, J.A.; Mei, B.; Shahan, M.N.; Raines, R.T. First-in-human phase I clinical trial of QBI-139, a human ribonuclease variant, in solid tumors. Am. Soc. Clin. Oncol. 2012. [Google Scholar] [CrossRef]
- Russo, N.; Shapiro, R.; Acharya, K.R.; Riordan, J.F.; Vallee, B.L. Role of glutamine-117 in the ribonucleolytic activity of human angiogenin. Proc. Natl. Acad. Sci. USA 1994, 91, 2920–2924. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, R.; Vallee, B.L. Human placental ribonuclease inhibitor abolishes both angiogenic and ribonucleolytic activities of angiogenin. Proc. Natl. Acad. Sci. USA 1987, 84, 2238–2241. [Google Scholar] [CrossRef] [PubMed]
- Dickson, K.A.; Kang, D.K.; Kwon, Y.S.; Kim, J.C.; Leland, P.A.; Kim, B.M.; Chang, S.I.; Raines, R.T. Ribonuclease inhibitor regulates neovascularization by human angiogenin. Biochemistry 2009, 48, 3804–3806. [Google Scholar] [CrossRef] [PubMed]
- Bochicchio, A.; Jordaan, S.; Losasso, V.; Chetty, S.; Perera, R.C.; Ippoliti, E.; Barth, S.; Carloni, P. Designing the Sniper: Improving Targeted Human Cytolytic Fusion Proteins for Anti-Cancer Therapy via Molecular Simulation. Biomedicines 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Cremer, C.; Hehmann-Titt, G.; Schiffer, S.; Melmer, G.; Carloni, P.; Barth, S.; Nachreiner, T. Engineered Versions of Granzyme B and Angiogenin Overcome Intrinsic Resistance to Apoptosis Mediated by Human Cytolytic Fusion Proteins, in Resistance to Immunotoxins in Cancer Therapy; Springer: Berlin, Germany, 2015; pp. 185–219. [Google Scholar]
- Cong, X.; Cremer, C.; Nachreiner, T.; Barth, S.; Carloni, P. Engineered human angiogenin mutations in the placental ribonuclease inhibitor complex for anticancer therapy: Insights from enhanced sampling simulations. Protein Sci. 2016, 25, 1451–1460. [Google Scholar] [CrossRef] [PubMed]
- Erickson, H.A.; Jund, M.D.; Pennell, C.A. Cytotoxicity of human RNase-based immunotoxins requires cytosolic access and resistance to ribonuclease inhibition. Protein Eng. Des. Sel. 2006, 19, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.R. Fusion Protein Technologies for Biopharmaceuticals: Applications and Challenges; John Wiley & Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
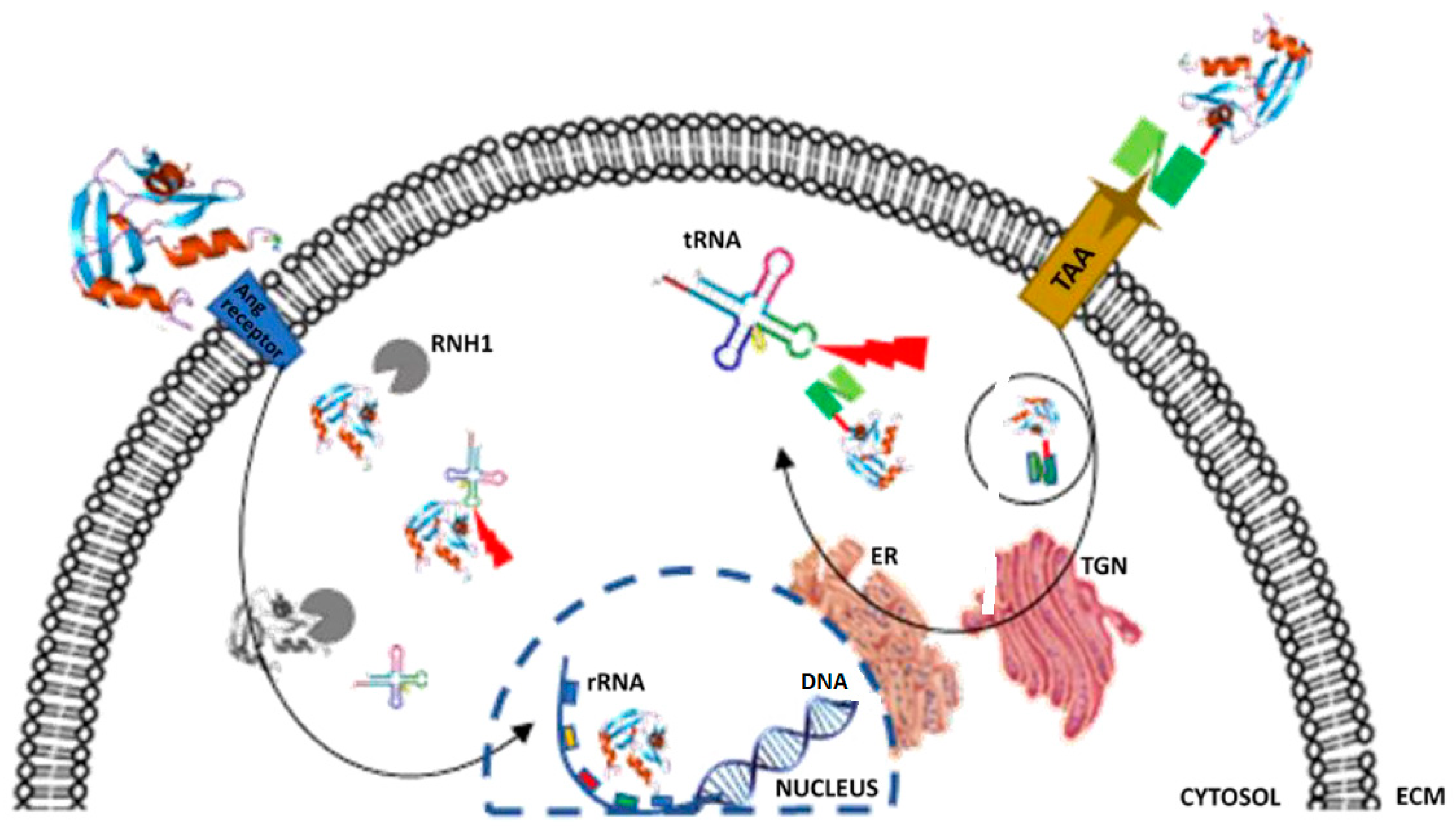

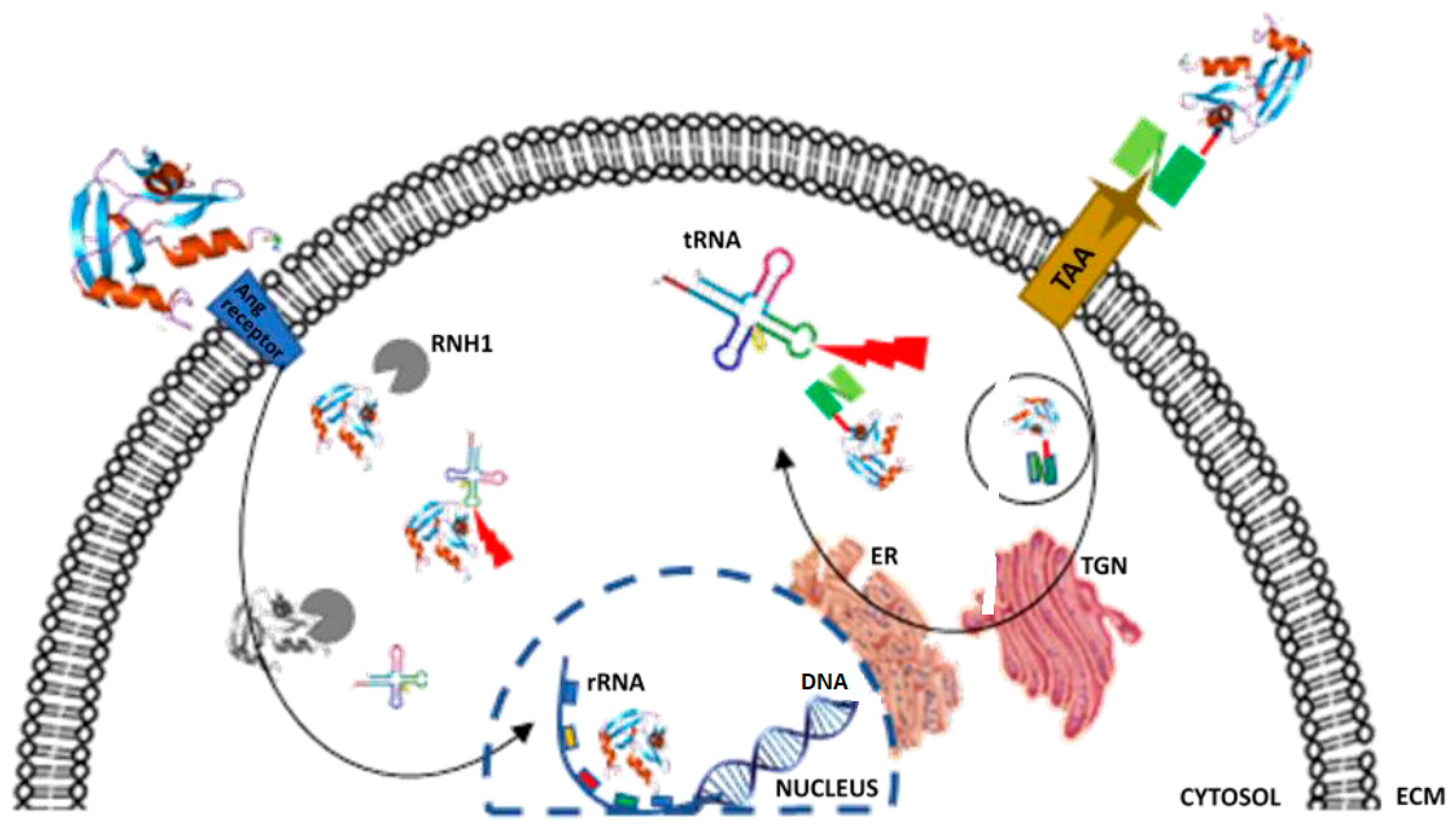
{kind=link}
{kind=link}
| ImmunoRNase ᵠ | Target Antigen | Effector RNase | Mode of Conjugation | Cancers Tested * | IC50 (nM) | Ref. |
|---|---|---|---|---|---|---|
| 454A12-RNase | Transferrin receptor | RNase A | SPDP coupling | GB, Leuk | <260 | [12] |
| 5E-9-RNase | Transferrin receptor | RNase A | SPDP coupling | GB, Leuk | <260 | [12] |
| T101-RNase | T-cell antigen CD5 | RNase A | SPDP coupling | GB, Leuk | ~120 | [12] |
| EGF-RNase | EGF receptor | RNase A | SPDP coupling | SCC, BC, SCLC | 300–1000 | [35] |
| RapLRI-SGIII(scFv) | CD22 | R.pipiens RNaseI | Rec. fusion | Burkitt’s Lym | 132–185 | [36] |
| RapLRI-SGIII (diabody) | CD22 | R.pipiens RNaseI | Rec. fusion | Burkitt’s Lym | 3–20 | [36] |
| 4D5(scFv)-dibarnase | HER2 | Barnase | Rec. fusion | BC | 2.4–4.1 | [37] |
| scFvA33T1 | GPA33 | RNase T1 | Rec. fusion | CC, PC | 300 | [38] |
| Ranpirnase- αEGFR(scFv) | EGF receptor | Ranpirnase | Rec. fusion | SCC | 120–>360 | [28] |
| ImmunoRNase | Target | Effector RNase | Mode of Conjugation Δ | Cancers Tested * | IC50 (nM) | Ref |
|---|---|---|---|---|---|---|
| EDN-sFv | Transferrin receptor | EDN | Rec. fusion | Leuk. | 0.2–1.0 | [17] |
| EDN- CD71 | Transferrin receptor | EDN | Rec. fusion | Mel, RCC, BC | 1.2–8 | [40] |
| RNase1-CD71 | Transferrin receptor | HP-RNase1 | Rec. fusion | Mel, RCC, BC | 5–10 | [40] |
| Ang-E6 | Transferrin receptor | Angiogenin | Rec. fusion | Glioma, TNBC | 15, 45 | [41] |
| EGF-Ang | EGF receptor | Angiogenin | Rec. fusion | SCC | 12.5–45 | [42] |
| CL-RFN89 | FGF receptor | HP-RNase1 | Insert. fusion | Mel | 60–460 | [43] |
| hpRNase1-hIL-2 | IL-2 receptor | HP-RNase1 | Rec. fusion | act. T lymphocytes | 20 | [44] |
| hERB2-hRNase | ErbB-2 receptor | HP-RNase1 | Rec. fusion | BC | 12.5–60 | [2] |
| MJ7(scFv)-Ang | CD22 | Angiogenin | Rec. fusion | Burkitt’s Lym | <1000 | [45] |
| MLT7(dsFv)-Ang | CD22 | Angiogenin | Rec. fusion | Burkitt’s Lym | ~100 | [45] |
| αCD30(scFv-Fc)-RNase | CD30 | HP-RNase1 | Rec. fusion | Lymphoma | 3.3 | [46] |
| H22(scFv)-Ang | CD64 | Angiogenin | Rec. fusion | Leuk, M1 macrophages | 10 ± 2.7 | [47,48] |
| 4LB5-HP-RNase | NCL | HP-RNase | Rec. fusion | TNBC | 20–70 | [49] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jordaan, S.; Akinrinmade, O.A.; Nachreiner, T.; Cremer, C.; Naran, K.; Chetty, S.; Barth, S. Updates in the Development of ImmunoRNases for the Selective Killing of Tumor Cells. Biomedicines 2018, 6, 28. https://doi.org/10.3390/biomedicines6010028
Jordaan S, Akinrinmade OA, Nachreiner T, Cremer C, Naran K, Chetty S, Barth S. Updates in the Development of ImmunoRNases for the Selective Killing of Tumor Cells. Biomedicines. 2018; 6(1):28. https://doi.org/10.3390/biomedicines6010028
Chicago/Turabian StyleJordaan, Sandra, Olusiji A. Akinrinmade, Thomas Nachreiner, Christian Cremer, Krupa Naran, Shivan Chetty, and Stefan Barth. 2018. "Updates in the Development of ImmunoRNases for the Selective Killing of Tumor Cells" Biomedicines 6, no. 1: 28. https://doi.org/10.3390/biomedicines6010028